De Novo Nucleic Acids: A Review of Synthetic Alternatives to DNA and RNA That Could Act as Bio-Information Storage Molecules †



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
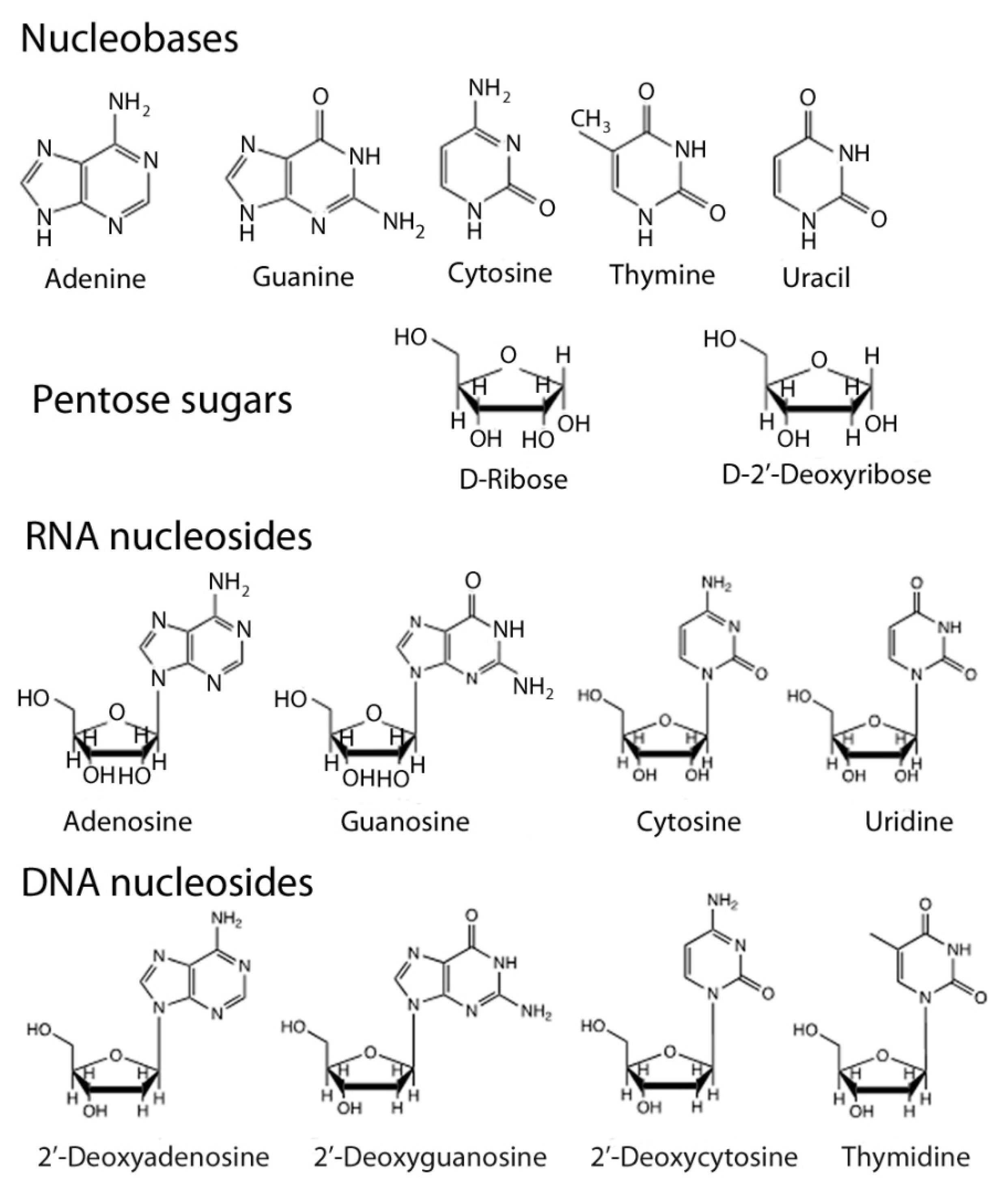
1.1. Nucleic Acid Structure
1.2. Genetic Molecular Constraints
1.3. Catalytic Nucleic Acids and the Origin of Life
1.4. Catalytic DNA Molecules: DNAzymes (Deoxyribozymes)
2. Synthetic Organic Modifications of Nucleic Acid Structure
2.1. Nucleic Acids with Uncharged Backbones
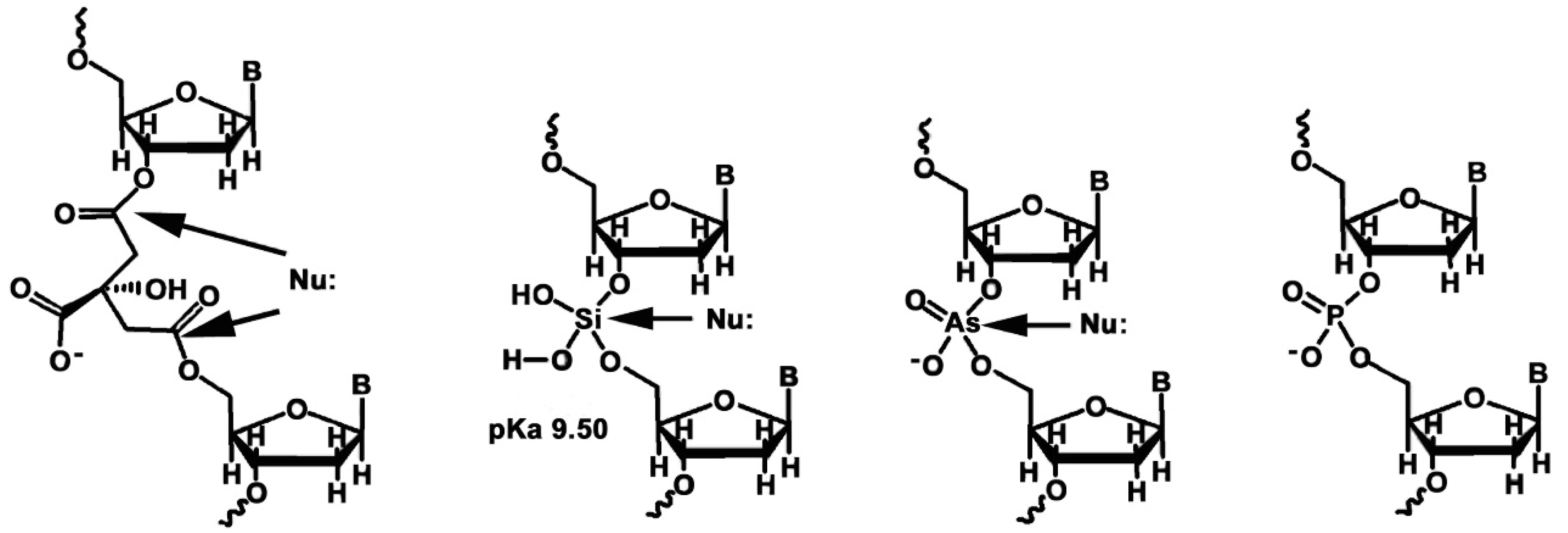
2.2. The Importance of Phosphates
2.3. Nucleic Acids Containing Acyclic and Non-Ribose Sugar-Phosphate Backbones
2.3.1. Acyclic 3′-1′-glycerol Phosphates
2.3.2. Hexose Sugar-Phosphates
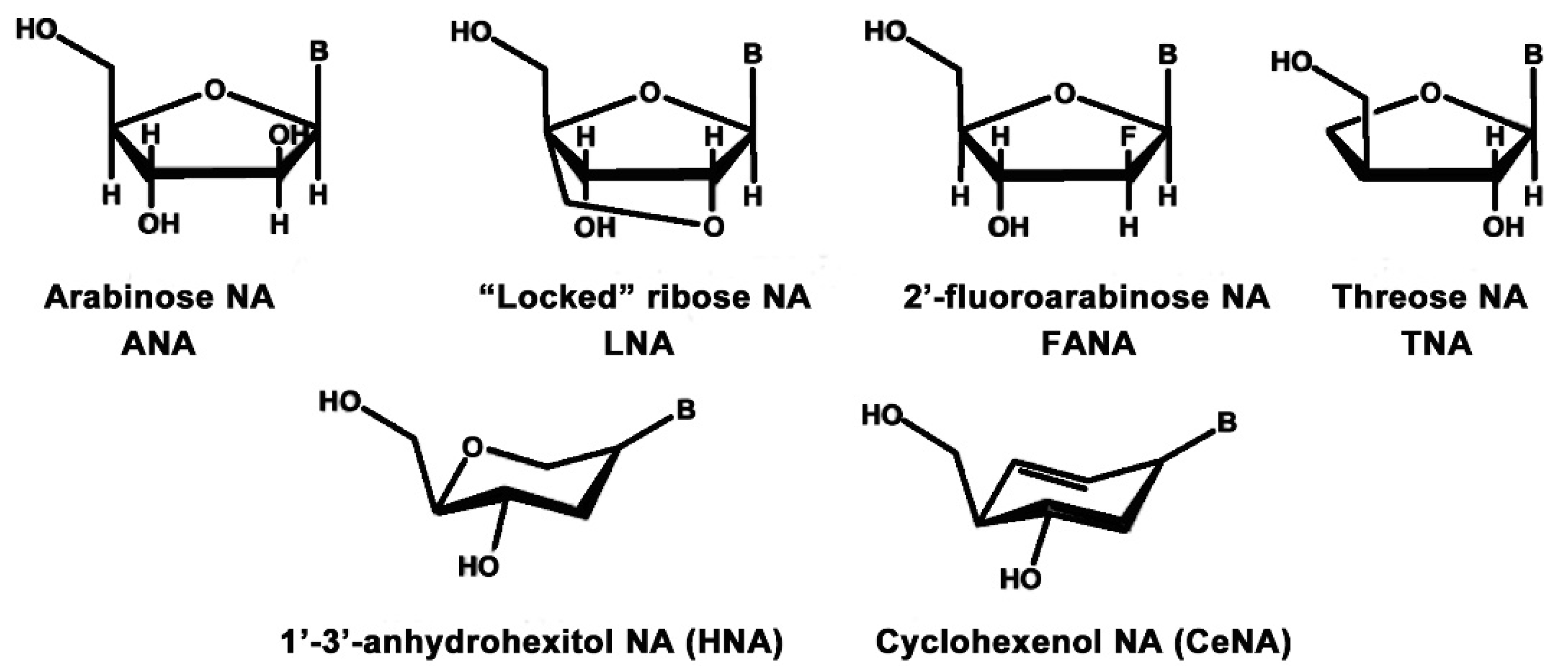
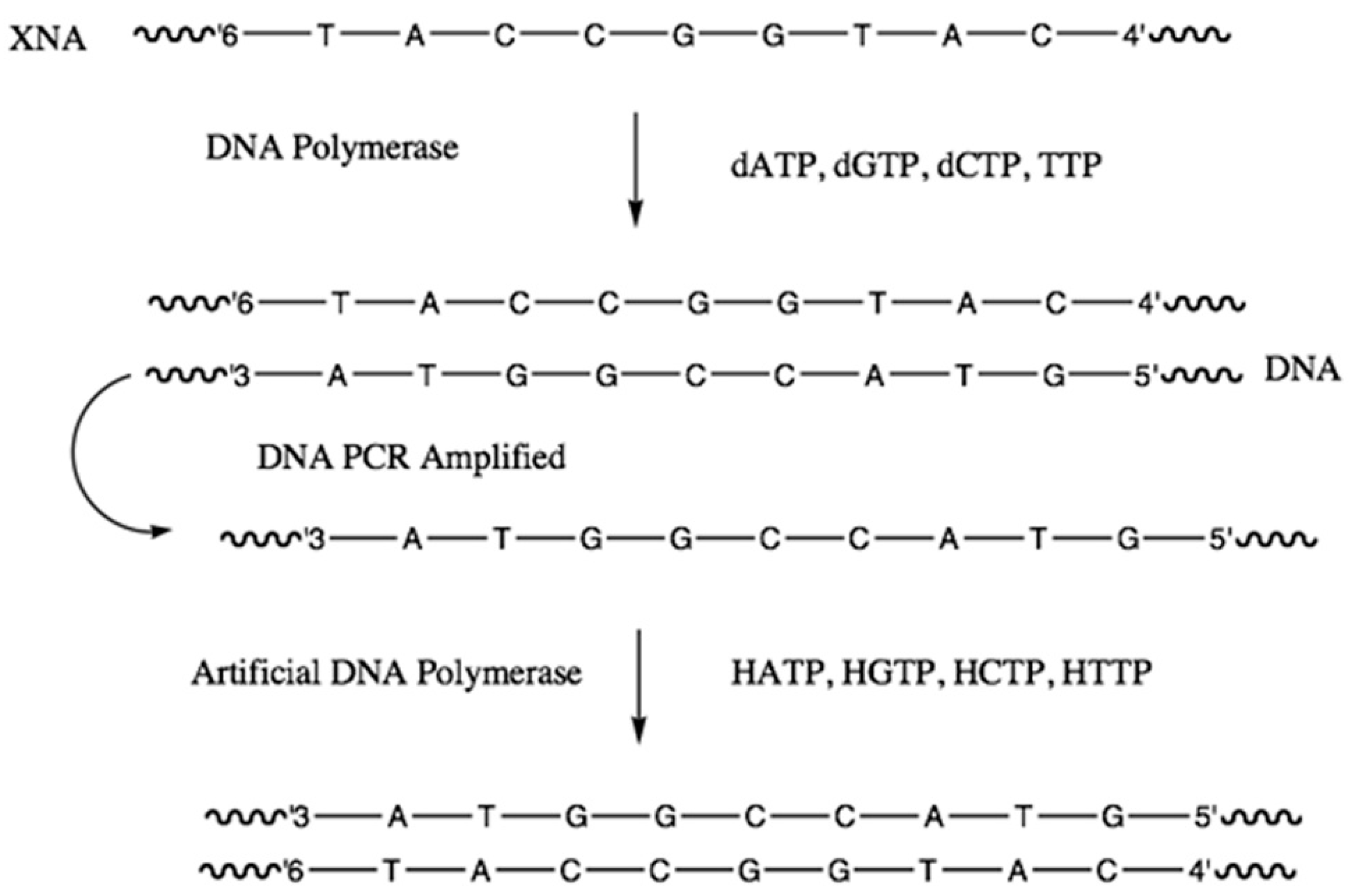
2.3.3. Alternative Ribose and Hexose-Sugar Phosphates; XNAs
2.3.4. Catalytic XNAs: Xenoribozymes (XNAzymes)
2.4. Nucleic Acids Containing Non-Standard Nucleobases
2.4.1. Nucleic Acids Containing Conventional Non-Standard Bases
2.4.2. Unconventional Non-Standard Bases
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Watson, J.D.; Crick, F.H.C. Molecular structure of Nucleic Acids. Nature 1953, 171, 737–738. [Google Scholar] [CrossRef]
- Hoogsteen, K. The crystal and molecular structure of a hydrogen-bonded complex between 1-methylthymine and 9-methyladenine. Acta Crystallogr. 1963, 16, 907–916. [Google Scholar] [CrossRef]
- Zagryadskaya, E.I.; Doyon, F.R.; Steinberg, S.V. Importance of the reverse Hoogsteen base pair 54-58 for tRNA function. Nucleic Acids Res. 2003, 31, 3946–3953. [Google Scholar] [CrossRef]
- Herbert, A. Z-DNA and Z-RNA in human disease. Nat. Commun. Biol. 2019, 2, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Benner, S.A. Size Limits of Very Small Microorganisms: Proceedings of a Workshop; Steering Group on Astrobiology of the Space Studies Board; National Research Council: Washington, DC, USA, 1999; pp. 126–135. [Google Scholar]
- Altman, S. Aspects of biochemical catalysis. Cell 1984, 36, 237–239. [Google Scholar] [CrossRef]
- Guerrier-Takada, C.; Gardiner, K.; Marsh, T.; Pace, N.; Altman, S. The RNA moiety of ribonuclease P is the catalytic subunit of the enzyme. Cell 1983, 35, 849–857. [Google Scholar] [CrossRef]
- Guerrier-Takada, C.; Altman, S. Catalytic activity of an RNA molecule prepared by transcription in vitro. Science 1984, 223, 285–286. [Google Scholar] [CrossRef]
- Zaug, A.J.; Cech, T.R. The intervening sequence RNA of Tetrahymena is an enzyme. Science 1986, 231, 470–475. [Google Scholar] [CrossRef]
- Breaker, R.R.; Joyce, G.F. A DNA enzyme that cleaves RNA. Chem. Biol. 1994, 1, 223–229. [Google Scholar] [CrossRef]
- Hollenstein, M. DNA Catalysis: The Chemical Repertoire of DNAzymes. Molecules 2015, 20, 20777–20804. [Google Scholar] [CrossRef]
- Zamecnik, P.C.; Stephenson, M.L. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc. Natl. Acad. Sci. USA 1978, 75, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Richert, C.; Roughton, A.L.; Benner, S.A. Nonionic Analogs of RNA with Dimethylene Sulfone Bridges. J. Am. Chem. Soc. 1996, 118, 4518–4531. [Google Scholar] [CrossRef]
- Nielsen, P.; Egholm, M.; Berg, R.; Buchardt, O. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science 1991, 254, 1497–1500. [Google Scholar] [CrossRef] [PubMed]
- Egholm, M.; Buchardt, O.; Nielsen, P.E.; Berg, R.H. Peptide nucleic acids (PNA). Oligonucleotide analogs with an achiral peptide backbone. J. Am. Chem. Soc. 1992, 114, 1895–1897. [Google Scholar] [CrossRef]
- Rasmussen, H.; Kastrup, J.S.; Nielsen, J.N.; Nielsen, J.M.; Nielsen, P.E. Crystal Structure of a peptide nucleic acid (PNA) duplex at 1.7 Å resolution. Nat. Struct. Biol. 1997, 4, 98–101. [Google Scholar] [CrossRef]
- Brown, S.C.; Thomson, S.A.; Veal, J.M.; Davis, D.G. NMR solution structure of a peptide nucleic acid complexed with RNA. Science 1994, 265, 777–780. [Google Scholar] [CrossRef]
- Eriksson, M.; Nielsen, P.E. PNA-nucleic acid complexes. Structure, stability and dynamics. Nat. Struct. Biol. 1996, 3, 410–413. [Google Scholar] [CrossRef]
- Westheimer, F.H. Why nature chose phosphates. Science 1987, 235, 1173–1178. [Google Scholar] [CrossRef]
- Schneider, K.C.; Benner, S.A. Oligonucleotides containing flexible nucleoside analogs. J. Am. Chem. Soc. 1990, 112, 453–455. [Google Scholar] [CrossRef]
- Eschenmoser, A.; Loewenthal, E. Chemistry of potentially prebiological natural products. Chem. Soc. Rev. 1992, 21, 1–16. [Google Scholar] [CrossRef]
- Pitsch, S.; Wendeborn, S.; Jaun, B.; Eschenmoser, A. Why Pentose-and Not Hexose-Nucleic Acids? Part VII. Pyranosyl-RNA (‘p-RNA’). Preliminary communication. Helvitica Chim. Acta 1993, 76, 2161–2183. [Google Scholar] [CrossRef]
- Eschenmoser, A. Towards a chemical etiology of the structure of nucleic acids. Chem. Biol. 1994, 1. [Google Scholar] [CrossRef]
- Egli, M.; Pallan, P.S.; Pattanayek, R.; Wilds, C.J.; Lubini, P.; Minasov, G.; Dobler, M.; Leumann, C.J.; Eschenmoser, A. Crystal structure of homo-DNA and nature’s choice of pentose over hexose in the genetic system. J. Am. Chem. Soc. 2006, 128, 10847–10856. [Google Scholar] [CrossRef] [PubMed]
- Egli, M.; Lubini, P.; Pallan, P.S. The long and winding road to the structure of homo-DNA. Chem. Soc. Rev. 2007, 36, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, V.B.; Taylor, A.I.; Cozens, C.; Abramov, M.; Renders, M.; Zhang, S.; Chaput, J.C.; Wengel, J.; Peak-Chew, S.-Y.; McLaughlin, S.H.; et al. Synthetic Genetic Polymers Capable of Heredity and Evolution. Science 2012, 336, 341–344. [Google Scholar] [CrossRef]
- Van, A.A.; Verheggen, I.; Hendrix, C.; Herdewijn, P. 1,5-Anhydrohexitol Nucleic Acids, a New Promising Antisense Construct. Angew. Chem. Int. Ed. 1995, 34, 1338–1339. [Google Scholar] [CrossRef]
- Mullis, K.; Faloona, F.; Scharf, S.; Saiki, R.; Horn, G.; Erlich, H. Specific Enzymatic Amplification of DNA In Vitro: The Polymerase Chain Reaction. Cold Spring Harb. Symp. Quant. Biol. 1986, 51, 263–273. [Google Scholar] [CrossRef]
- Arnold, F.H. Directed evolution: Creating biocatalysts for the future. Chem. Eng. Sci. 1996, 51, 5091–5102. [Google Scholar] [CrossRef]
- Taylor, A.I.; Pinheiro, V.B.; Smola, M.J.; Morgunov, A.S.; Peak-Chew, S.; Cozens, C.; Weeks, K.M.; Herdewijn, P.; Holliger, P. Catalysts from synthetic genetic polymers. Nature 2015, 518, 427–430. [Google Scholar] [CrossRef]
- Collins, M.L.; Irvine, B.; Tyner, D.; Fine, E.; Zayati, C.; Chang, C.-A.; Horn, T.; Ahle, D.; Detmer, J.; Shen, L.-P.; et al. A branched DNA signal amplification assay for quantification of nucleic acid targets below 100 molecules/ml. Nucleic Acids Res. 1997, 25, 2979–2984. [Google Scholar] [CrossRef]
- Gleaves, C.A.; Welle, J.; Campbell, M.; Elbeik, T.; Ng, V.; Taylor, P.E.; Kuramoto, K.; Aceituno, S.; Lewalski, E.; Joppa, B.; et al. Multicenter evaluation of the Bayer VERSANT™ HIV-1 RNA 3.0 assay: Analytical and clinical performance. J. Clin. Virol. 2002, 25, 205–216. [Google Scholar] [CrossRef]
- Elbeik, T.; Surtihadi, J.; Destree, M.; Gorlin, J.; Holodniy, M.; Jortani, S.A.; Kuramoto, K.; Ng, V.; Valdes, R.; Valsamakis, A.; et al. Multicenter Evaluation of the Performance Characteristics of the Bayer VERSANT HCV RNA 3.0 Assay (bDNA). J. Clin. Microbiol. 2004, 42, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Glushakova, L.G.; Sharma, N.; Hoshiko, S.; Bradley, A.; Bradley, K.M.; Yang, Z.; Benner, S.A. Detecting respiratory viral RNA using expanded genetic alphabets and self-avoiding DNA. Anal. Biochem. 2015, 489, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Benner, S.A.; Kim, H.-J.; Yang, Z. RNA Worlds: From Life’s Origins to Diversity in Gene Regulation; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2011. [Google Scholar]
- Benner, S.A.; Hoshika, S.; Leal, N.A.; Kim, M.-J.; Kim, M.-S.; Kim, H.-J.; Karalkar, N.B.; Bates, A.M.; Watkins, N.E., Jr.; SantaLucia, H.A.; et al. Hachimoji DNA and RNA: A genetic system with eight building blocks. Science 2019, 363, 884–887. [Google Scholar]
- Benner, S.A.; Hoshika, S.; Singh, I.; Switzer, C.; Molt, R.W.; Leal, N.A.; Kim, M.-J.; Kim, M.-S.; Kim, H.-J.; Georgiadis, M.M. “Skinny” and “Fat” DNA: Two New Double Helices. J. Am. Chem. Soc. 2018. [Google Scholar] [CrossRef]
- Schweitzer, B.A.; Kool, E.T. Aromatic Nonpolar Nucleosides as Hydrophobic Isosteres of Pyrimidines and Purine Nucleosides. J. Org. Chem. 1994, 59, 7238–7242. [Google Scholar] [CrossRef]
- Dunitz, J.D.; Taylor, R. Organic Fluorine Hardly Ever Accepts Hydrogen Bonds. Chem. A Eur. J. 1997, 3, 89–98. [Google Scholar] [CrossRef]
- Liu, D.; Moran, S.; Kool, E.T. Bi-stranded, multisite replication of a base pair between difluorotoluene and adenine: Confirmation by ‘inverse’ sequencing. Chem. Biol. 1997, 4, 919–926. [Google Scholar] [CrossRef]
- Evans, T.A.; Seddon, K.R. Hydrogen bonding in DNA—A return to the status quo. Chem. Commun. 1997, 21, 2023–2024. [Google Scholar] [CrossRef]
- Kool, E.T.; Sintim, O.H. The difluorotoluene debate—A decade later. Chem. Commun. 2006, 35, 3665–3675. [Google Scholar] [CrossRef]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Biochemistry, 5th ed.; W.H. Freeman: New York, NY, USA, 2002; 751p. [Google Scholar]
- Morales, J.C.; Kool, E.T. Efficient replication between non-hydrogen-bonded nucleoside shape analogs. Nat. Genet. 1998, 5, 950–954. [Google Scholar] [CrossRef] [PubMed]
- Hirao, I. Unnatural base pair systems for DNA/RNA-based biotechnology. Curr. Opin. Chem. Biol. 2006, 10, 622–627. [Google Scholar] [CrossRef] [PubMed]
- Yamashige, R.; Kimoto, M.; Takezawa, Y.; Sato, A.; Mitsui, T.; Yokoyama, S.; Hirao, I. Highly specific unnatural base pair systems as a third base pair for PCR amplification. Nucleic Acids Res. 2012, 40, 2793–2806. [Google Scholar] [CrossRef] [PubMed]
- Hirao, I.; Kimoto, M.; Lee, K.-H. DNA aptamer generation by ExSELEX using genetic alphabet expansion with a mini-hairpin DNA stabilization method. Biochimie 2018, 145, 15–21. [Google Scholar] [CrossRef]
- Ogawa, A.K.; Wu, Y.; McMinn, D.L.; Liu, J.; Schultz, P.G.; Romesberg, F.E. Efforts toward the expansion of the genetic alphabet: Information storage and replication with unnatural hydrophobic base pairs. J. Am. Chem. Soc. 2000, 122, 3274–3287. [Google Scholar] [CrossRef]
- Malyshev, D.A.; Pfaff, D.A.; Ippoliti, S.I.; Hwang, G.T.; Dwyer, T.J.; Romesberg, F.E. Solution Structure, Mechanism of Replication, and Optimization of an Unnatural Base Pair. Chem. A Eur. J. 2010, 16, 12650–12659. [Google Scholar] [CrossRef]
- Malyshev, D.A.; Dhami, K.; Lavergne, T.; Chen, T.; Dai, N.; Foster, J.M.; Correa, I.R.; Romesberg, F.E. A semi-synthetic organism with an expanded genetic alphabet. Nature 2014, 509, 385–388. [Google Scholar] [CrossRef]
- Zhang, Y.; Lamb, B.M.; Feldman, A.W.; Zhou, A.X.; Lavergne, T.; Lingjun, L.; Romesberg, F.E. A semisynthetic organism engineered for the stable expansion of the genetic alphabet. Proc. Natl. Acad. Sci. USA 2017, 114, 1317–1322. [Google Scholar] [CrossRef]
- Zhang, Y.; Ptacin, J.L.; Fischer, E.C.; Aerni, H.R.; Caffro, C.E.; San Jose, K.; Feldman, A.W.; Turner, C.R.; Romesberg, F.E. A semi-synthetic organism that stores and retrieves increased genetic information. Nature 2017, 551, 644–647. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Devine, K.G.; Jheeta, S. De Novo Nucleic Acids: A Review of Synthetic Alternatives to DNA and RNA That Could Act as Bio-Information Storage Molecules. Life 2020, 10, 346. https://doi.org/10.3390/life10120346
Devine KG, Jheeta S. De Novo Nucleic Acids: A Review of Synthetic Alternatives to DNA and RNA That Could Act as Bio-Information Storage Molecules. Life. 2020; 10(12):346. https://doi.org/10.3390/life10120346
Chicago/Turabian StyleDevine, Kevin G, and Sohan Jheeta. 2020. "De Novo Nucleic Acids: A Review of Synthetic Alternatives to DNA and RNA That Could Act as Bio-Information Storage Molecules" Life 10, no. 12: 346. https://doi.org/10.3390/life10120346
APA StyleDevine, K. G., & Jheeta, S. (2020). De Novo Nucleic Acids: A Review of Synthetic Alternatives to DNA and RNA That Could Act as Bio-Information Storage Molecules. Life, 10(12), 346. https://doi.org/10.3390/life10120346