3.1. Anticodon Preference Rules Govern Filling the Genetic Code
Amino acids appear to enter the genetic code according to ordered rules for tRNA anticodon preference. In the 2nd and 3rd anticodon positions, the preference appears to be C > G > U >> A [
1]. Therefore, 2nd and 3rd anticodon position C is favored. We posit that C is favored in the anticodon because it is a pyrimidine (a smaller base), and C forms three hydrogen bonds to G in mRNA. G and U are approximately equally favored, but amino acids with anticodon 3rd position G enter the code before amino acids with 3rd position U, indicating a preference for anticodon G over U (see below). The explanation we offer is that G forms three hydrogen bonds, but is a purine (a larger base), and U forms two hydrogen bonds but is a pyrimidine (a smaller base). A is strongly disfavored, particularly in the 3rd anticodon position. A is disfavored because it forms two hydrogen bonds and is a purine (a larger base). Of course, despite preferences, single base recognition (A, G, C, and U) was essential in the 2nd and 3rd anticodon positions to evolve a complex code. The preferences we identify for tRNA anticodons could potentially be preferences for the complementary bases in mRNA. Perhaps insight could be obtained from molecular dynamics or quantum mechanical simulations of codon-anticodon pairs on the ribosome to help judge the precise chemistry underlying these base selections.
Preferences in the anticodon wobble position appear to be G > (U/C) >>>> A. Only purine versus pyrimidine resolution is achieved for the tRNA wobble position, limiting the size of the code [
1]. This is why the genetic code is divided into mostly 4-codon sectors and 2-codon sectors, with few 1-codon sectors. In the wobble position, anticodon G reads mRNA codon C, as a Watson–Crick pair, and U, as a wobble pair. Anticodon U reads codon A, as a Watson–Crick pair, and G, as a wobble pair. Without modification, anticodon C reads mRNA codon G with reasonable fidelity. In evolution, anticodon wobble U and C are rarely or never split in coding two amino acids, as we discuss below. Anticodon wobble A is essentially never used in Archaea and Bacteria [
14,
15]. When A is modified to inosine, however, A may be encoded in tRNA in Bacteria and Eukarya.
3.2. Guidelines for Placements of Amino Acids into the Code
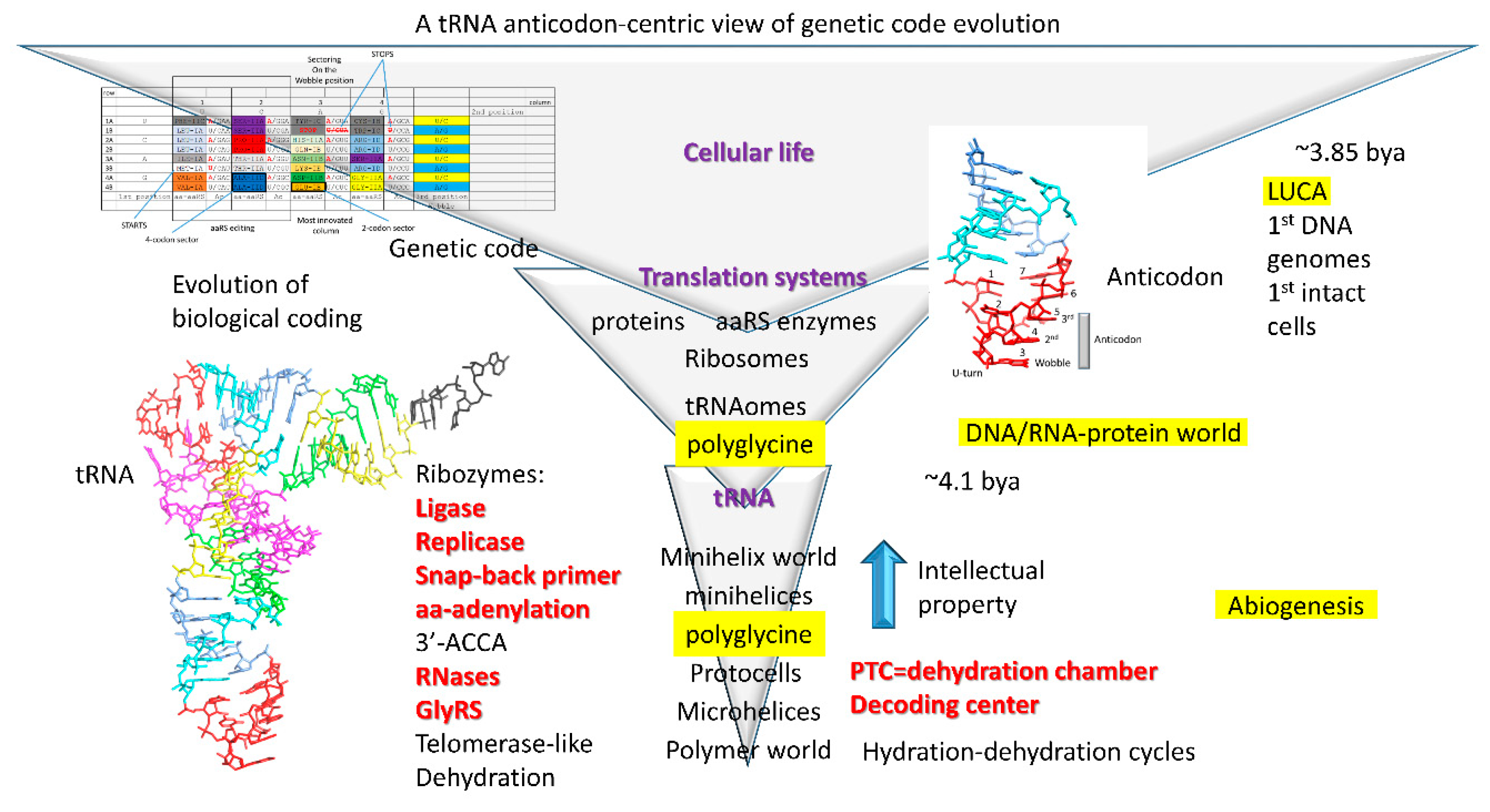
A highly detailed description of genetic code evolution is provided in this report. The working model is built on a simple set of ideas: (1) glycine was the first encoded amino acid, and at an early stage, all anticodons and all codons encoded glycine; (2) amino acids were added to the code via invasion of occupied sectors; (3) the genetic code evolved around the tRNA anticodon; (4) the final sectoring structure of the code depended strongly on the order of additions of amino acids; and (5) the code was highly structured according to its evolutionary history, and code structure was largely maintained through subsequent evolution. That glycine was the first encoded amino acid has also been hypothesized by others [
16,
17,
18]. We posit this idea mostly based on the observation that tRNA
Pri (a primordial tRNA) is closest in sequence to tRNA
Gly in ancient Archaea [
1,
3]. Also, Gly occupies what appears to be the most favored anticodon positions in the code (tRNA
Gly (GCC, UCC, CCC); favored column 4 (2nd anticodon position C); favored row 4 (3rd anticodon position C)).
We posit that Gly, Asp, Glu, Ala, and Val are among the first encoded amino acids. All of these amino acids are found in favored row 4 of the code (3rd anticodon position C). Furthermore, Phe, Tyr, Trp, and Cys appear to be among the last amino acids encoded (disfavored 3rd anticodon position A) [
19,
20]. Also, stop codons are recognized in mRNA by protein translation release factors [
21], so stop codons do not follow rules for tRNA anticodons. Phe, Tyr, Trp, Cys, and stop codons are all located in row 1 of the genetic code, indicating that row 1 (3rd anticodon position A) is disfavored. We conclude that the position of amino acids in the code reflects a Darwinian tRNA anticodon preference selection and also the order of amino acid additions to the code. The genetic code is highly structured according to its evolutionary history, and structure is demonstrated best by evolution of aminoacyl-tRNA synthetases (aaRS; i.e., GlyRS) [
1,
14]. We discuss the evidence below.
3.5. Evolution of tRNA
Because life evolved around tRNA, solving the evolution of tRNA is a core issue in the evolution of life [
1,
2,
3,
4,
13]. Fortunately, the evolution of tRNA is a simple problem. tRNA arose from ordered sequences: repeats (GCG, CGC and UAGCC) and inverted repeats (~CCGGGUUAAAAACCCGG). Despite some controversy [
23,
24,
25], the evolution of tRNA is a known and solved problem [
4]. Specifically, tRNA evolved from ligation of three 31-nt minihelices followed by 9-nt internal deletion(s). A single internal 9-nt deletion generated a type II tRNA (initially 84-nt) with an expanded variable loop. Two internal 9-nt deletions generated a type I tRNA (initially 75-nt). There are other opinions about tRNA evolution [
23,
24,
25,
26,
27,
28,
29,
30] that are inconsistent with tRNA sequences [
4]. The three minihelix model for tRNA evolution, by contrast, fully describes genesis of tRNA from the beginning of the 5’-acceptor stem to the end of the 3’-acceptor stem [
4]. The 3’-ACCA (initial sequence), to which the amino acid is bound, may have attached by ligation. The evolution of tRNA was solved as a puzzle, and this is a puzzle that anyone can solve. Because tRNA evolved from minihelices of known sequence, information is obtained about a minihelix world and a polymer world that preceded tRNA (
Figure 1).
3.6. The Minihelix and Polymer Worlds
We posit that polyglycine was generated in a pre-life world, as a cross-linking agent to stabilize protocells. Polyglycine (i.e., Gly
5) is a component of bacterial peptidoglycan cell walls [
31,
32,
33]. Gly
5 is a cross-linker connecting short peptide chains (i.e., L-Ala-D-Glu-L-Lys-D-Ala) that are linked to glycan chains (i.e., (N-acetylglucosamine-N-acetylmuramic acid)
n). Gly
5 cross-links L-Lys on one peptide chain to D-Ala on another. We posit that, in the pre-life world, an analogous peptidoglycan coat was synthesized using ribozyme catalysts and assembled on protocells. One prediction of our model is that peptidoglycan coats enhance the functions of protocells for pre-biotic chemistry.
Because aspects of the more ancient minihelix and polymer worlds are known from current tRNA sequences [
1,
3,
4,
13], what were the functions of these ancient repeats and inverted repeats? We posit that a large set of 31-nt minihelices was utilized with RNA templates of diverse sequences to synthesize polyglycine, which stabilizes cells or protocells as a component of peptidoglycan [
31,
32,
33]. In tRNA sequences, only two minihelices survive: a D loop minihelix (GCGGCGGUAGCCUAGCCUAGCCUACCGCCGC) and two copies of an anticodon/T loop minihelix (~GCGGCGGCCGGGUUAAAAACCCGGCCGCCGC). The only sequence ambiguity in the anticodon and T loop minihelices is in the ~UUAAAAA loops. There is no ambiguity in the 5’- (GCGGCGG) and 3’-acceptor stems (CCGCCGC) or in the 5-nt stems (5’ CCGGG and 3’ CCCGG). We posit that minihelices evolved to synthesize polyglycine using mixed mRNA templates, and mixed 31-nt minihelices, including some no longer known. A D loop minihelix would be expected to project a single C to recognize G in a mRNA (1-nt code). An anticodon/T loop minihelix would be expected to project ~AAA to recognize ~UUU in a mRNA (3-nt code). Although only two minihelix sequences survived in tRNAs, it is expected that many minihelices with different core sequences than those described here helped a primitive translation system to recognize different mRNA sequences. In order to attach glycine using a ribozyme GlyRS (GlyRS-RBZ), we posit that ACCA was ligated to 31-nt minihelices. A primitive decoding center scaffold and a mobile peptidyl transferase center (PTC) appear sufficient to serve as an ancient ribosome [
7].
Because minihelices were generated from GCG, CGC, and UAGCC repeating polymers, a mechanism must have existed to generate these repeats. Although there may be other possibilities, we posit that RNA repeats may have been synthesized via a telomerase-like ribozyme with a guide RNA template. These and other RNAs may have been replicated via a template-dependent ribozyme replicase (
Figure 1). A mechanism must have existed to generate RNA fragments of defined lengths (i.e., 7-nt (acceptor stems) and 17-nt (minihelix cores)). To attach fragments of RNA, a ribozyme ligase was required. A ribozyme replicase must have existed to utilize and replicate inverted repeats. A 31-nt minihelix ligated to an RNA can be utilized as a snap-back primer for replication of the complementary strand. A ribozyme GlyRS must have existed, but no other aminoacyl transferases were initially required. The system requires RNA scaffolds to act as a primitive decoding center. The system further requires a mobile peptidyl-transferase center [
34]. We posit that the primitive ribosome was approximately built on this model [
7]. Such an ancient minihelix world could be reconstructed in a laboratory and/or generated computationally to challenge these ideas. All of the requisite ribozymes have been generated or approximated through selection in vitro [
34,
35,
36,
37,
38,
39,
40,
41,
42,
43,
44]. Although not discussed here, similar hypotheses can be posited for the more ancient polymer world. It was initially a surprise to us that the central functions of life appear to have been generated from ordered, rather than random polymer sequences. Existing tRNA sequences show that life evolved, at least in part, from ordered sequences (repeats and inverted repeats).
So, we posit that the Darwinian selection for the minihelix world was primarily to synthesize polyglycine to stabilize protocells (
Figure 1). Protocells were necessary to generate membrane potentials to harness redox energy. Initially, tRNA evolved from minihelices as an improved mechanism to generate polyglycine. Evolution of tRNA eventually established the 3-nt genetic code. From this modest beginning, the genetic code evolved, driven by Darwinian selection.
3.7. The Genetic Code Has Order
A number of models have been advanced to describe the evolution of the genetic code [
6,
28,
45,
46,
47,
48,
49,
50,
51,
52]. With the exception of our model, we find these alternate opinions to be potentially flawed. Specifically, alternate models show the genetic code in terms of mRNA codons, rather than tRNA anticodons. As we have shown, however, the genetic code complexity was determined by how the tRNA anticodon was read on the ribosome. Reading of the tRNA anticodon, therefore, limited the size and complexity of the genetic code [
1,
3,
13,
14]. Sixty-four codons are recognized in mRNA (4 × 4 × 4), but, in tRNA, the maximal complexity of the genetic code is 32 anticodons (2 × 4 × 4), explaining why only 20 amino acids and stop codons were encoded. Most notably, only pyrimidine versus purine discrimination was initially permitted at the anticodon wobble position, limiting the size of the code in tRNA.
Because tRNA limits the size of the genetic code, we show the code as a 32-assignment, codon-anticodon table (
Figure 2 and
Figure 3).
Figure 2 is annotated to describe important features of the code (i.e., start and stop codons). Because of the history of code evolution, the genetic code is primarily broken into 2-codon and 4-codon sectors. Because of tRNA anticodon wobble ambiguity, separating 1-codon sectors to encode two amino acids poses a challenge. Trp (CCA) is a special case because stop codon (UGA; anticodon UCA) is recognized only as a codon in mRNA by a protein translation release factor [
21], which does not obey tRNA anticodon rules. In Archaea, Ile (CAU) and Met (CAU) anticodons co-occupy a sector and are discriminated by distinct anticodon wobble C modifications [
53,
54,
55]. In order to reduce translation errors, tRNA
Ile (UUA) anticodons are rarely used in prokaryotes. Ile (CAU) and Met (CAU) anticodons provide insight into how amino acids invaded the code. Therefore, apparent 1-codon sectors are special cases that can be ignored in the initial establishment of the code. Because the code does not divide easily into 1-codon sectors, the initial maximal code complexity must be 32-assignments as shown in
Figure 2 and
Figure 3.
3.8. History of Genetic Code Evolution is Largely Preserved in aaRS Evolution Patterns
Aminoacyl-tRNA synthetases (aaRS; i.e., GlyRS) belong to two structural classes (class I and class II; i.e., GlyRS-IIA) with many structural subclasses (A–E) (
Figure 2 and
Figure 3). Class I and class II aaRS have incompatible folds. Sometimes, structural subclasses do not adequately reflect evolutionary relatedness of aaRS enzymes, but these relationships have recently been clarified (
Figure 3A) [
1]. Class II aaRS are older than class I aaRS. GlyRS-IIA is the primordial aaRS from which all aaRS enzymes were derived. As we have shown, GlyRS-IIA is a simple sequence homolog of ValRS-IA and IleRS-IA [
1,
14]. GlyRS-IIA was refolded into (probably) ValRS-IA from which other class I aaRS enzymes radiated [
1]. Much of the structure of the genetic code and the history of genetic code evolution can be inferred from the relatedness of aaRS enzymes. The distributions of related aaRS enzymes indicate that much of the evolution of the genetic code occurred within code columns (
Figure 3C). Code columns relate to the middle position of the tRNA anticodon, which is the most important position for translational accuracy (
Figure 3B). Because the genetic code has a highly conserved structure, a history of genetic code evolution is preserved, particularly in aaRS evolution patterns.
3.9. The “Frozen Accident”
The genetic code is highly structured, indicating that a history of its evolution is recorded and preserved (
Figure 2 and
Figure 3). Conservation of structure in the code is reminiscent of Francis Crick’s “frozen accident” [
1,
45,
46,
57,
58,
59], which we interpret as a rapid evolution to an enduring form with very little subsequent movement or replacement of amino acids within the code. Our approach to describe the sectoring of the code has been explained in some detail, but in this report, we improve and extend our previous model. Notably, making a small number of simplifying assumptions, the entire sectoring of the code can be described via Darwinian selection. Although we relate events from ~4 billion years ago, we consider these detailed working models to be very useful to strengthen our understanding of the evolution of the code.
Establishment of the genetic code requires consideration of a challenging “chicken and egg” problem: that is, how do you establish the functions and structural sub-classes of aaRS enzymes before the code evolves to provide complex proteins [
1,
12]? We posit that this is part of the frozen accident. We posit that the code was sectored initially by ribozyme aaRS enzymes. Once the code was sufficiently established, protein aaRS enzymes replaced their ribozyme analogues. aaRS enzyme divergence was driven by the pressures caused by the increasing requirements for accurate coding. Such a model requires very rapid and almost irreversible establishment of the genetic code, consistent with conserved code structure (
Figure 2 and
Figure 3). We reiterate that there is very little evidence for migration of amino acids in the code. Ser and Arg may be exceptions.
Analysis of tRNAomes indicates much higher order in more ancient species such as ancient Archaea [
2]. By contrast, bacterial tRNAomes are much more derived. We posit, therefore, that tRNAomes are highly dynamic in evolution, and that much of the initial patterning of tRNAomes has been lost, as tRNAs mutate, in order for, for instance, aaRS enzymes to distinguish one tRNA from another. tRNAomes occupy a limited evolutionary space. Because tRNAs are small RNAs that must maintain their characteristic structure, tRNAomes undergo both divergent and convergent evolution within a constrained Darwinian space. Therefore, the frozen accident, which was initially established in the tRNAome, is now more apparent in aaRS sequences and structures than in tRNAomes. We posit that during genetic code sectoring, tRNAomes were more highly structured, and that, when proteins gained sufficient complexity, aaRS evolution tracked the highly ordered tRNAome. After sectoring of the code and aaRS enzymes was mostly complete, Darwinian selection drove diversification of tRNAomes to enhance translational accuracy. Consistent with this view, we observe that archaeal tRNAomes are more highly structured (and more similar to LUCA) than bacterial tRNAomes, which are more derived [
3]. We posit that the ~8→~16 amino acid transition (see below) provides proteins of sufficient complexity to evolve the patterned code.
The tRNAome is an enclosed evolutionary space because tRNAs must maintain their shape and structure. Analysis of tRNAomes indicates both convergent and divergent evolution of tRNAomes. For instance, for the most part, the
Pyrococcus furiosis (an ancient Archaea) tRNAome resembles a highly ordered LUCA tRNAome. By contrast,
Escherichia coli (a highly derived Bacterium) has a much more scrambled tRNAome with little apparent evolutionary order. As evidence of continuing tRNA evolution, we have presented evidence in Archaea for recruitment of a duplicated tRNA
Pro to become reassigned as a tRNA
Trp and a duplicated tRNA
Trp to become reassigned as a tRNA
Phe, indicating how tRNAs diverge, converge, and jump within a tRNAome [
3]. To avoid errors in coding, tRNAs sometimes jump to a neighboring column within a genetic code row, in contrast to aaRS enzymes that tend to evolve within columns (
Figure 3). We posit that initially, tRNAomes mostly evolved within columns, followed by tRNAome divergence that includes tRNA jumping to a neighboring column but often maintaining the original row.
3.11. Evolution of the Genetic Code (A Working Model)
We posit that the genetic code initially evolved primarily to synthesize polyglycine used as a cross-linking agent to stabilize protocells (
Figure 1). What this indicates is that, after evolution of tRNA, the tRNA anticodon mutated rapidly to all possible sequences. We imagine a primordial world in which tRNA can replicate and mutate rapidly. tRNA sequences other than the anticodon posed difficulties to mutate because alterations in stems or in the “elbow” (where the D loop, V loop, and T loop interact; the bend of the tRNA “L” shape) likely require compensatory mutations to stabilize the tRNA fold. After generating many anticodon mutations, all anticodons and all mRNA codons encoded glycine to synthesize polyglycine. We posit that a GlyRS ribozyme charged these tRNAs (mostly) with glycine. Mistakes in aminoacyl-tRNA charging and modifications of amino acids linked to tRNAs drove the evolution of the code.
Because tRNA has a specialized 7-nt anticodon loop, the 3-nt tRNA anticodon projects to recognize a 3-nt mRNA codon. On a primitive ribosome, reading 3-nt codons, however, presented some difficulties that can still be recognized in wobble anticodon-codon interactions (
Figure 3B). On the ribosome, the accuracy of the anticodon-codon interaction is monitored by the EF-Tu latch [
60,
61,
62,
63]. EF-Tu is a GTPase that enforces Watson–Crick geometry on the anticodon-codon interaction at the 2nd and 3rd anticodon positions. By contrast, the 1st anticodon wobble position is monitored by the latch, but, at the wobble position, only pyrimidine-purine recognition is initially achieved. The 2nd anticodon position is recognized with the highest accuracy. The 3rd anticodon position also requires Watson–Crick geometry. Rarely, in translation errors, G~U wobble pairs occur at the 3rd anticodon position, but G~U wobble pairs require tautomerization of either the anticodon or codon base to allow Watson–Crick geometry. Because of the EF-Tu latch, generally, unstable wobble pairs in the 3rd anticodon position are rejected before amino acid misincorporation.
The genetic code evolved according to these tRNA anticodon recognition rules (2nd > 3rd > 1st (wobble) anticodon positions). To generate polyglycine, little translational accuracy in reading the anticodon is initially required because all anticodons encoded Gly. As amino acids invaded the code, we posit the following. The code generally sectored first around the 2nd anticodon position (most important), followed mostly by the 3rd position (next most important), followed by the wobble position (generally least important) [
1]. Therefore, the genetic code evolved mostly around the tRNA anticodon, generating a clear working model for evolution of the code. In the model advocated here, the entire code is first populated with Gly. As other amino acids enter the code, Gly is displaced. As Gly is displaced, however, Gly retains the most-favored anticodons, based on a clear set of selection rules, and Gly gives up less favored anticodons to incoming amino acids [
1]. According to such a model, the placements of all amino acids in the genetic code can be readily described. Alternate models for populating the genetic code are more problematic. Any model that attempts to populate the code from a single sector followed by migrating into other sectors appears unlikely [
28,
64,
65]. For such an alternate model, how can a Darwinian selection be imagined for matching assignments of codons and anticodons? In our model, codon and anticodon specifications coevolve, and evolution of the tRNA anticodon drives evolution of codons. We find alternate models to make few useful predictions. By contrast, our model makes many predictions, many of which are justified.
In
Figure 4, we show a proposed order of addition for amino acids into the genetic code. We posit that the genetic code evolved from a 1-amino acid code (Gly) to a 4-amino acid code (Val, Ala, Asp, Gly) to an 8-amino acid code (Val, Leu, Ala, Pro, Asp, Glu, Arg, Gly). The selection to add amino acids to the code was to generate novel polypeptide products. Sectoring to a 4-amino acid code required single-base recognition of the 2nd anticodon position. Evolution to an 8-amino acid code required recognition of two anticodon positions, but in the absence of the EF-Tu latch [
60,
61,
62,
63], recognition of the 1st wobble base and the 3rd anticodon position were both wobbly, and only pyrimidine versus purine recognition was initially achieved. Therefore, to advance beyond an 8-amino acid code required evolution of the EF-Tu latch to confer single-base recognition at the 2nd and 3rd anticodon positions. At the 8-amino acid stage, we posit that genetic code columns 1, 2, and 4 sectored by one mechanism, and column 3 sectored by a slightly different mechanism. Columns 1, 2, and 4 sectored according to the 3rd anticodon position (2nd + 3rd anticodon positions). Column 3 sectored according to the 1st (wobble) position (2nd + 1st anticodon positions). Essentially, in order to expand the genetic code, the primitive ribosome and translation system were “learning” (teaching themselves) to read just two out of three anticodon bases. The evolutionary history of column 3, sectoring on the 1st anticodon wobble base rather than the 3rd anticodon position, caused column 3 to become the most innovated column in the code.
For the code to continue to sector requires evolution of the EF-Tu GTPase anticodon-codon latch (
Figure 4). From the 8-amino acid code, we posit that the code sectored to approximately a 16-amino acid code (i.e., ~Gly, Arg, Asp, Asn, Glu, Gln, Lys, His, Ala, Thr, Pro, Ser, Val, Ile, and Leu). This transition requires evolution of the EF-Tu latch because the 2nd and 3rd anticodon positions must be read with single-base resolution to achieve an ~16-amino acid code, and in the absence of the latch, the 3rd anticodon position is read essentially as a wobble position, with more difficulty than the 2nd position. At the ~16 amino acid stage of evolution, additional amino acids can be added, but as the system evolves enhanced translational accuracy, fidelity restrictions create an ever-more stringent barrier to encoding additional amino acids. The reason for this limitation is that amino acids are added to the code through mischarging of tRNAs followed by selection for a more complex and innovated code. Therefore, addition of amino acids to the code is positively selected, because a richer code makes encoded proteins more complex, but negatively selected because additions increase tRNA-charging and translation errors, forcing additional challenges to the system. Potentially, a 32-assignment code could have evolved (i.e., 29 amino acids + 3 stop codons). We posit that some sectors of the code are difficult to split, however, from 4-codon sectors to 2-codon sectors because these divisions cause error catastrophe, particularly for the left half of the genetic code. Hydrophobic and neutral amino acids encoded by the left half of the code have little amino acid side chain character for recognition by aaRS enzymes, so splitting a 4-codon sector into two 2-codon sectors might cause too frequent tRNA charging errors [
1,
14].
3.12. Evolution of the Genetic Code within Columns
Because genetic code columns appear to dominate evolution of the code (
Figure 2 and
Figure 3), how can this be explained? We posit that evolution of the genetic code is dominated by the tRNA anticodon. The second anticodon position, which is most important for translational accuracy, is represented by genetic code columns (
Figure 3B). Therefore, the genetic code evolved primarily within columns, rather than within rows, or according to a random distribution. Based on this reasoning, a simple, step-by-step model for genetic code evolution is proposed in
Figure 5,
Figure 6,
Figure 7,
Figure 8 and
Figure 9.
Figure 5 shows the initial evolution of the code to encode polyglycine.
Figure 6 shows the 4-amino acid code to encode Gly, Asp, Ala, and Val [
1,
6,
46,
47].
Figure 7 shows wobbly sectoring to an 8-amino acid code.
Figure 8 shows evolution to a ~16-amino acid code, after evolution of the EF-Tu latch [
60,
61,
62,
63].
Figure 9 shows evolution to the standard genetic code (21 assignments: 20-amino acids + stops).
3.13. Column 2
Column 2 of the genetic code sectored evenly into four 4-codon sectors encoding Ala, Thr, Pro, and Ser (
Figure 8 and
Figure 9). Because of early wobbly recognition of the 3rd anticodon position, however, we posit that column 2 may have initially sectored into two 8-codon sectors encoding Ala and Pro (
Figure 7). Essentially, the idea is that, as the ribosome evolves to read the 3rd anticodon position, initially only pyrimidine versus purine discrimination was possible. Only after evolution of the EF-Tu anticodon-codon latch, is single base recognition at the 3rd anticodon position achieved (
Figure 8). We posit that Ala, which we posit entered the code before Pro, protected the more favored sectors, according to the 2nd and 3rd position anticodon rules C > G > U >> A. Because the 4th row of the code is favored (3rd position C), Ala protected favored sectors from invasion by Pro. After evolution of the EF-Tu latch, the entire anticodon could be read on the ribosome. Thus, Thr could invade the Ala sector with Ala retaining the most favored rows (row 4; 3rd anticodon position C). Ser could invade the Pro sector. Amino acids that entered the code first, therefore, retained the most favored rows in the code. Because column 2 has no 2-codon sectors, column 2 has no sectoring to discriminate amino acids according to the wobble position. In part, we posit that column 2 amino acids (Ala, Thr, Pro, and Ser) have too little character to be more accurately distinguished within their aaRS active sites, limiting further innovation in column 2 from 4-codon sectors to 2-codon sectors [
1,
14]. Interestingly, ThrRS-IIA, ProRS-IIA, and SerRS-IIA are closely related aaRS enzymes by structure and sequence (
Figure 3). The similarity of these enzymes indicates evolution within genetic code column 2.
3.14. Columns 1 and 4
Columns 1 and 4 are posited to be more chaotic versions of sectoring, similar to the sectoring of column 2. Column 1 is posited to have sectored first between Val (8-codon sector) and Leu (8-codon sector) (
Figure 7). Evolution of the EF-Tu anticodon-codon latch was necessary to sector further. Val was invaded by Ile, with Val retaining the most favored sectors (row 4; 3rd anticodon position C). We posit that, prior to Met invasion, there were three tRNA
Ile anticodons (GAU, UAU, and CAU) (in Archaea, AAU is rarely or never used). Ile (CAU) was invaded by Met (CAU). To retain Met in the code, Ile (UAU) was selected against, because tRNA
Ile (UAU) potentially reads both Ile (AUA) and Met (AUG) codons. Different modification enzymes evolved to alter the wobble base of tRNA
Ile (CAU) and tRNA
Met (CAU) so these tRNAs can be accurately discriminated, specifically reading Ile (AUA) and Met (AUG) codons. At an early time in evolution (i.e., before LUCA), therefore, two tRNA
Met (CAU), 1 elongator tRNA
Met (CAU) and 1 initiator tRNA
Met (CAU), and two tRNA
Ile (GAU and CAU) were utilized [
53,
54]. There are minor alterations to this common situation in some organisms. In eukaryotes, more tRNA
Ile anticodons are utilized (UAU and IAU; I for inosine (adenine→inosine)), and additional rules apply [
14]. Phe was a late addition to the code invading a Leu sector. As a late invader, Phe is relegated to disfavored row 1 (3rd anticodon position A). In previous publications, our laboratory has somewhat confused discussion of Ile and Met sectoring. We apologize. Ile and Met sectoring is a fundamental but simple story in structuring of the code. Essentially, Met invaded a 4-codon Ile sector, but the invasion was never fully resolved and now can never be.
Interestingly, in column 1, Val, Ile, and Leu are hydrophobic amino acids. Furthermore, ValRS-IA, IleRS-IA, MetRS-IA, and LeuRS-IA are closely related enzymes by sequence and structure (
Figure 3). We posit that amino acid hydrophobicity and conserved aaRS enzymes demonstrate evolution of the genetic code within column 1. We posit, further, that evolution of the genetic code within columns relates to the central importance of tRNA anticodon position 2. Met is in some ways a special case because Met is a late addition to the code and Met utilizes two tRNA
Met (CAU) at the base of code evolution (1 elongator and 1 initiator tRNA
Met). Phe is also a late entry into the code. Phe is an aromatic and hydrophobic amino acid. Interestingly, PheRS-IIC helps to discriminate charging of tRNA
Phe (GAU) from charging by the class IA enzymes for other column 1 amino acids. All of the aaRS enzymes in column 1 have an editing active site to enhance the accuracy of tRNA charging [
56]. Editing is posited to be important for these aaRS enzymes because hydrophobic amino acids have few discriminating features within the aaRS active site for accurate charging to tRNA. To protect against charging errors, column 1 has 4-codon sectors for Val and Leu. Ile was reduced to a 3-codon sector because of invasion by Met. Met (CAU) and Ile (CAU) share a 1 codon sector, but Met is required for translation starts, and Met was a late addition to the code. Although Met invading the Ile sector was never fully resolved during code establishment, Met invading the Ile sector suggests a mechanism for the previous successful invasions of other sectors of the code by other amino acids.
Column 4 may have sectored as follows. Initially, column 4 split between Gly and Arg (8-codon sectors), with Gly occupying the favored rows 3 and 4 (
Figure 7). With evolution of the anticodon-codon EF-Tu latch, Arg invaded row 3, displacing Gly. We posit that Ser invaded column 4 to attain a better anticodon with 2nd anticodon position C (column 4; anticodon GCU). Ser was able to invade column 4 because tRNA
Ser is a type II tRNA with an expanded variable loop, which is recognized as a determinant by SerRS-IIA for accurate amino acid placement [
56]. Other tRNAs generally depend strongly on recognition of their anticodon, for amino acid placement by their aaRS, making such a jump within the genetic code impossible, without inducing error catastrophe. The Arg sector could be invaded by Ser because ArgRS-ID cannot charge type II tRNA
Ser (GCU) with Arg. Ser is the only amino acid that is split between columns (in this case columns 2 and 4) within the genetic code table. We posit that Ser is the only amino acid to have jumped successfully in evolution of the code, and that only SerRS-IIA and type II tRNA
Ser could readily support such a jump. Because ArgRS-ID is a class I aaRS, invasion of the Arg sector by SerRS-IIA may have been facilitated. In column 4, Trp, Cys and a stop codon are late entries into the genetic code that are relegated to disfavored row 1 (3rd anticodon position A). Stop codons are recognized by proteins, not tRNA [
21], so stop codons do not follow tRNA anticodon selection rules. Aromatic amino acids Phe (column 1), Tyr (column 3), and Trp (column 4) are late entries into the genetic code, all found in disfavored row 1 (3rd anticodon position A).
3.15. Column 3
Column 3 is the most innovated column in the code. We posit that the reason for such high innovation is because column 3 sectored initially on the 2nd and 1st (wobble) anticodon positions (
Figure 7). Columns 1, 2, and 4, by contrast, sectored initially on the 2nd and 3rd anticodon positions, as described above. We posit that column 3 initially sectored between Asp and Glu, with Asp utilizing 1st wobble anticodon position G, and Glu utilizing 1st wobble anticodon position U/C, creating the striped pattern. Because we posit that Asp entered the code before Glu, we posit that a preference is indicated for selection of wobble anticodon position G over U/C. A possible reason for this preference is that only one tRNA (GUN) is necessary to recognize two mRNA codons, rather than two tRNAs (UUN, CUN). In the wobble position, G pairs with C as a Watson–Crick pair, and G forms a wobble pair with U. Full establishment of the three-nucleotide code required the EF-Tu latch. Once the latch evolved, other amino acids could invade column 3. Initially, Asp is posited to have occupied all sectors in column 3 (
Figure 6). After invasion by Glu, Asp may have occupied sectors 4A, 3A, 2A, and 1A (
Figure 7). Ultimately, Asp retained the most favored row 4A, abandoning all other sectors in column 3 (
Figure 9). Glu retained favored row 4B and abandoned less favored rows 3B, 2B, and 1B to invading amino acids. Asn invaded row 3A. Lys invaded row 3B. Gln invaded row 2B. His invaded row 2A. Tyr and stop codons were late entries into the code occupying disfavored row 1 (3rd anticodon position A).
As noted above, we posit that Archaea are most similar to LUCA for translation functions [
1,
2,
13,
22]. For instance, archaeal tRNAs are more ancient than bacterial tRNAs. Therefore, to understand the initial evolution of the code requires a focus on archaeal systems. Notably, in Archaea, AspRS-IIB, AsnRS-IIB, and HisRS-IIA enzymes are reasonably closely related in structure and sequence (
Figure 3A). Furthermore, in Archaea, GluRS-IB, LysRS-IC and GlnRS-IB are closely related enzymes in structure and sequence (
Figure 3A). Evolution of aaRS enzymes, therefore, strongly supports our proposed model for column 3 evolution. Interestingly, in Bacteria, LysRS-IIB is closely related to AspRS-IIB and AsnRS-IIB in structure and sequence. Therefore, we posit that LysRS-IIB in Bacteria was evolved from AspRS-IIB, within column 3, reinforcing the importance of evolution within columns. The switch from LysRS-IC (Archaea) to LysRS-IIB (Bacteria) likely occurred close to the root of the archaeal→bacterial divergence. Clearly, evolution of the genetic code within column 3 (tRNA anticodon position 2 U) is demonstrated.
3.16. Evolution within Genetic Code Columns
So, why did the genetic code evolve so clearly along code columns? The obvious answer is that the code initially evolved around the tRNA anticodon 2nd position, which is most important for translational accuracy. Assuming this idea is correct, what is the mechanism? One mechanism is indicated by translation systems in Archaea (and some Bacteria). We identify ancient Archaea, those most closely related to LUCA, as species with the least radiated tRNAomes from tRNA
Pri [
1,
3,
14]. From analysis of tRNAomes, examples of ancient archaeal families include
Pyrococcus,
Pyrobaculum,
Staphylothermus,
Aeropyrum, and
Sulfolobus. Ancient Archaea lack GlnRS-IIB. In place of GlnRS-IIB, these species charge tRNA
Gln with Glu using GluRS-IIB. Then a Glu-tRNA
Gln amidotransferase converts the complex to Gln-tRNA
Gln [
66,
67]. As described above, Met also invaded an Ile sector leading to the sharing of the CAU anticodon. Similar mechanisms appear to be common in sectoring the code within columns. A similar mechanism could potentially explain transitions leading to the occupation of the following code sectors: (1) Val→Leu; (2) Val→Ile; (3) Ile→Met; (4) Ala→Pro; (5) Ala→Thr; (6) Pro→Ser; (7) Asp→Asn; (8) Asp→His; and (9) Glu→Gln. In some cases, mischarging of tRNAs may simply occur by invasion of an amino acid from outside the code, as with Ile→Met, rather than metabolic modification of a charged tRNA, as with Glu→Gln and, we assume, Asp→Asn. Some of these sectoring events require additional steps, i.e., exchanging AlaRS-IIA with AlaRS-IID, a step that most likely occurred about the time of full sectoring of genetic code column 2. These posited events are best indicated from evolution of aaRS enzymes, which are often closely related within columns (
Figure 3). To a lesser extent, these events may be indicated by analysis of tRNAomes, particularly in ancient Archaea [
1,
3]. tRNAs that evolve within genetic code columns tend to mutate and/or accumulate modifications rapidly to establish their new identity and to prevent mischarging. In columns 1 and 2, aaRS editing also suppresses mischarging of tRNAs.
3.17. An Alternate Model for Sectoring of Column 2
Because there are some limits to our clairvoyance describing events from ~4 billion years ago, we posit an alternate model for sectoring of column 2 and for Ser jumping from column 2 to column 4. Column 2 may have initially split between Ala (8-codon sector; rows 3 and 4) and Ser (8-codon sector; rows 1 and 2). After evolution of the EF-Tu latch, Ser may then have invaded the Ala sector, resulting in Ser occupying column 2, rows 1, 2, and 3, and Ala retaining favored row 4. Ser (GGU) may then have jumped to Ser (GCU) (column 4) via a single base change in tRNASer (anticodon position 2). Subsequently, Pro invaded row 2, displacing Ser, and Thr invaded row 3, displacing Ser. Such a model has potential advantages over that described above. Notably, Ser jumping to favored column 4 is more easily described. Also, because of amino acid similarity, Ser→Thr appears to be a reasonable transition in row 3. Pro would displace Ser in row 2, leaving Ser in disfavored row 1, but this might not pose a difficulty if Ser had previously occupied favored column 4 row 3A. We give this example of an alternate model as a guide for understanding evolution of the genetic code. The model we advance is a working model. The detail in the model we find to be an advantage. Alternate pathways make slightly different predictions that potentially can be challenged computationally and/or by experiment. We know of no other model for code evolution that could potentially make such detailed or informative predictions.
3.20. Late Additions to the Genetic Code
Phe, Tyr, Trp, Cys, Met, and His are posited to be among the last additions to the genetic code [
19,
20,
68]. Stop codons, which are read by protein release factors rather than tRNAs, are also posited to be late additions to the code. Phe, Tyr, Trp, and Cys are all in disfavored row 1 of the code (3rd anticodon position A). Stop codons, which do not follow tRNA anticodon rules [
21], are in disfavored row 1 of the code. Met appears to have invaded the Ile sector as a late event. For tRNA
Met (CAU) and tRNA
Ile (CAU), wobble C covalent modifications, particularly to tRNA
Ile (agmatidine in Archaea; lysidine in Bacteria), are very important to discriminate tRNA
Met and tRNA
Ile. tRNA
Met acceptor stems (As) are important to discriminate elongator tRNA
Met (i.e., 5’-As GCCCGGG) from initiator tRNA
Met (i.e., 5’-As AGCGGGA). In Archaea, tRNA
Ile (CAU) (C→agmatidine) is commonly used to recognize AUA Ile codons without recognition of AUG Met codons. The tRNA
Ile (CAU) C~A wobble pair is recognized by modification of the C to agmatidine to achieve this discrimination [
53,
54,
55]. Only in eukaryotes is tRNA
Ile (UAU) commonly utilized. In eukaryotes, tRNA
Ile (IAU; I for inosine) can recognize the three Ile codons (AUC, AUU, AUA).
3.21. Alternate Genetic Code Models
Focusing on tight coevolution of metabolic systems to describe patterns of genetic code evolution is likely a mistake. We posit that apparent parallels between genetic code evolution and amino acid metabolism evolution (i.e., evolution of the genetic code within columns) occur primarily because of evolution of the genetic code according to the tRNA anticodon, as described above. If the tRNA anticodon is the central feature determining the history of evolution of the genetic code, the entire scenario makes sense according to Darwinian principles. Otherwise, the sectoring of the code makes little to no sense. That stated, we do accept that coevolution of metabolism and the genetic code occurred. In some cases, modification of amino acids on tRNAs is a mechanism for code sectoring (i.e., Asp→Asn and Glu→Gln). In other cases, amino acids appear to invade from outside the code (i.e., Ile→Met). We argue that, with invasions from outside the code, coevolution of metabolism and the code provided the amino acids that were available to invade the code rather than metabolism driving the sectoring of the code.
Another potential error that we perceive in genetic code models is to build up the code a sector at a time. We do not believe such models are reasonable. Initially, we made the assumption that the entire code (all anticodons and all codons) was populated with tRNA
Gly (
Figure 5). To our surprise, this simple assumption allowed a rich and highly detailed model for genetic code evolution with clear selection strategies to emerge. We were surprised because we commenced this effort thinking that, after ~4 billion years of evolution, development of such a detailed model was not possible. In support of our assumption, however, the tRNA anticodon is successfully mutated more rapidly than other tRNA sequences, which are required to support RNA stems and to stabilize tRNA structure. This is because the anticodon stem and loop sticks out from the tRNA. Other anticodon loop bases (7-nt loop positions 1, 2, 6, and 7) are highly constrained in sequence to maintain U-turn geometry, which is necessary to present a 3-nt anticodon. We imagined that tRNA initially was essential genetic evolutionary property (
Figure 1). That is to say that tRNA was of value to synthesize polyglycine to stabilize protocells. We imagined that, being of value, tRNA was replicated rapidly in an RNA world, providing many copies for modification and genetic alteration. The other advantage to our model was that the model brought mRNA in line with tRNA, because, initially, all codons encoded glycine. In this way, tRNA anticodons and mRNA codons could more easily coevolve to encode incoming amino acids. In other scenarios, anticodons and codons must converge to encode products, which is more difficult to imagine. As anticodons evolved within the code, mRNA codons coevolved to encode ever more complex products. By contrast, a GC→GCA→GCAU genetic code (based on codons rather than anticodons) [
52] evolutionary model does not adequately address this difficulty. Such a model begins within a sector and evolves to occupy other sectors. Such models have the critical flaw described above.
Another scheme for evolution of the genetic code is the GNC→SNS→standard code model (N = any base; S = G or C) [
6]. We find some agreement and some disagreement with this model. One objection that we would raise is that the scheme is presented as a codon-centric model rather than an anticodon-centric model. Working out from tRNA is a big advantage, as we hope we have shown. Also, why begin with codon GNC (also anticodon GNC) rather than, for instance, CNG? Anticodon GNC gives Gly, Asp, Ala, and Val as the first four amino acids, which agrees with our assessment (
Figure 4 and
Figure 6). GNC also supports 3rd anticodon position C, which we deem favorable because of the C > G > U >> A anticodon rule. However, initial selection of GNC rather than CNG appears arbitrary. SNS makes some sense because C/G should facilitate 1st and 3rd position anticodon reading before evolution of the EF-Tu anticodon-codon latch. The SNS phase brings Gly, Asp, Ala, Val, Leu, Pro, Glu, Gln, His, and Arg into the code, which is very similar to the recruitment order we favor. Possible limitations of the alternate GNC model include: (1) apparent favoring of codons over anticodons; (2) beginning the code arbitrarily with GNC, rather than SNS; (3) no consideration of the EF-Tu latch in anticodon-codon reading; (4) building up the code via sectors rather than filling in and invading; (5) awkward co-evolution of anticodons and codons; and (6) little consideration of why 4- and 2-codon sectors are favored over 1-codon sectors. A recent paper on this subject [
6] utilizes an incorrect tRNA evolution model and an incorrect aaRS evolution model. The model does not fully recognize the extent to which aaRS enzyme evolution parallels the establishment of the code (
Figure 3).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}