Optimization of Molecular Dynamics Simulations of c-MYC1-88—An Intrinsically Disordered System


Abstract
:1. Introduction
1.1. Optimized Force Fields for IDP Simulations
1.2. Optimizing Explicit and Implicit Water Models
1.3. Assessing Simulation Convergence
2. Materials and Methods
2.1. MD Set-up
2.2. Trajectory Analysis
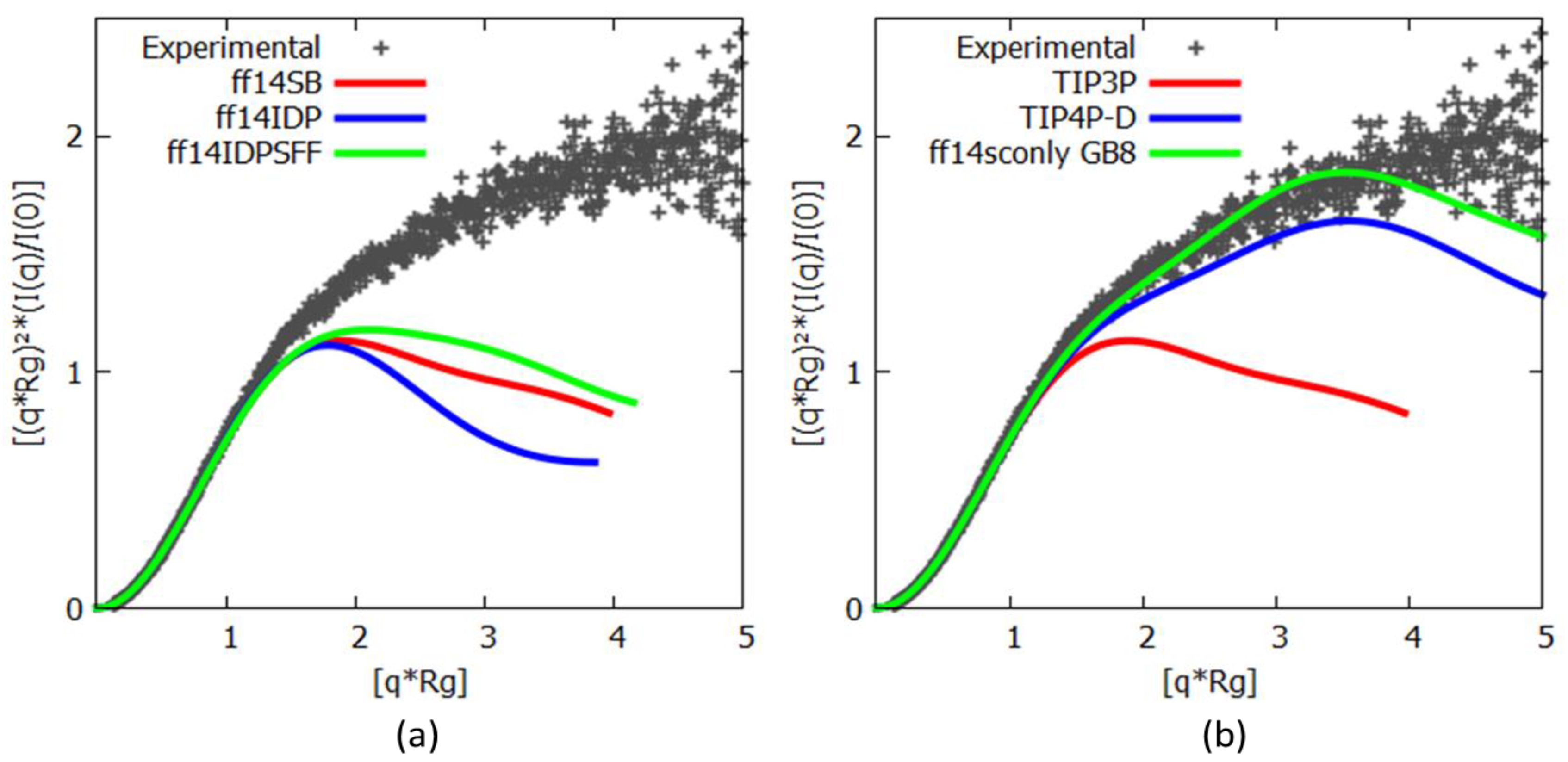
2.3. Experimental Data
2.4. Markov Chain Monte Carlo
3. Results
3.1. Histatin 5
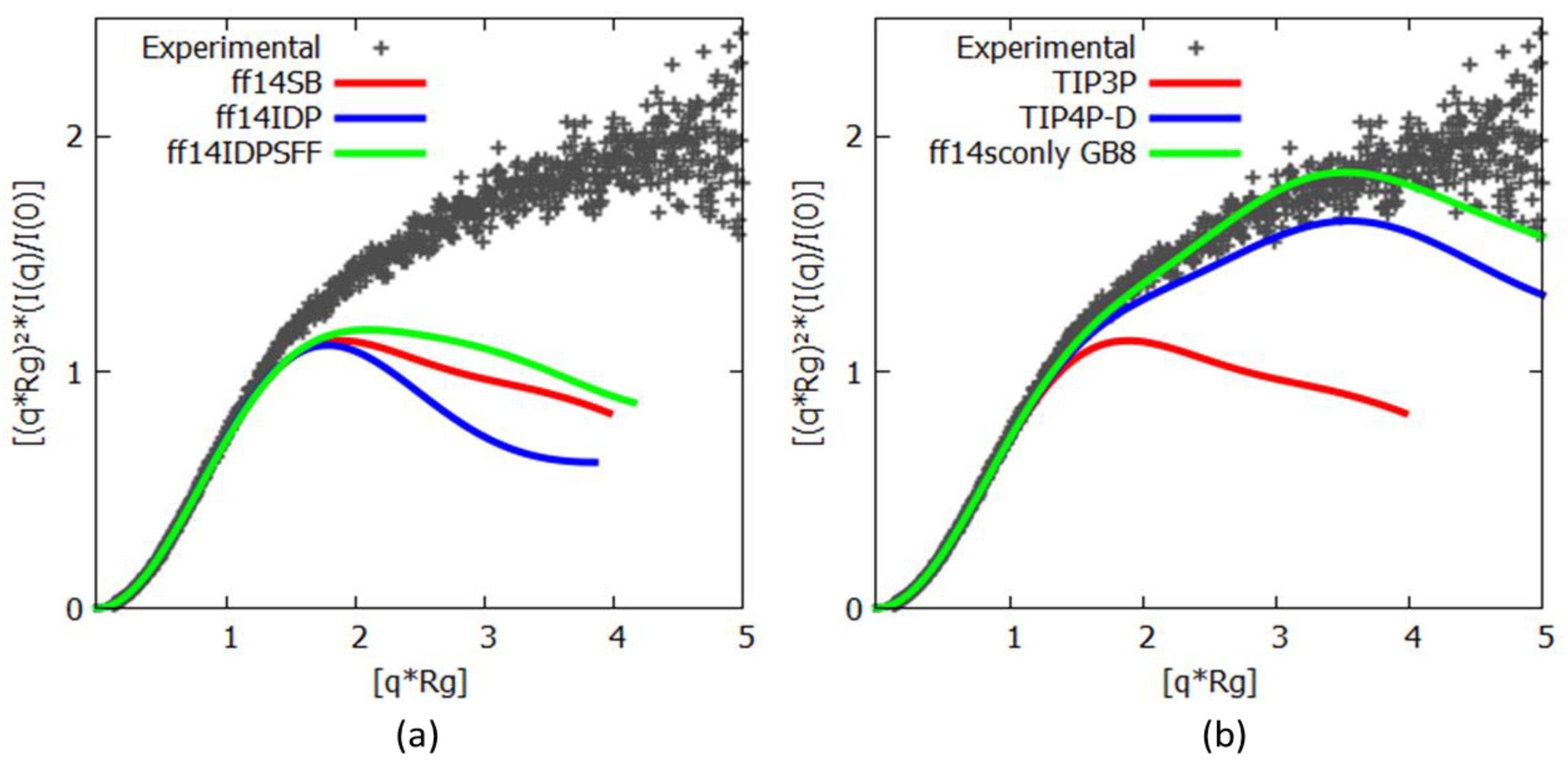
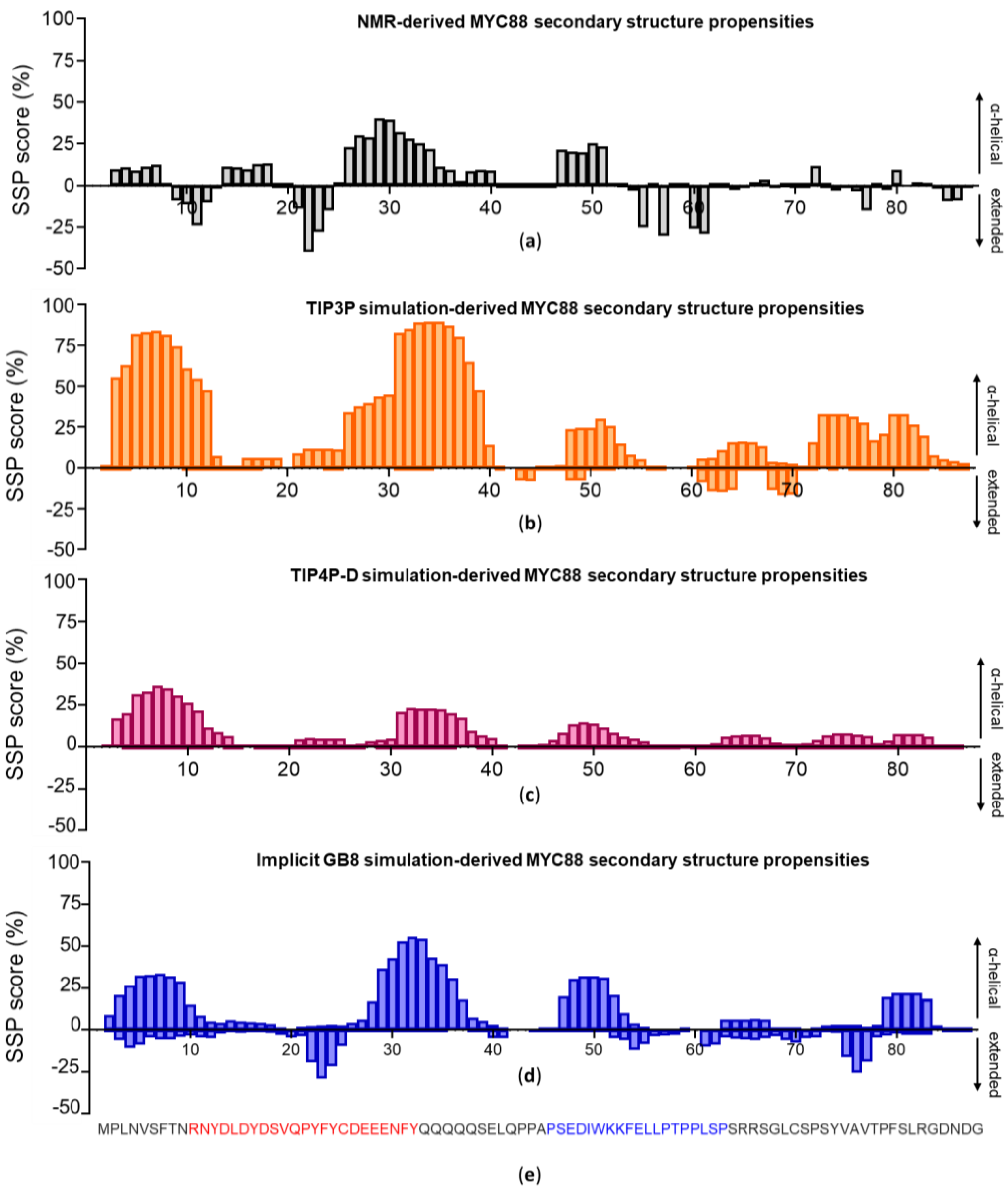
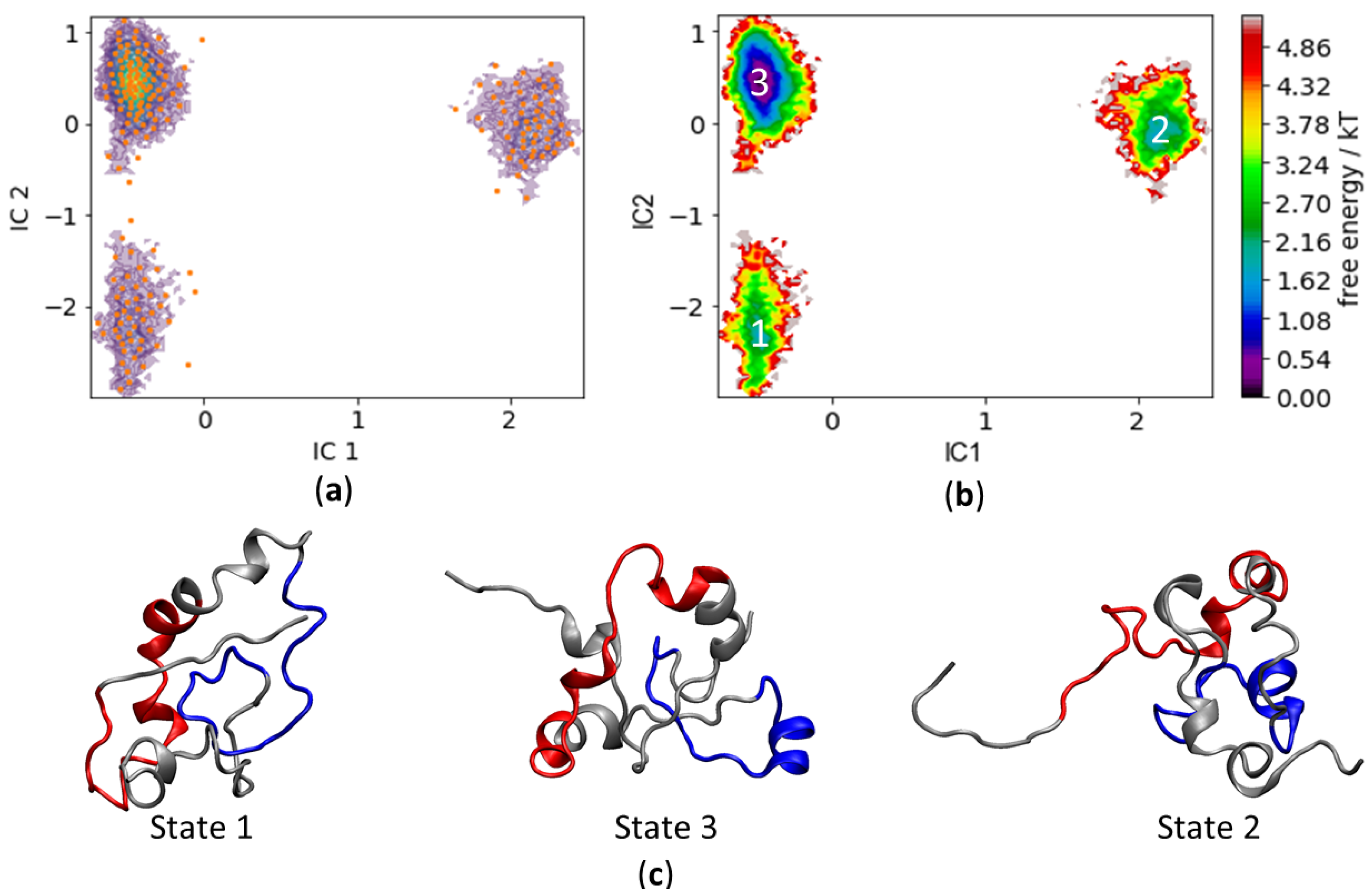
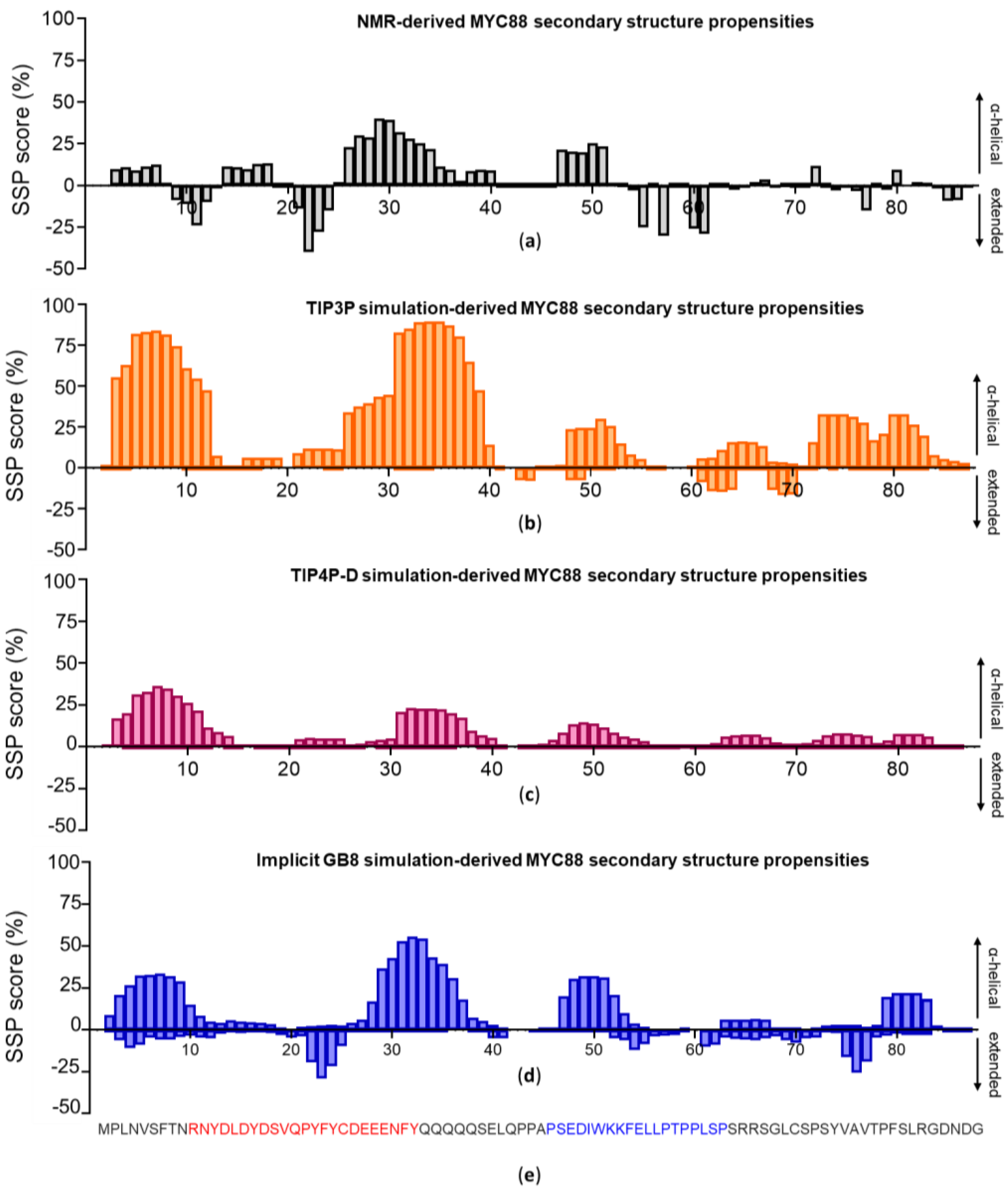
3.2. c-MYC1-88
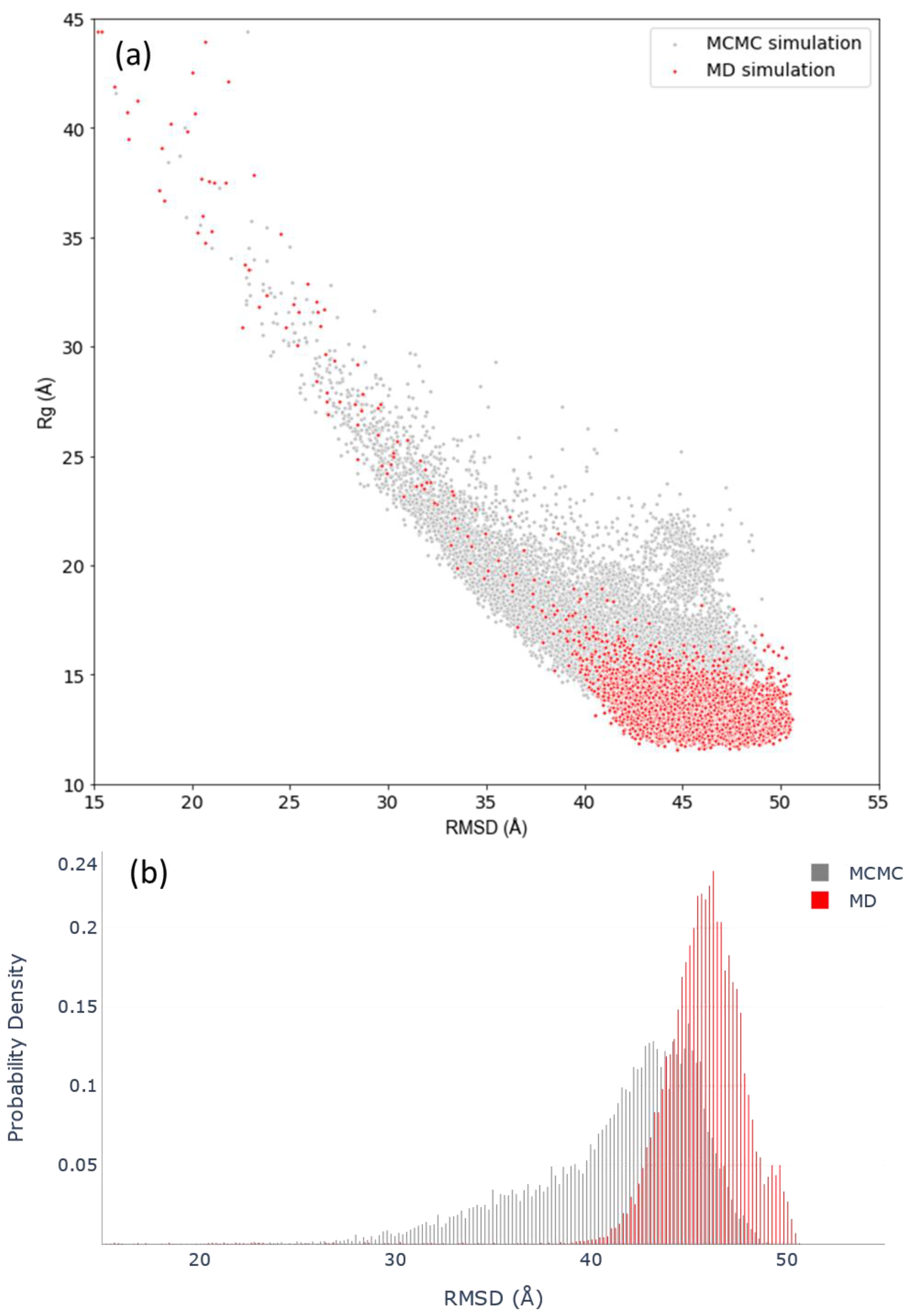
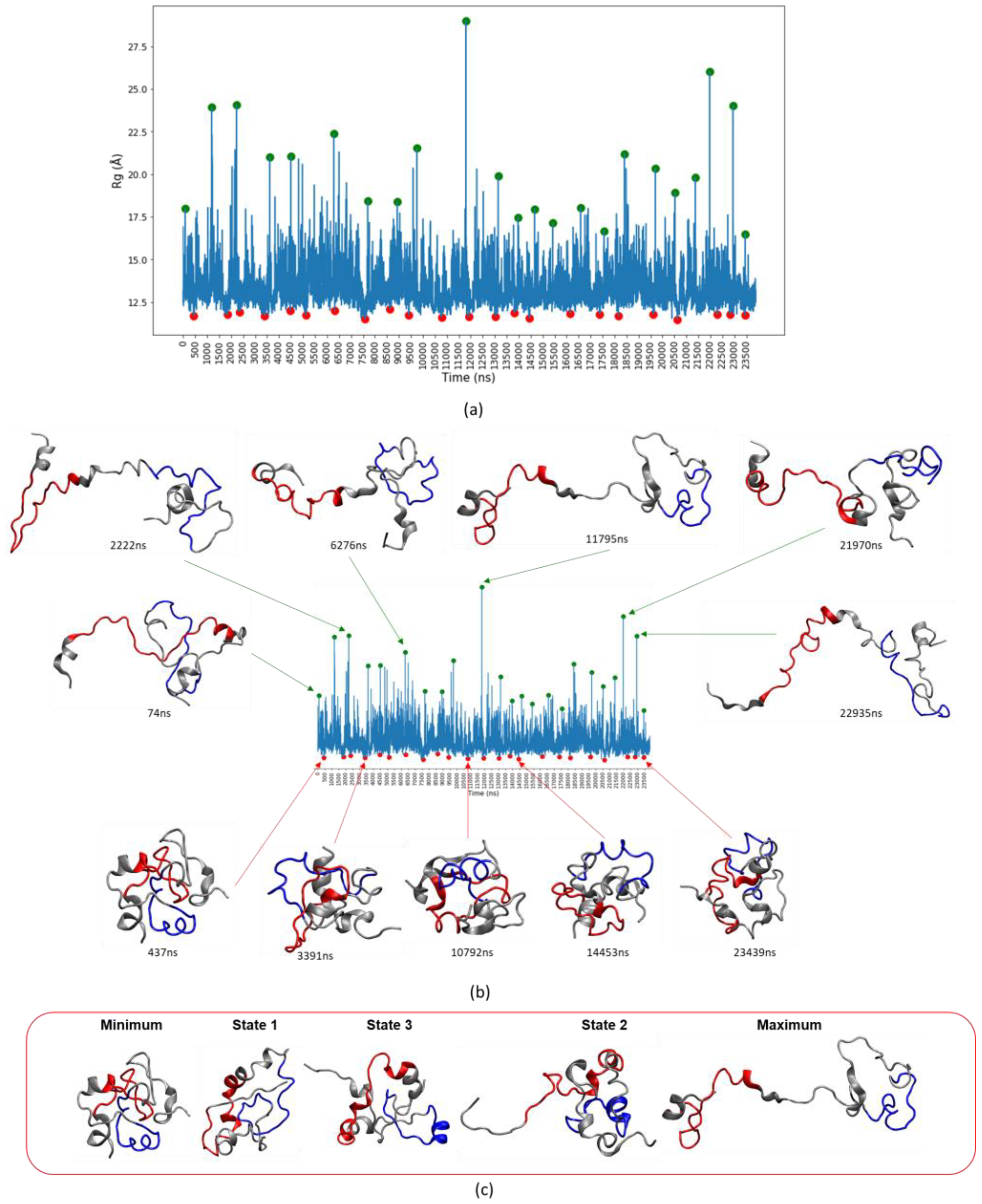
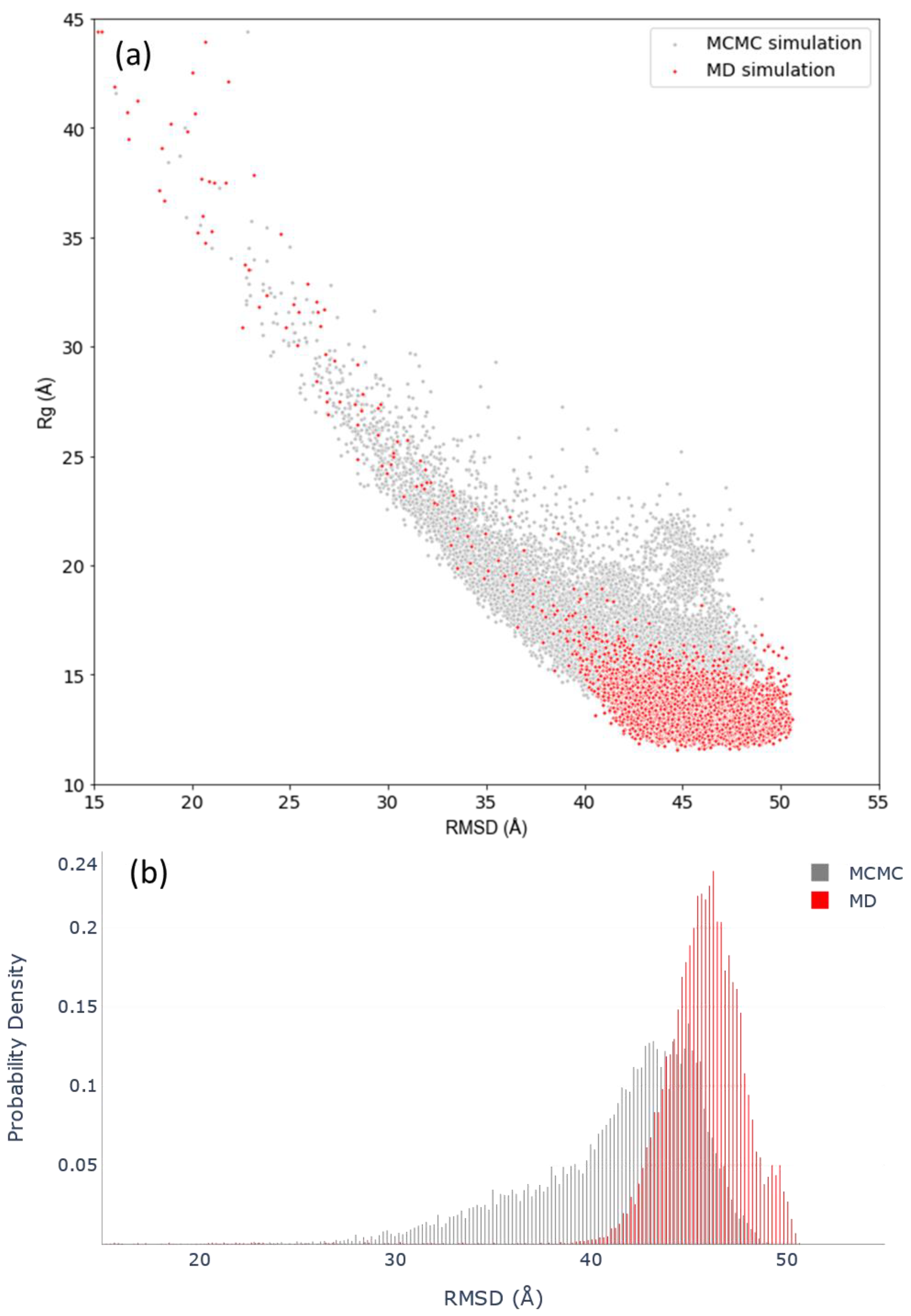
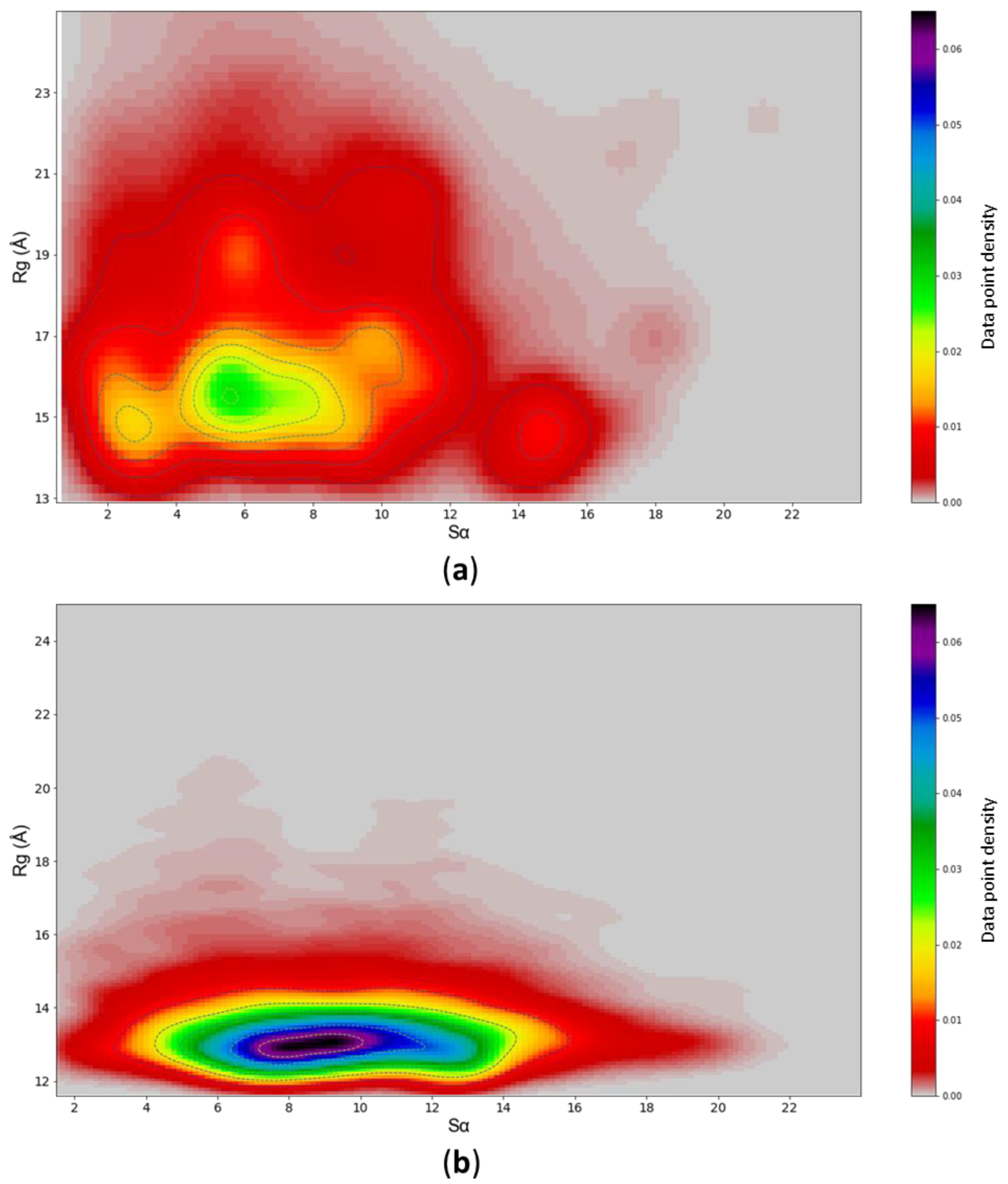
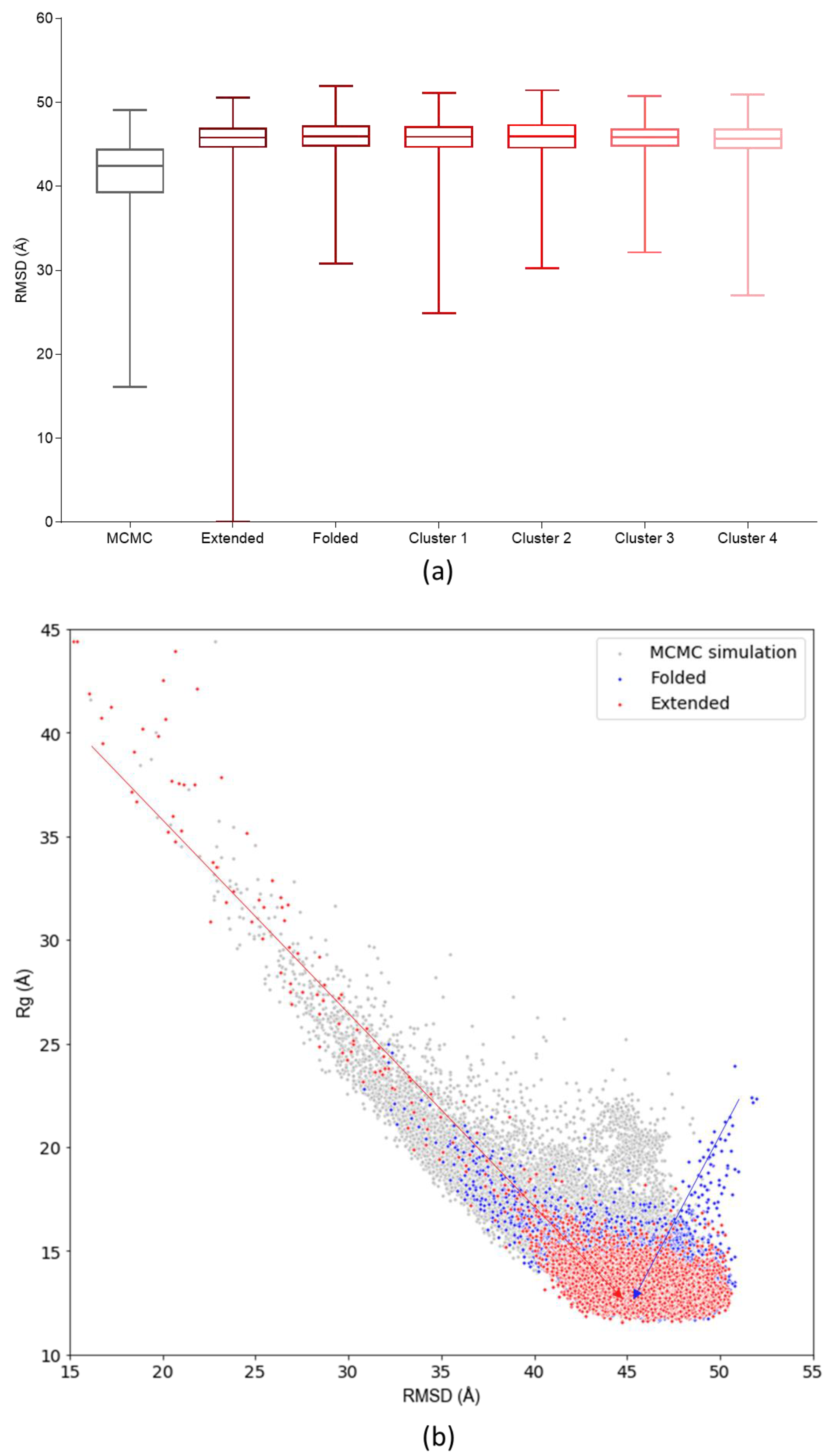
3.3. Assessing Convergence
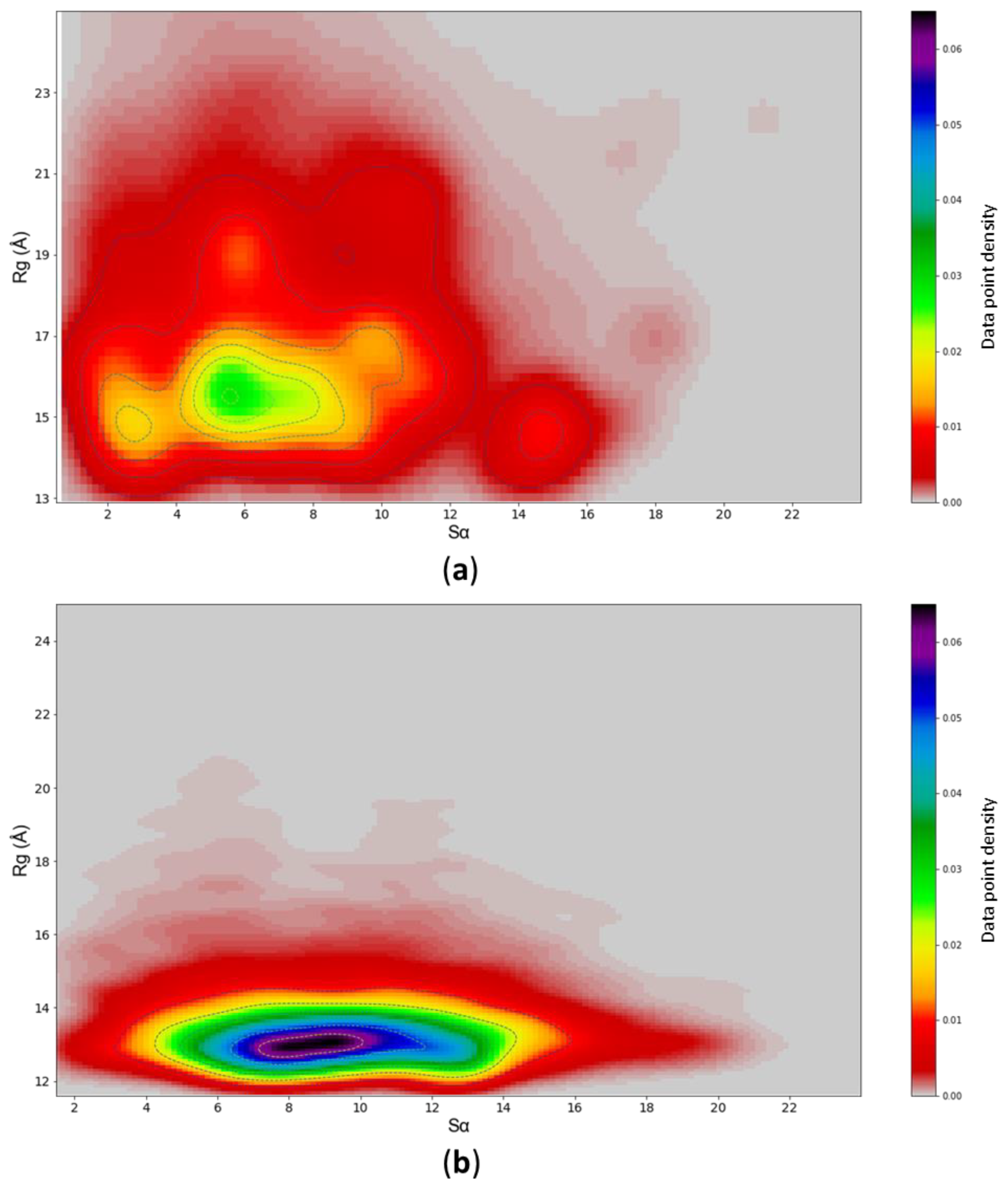
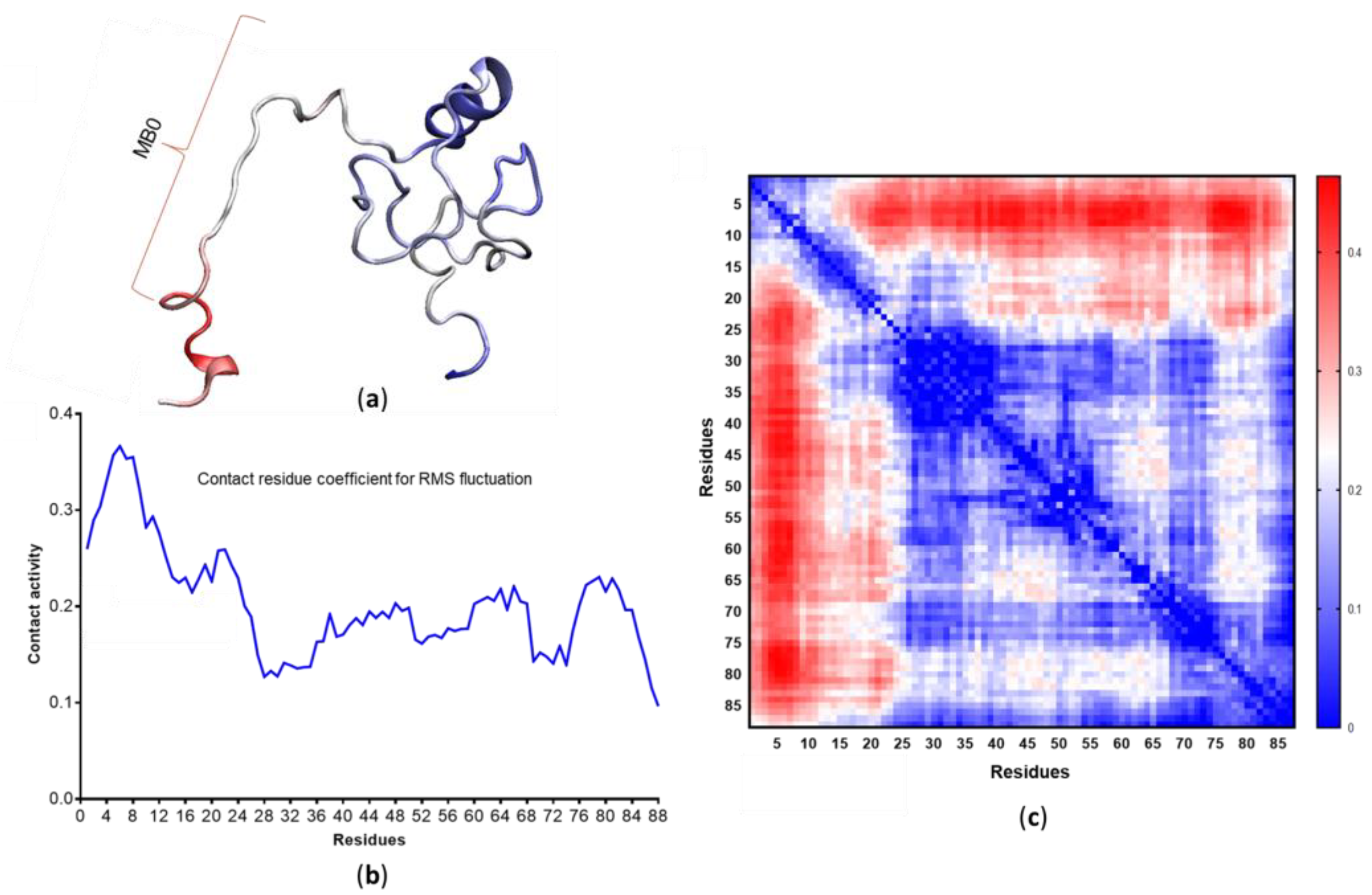
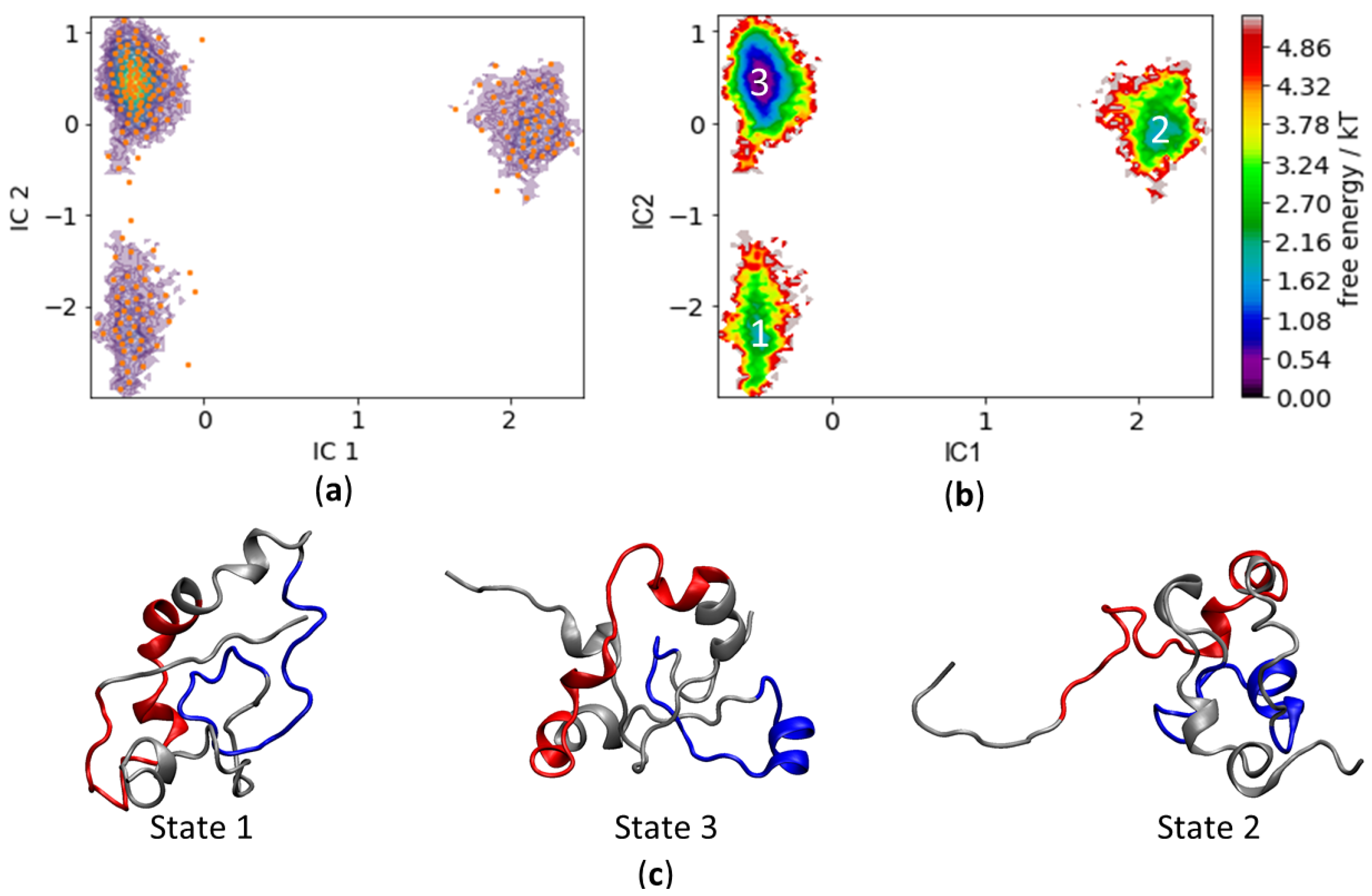
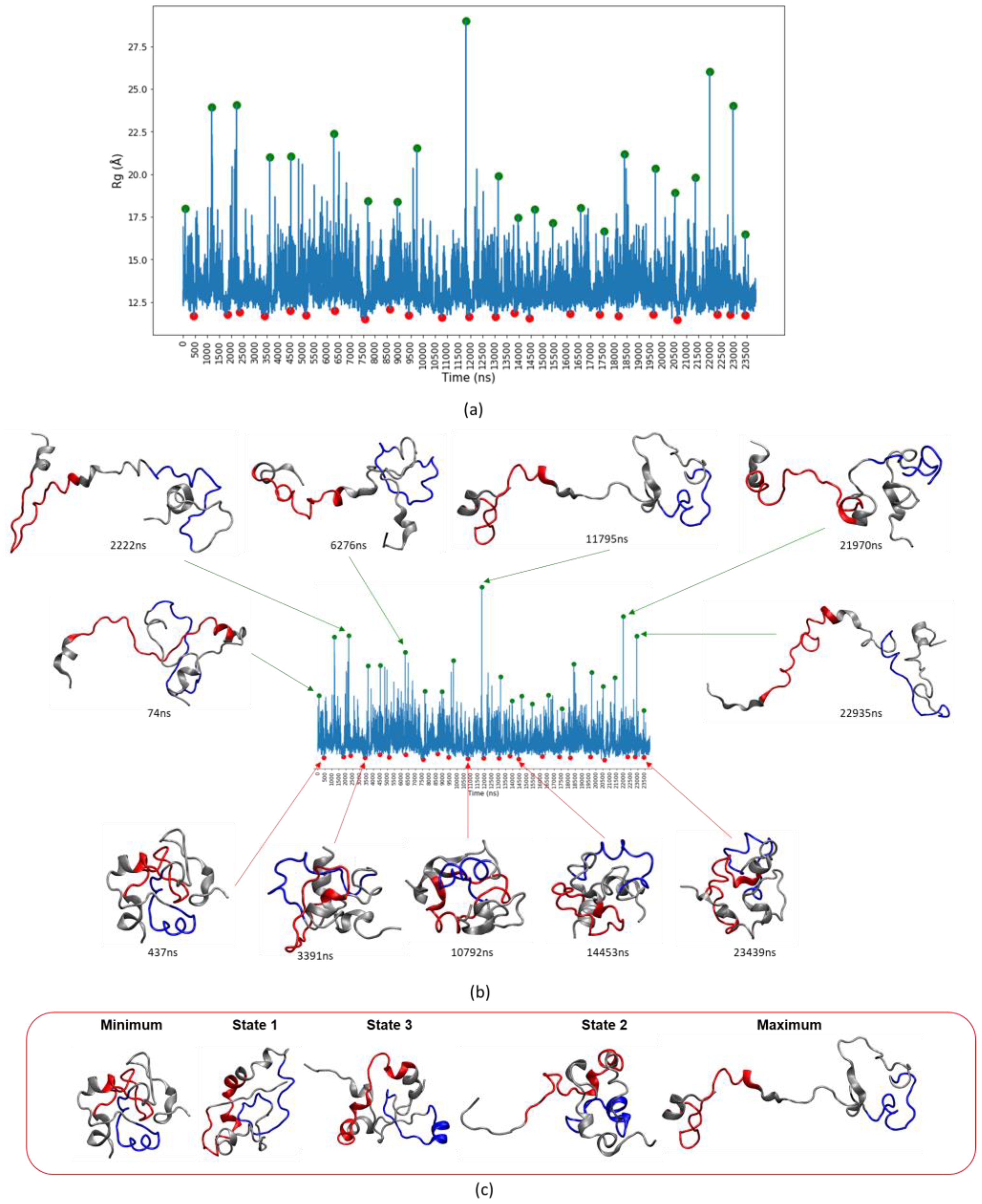
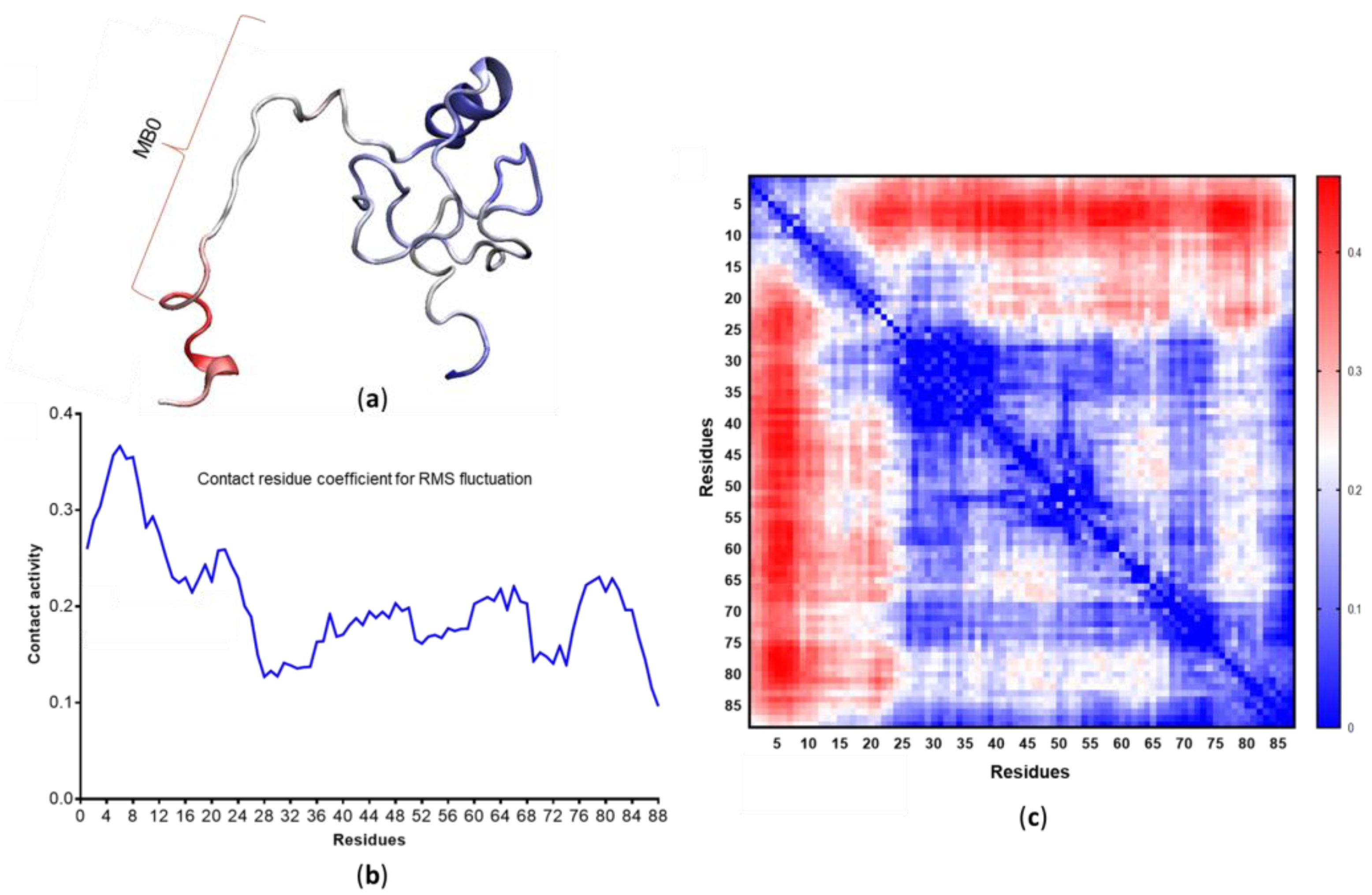
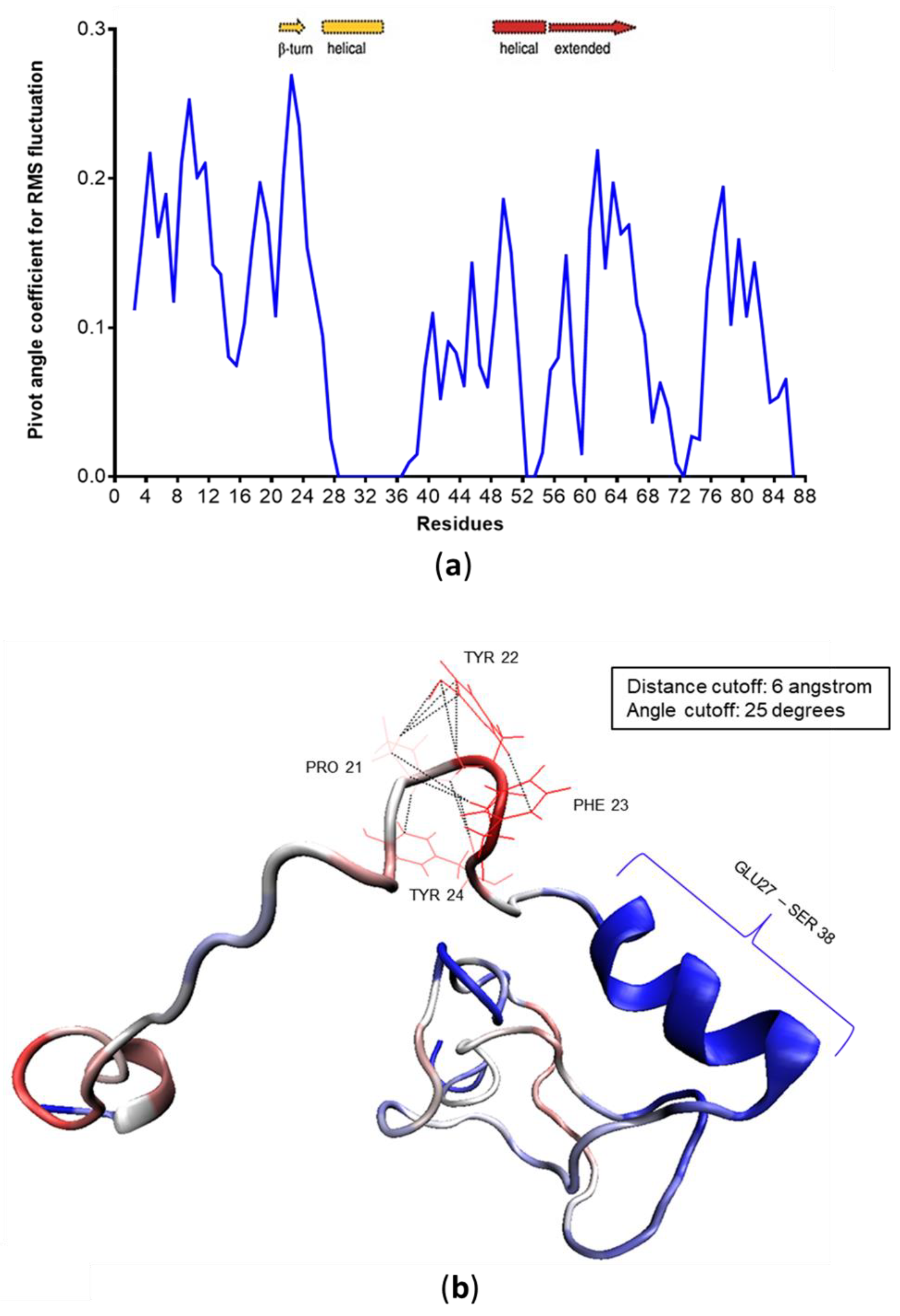
3.4. c-MYC1-88 Trajectory Analysis and Structural Insights
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Levine, Z.A.; Shea, J. Simulations of disordered proteins and systems with conformational heterogeneity. Curr. Opin. Struct. Biol. 2017, 43, 95–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babu, M.M. The contribution of intrinsically disordered regions to protein function, cellular complexity, and human disease. Biochem. Soc. Trans. 2016, 44, 1185–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salomon-Ferrer, R.; Gotz, A.W.; Poole, D.; Grand, S.L.; Walker, R.C. Routine Microsecond Molecular Dynamics Simulations with AMBER on GPUs. 2. Explicit Solvent Particle Mesh Ewald. J. Chem. Theory Comput. 2013, 9, 3878–3888. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.E.; Hallock, M.J.; Phillips, J.C.; Peterson, J.R.; Schulten, Z.L.; Schulten, K. Evaluation of emerging energy-efficient heterogeneous computing platforms for biomolecular and cellular simulation workloads. In Proceedings of the 2016 IEEE International Parallel and Distributed Processing Symposium Workshops (IPDPSW), Chicago, IL, USA, 23–27 May 2016; IEEE: Piscataway, NJ, USA, 2016; pp. 89–100. [Google Scholar]
- Hollingsworth, S.A.; Dror, R.O. Molecular dynamics simulation for all. Neuron 2018, 99, 1129–1143. [Google Scholar] [CrossRef] [Green Version]
- Beauchamp, K.A.; Lin, Y.-S.; Das, R.; Pande, V.S. Are Protein Force Fields Getting Better? A Systematic Benchmark on 524 Diverse NMR Measurements. J. Chem. Theory Comput. 2012, 8, 1409–1414. [Google Scholar] [CrossRef] [Green Version]
- Chong, S.-H.; Chatterjee, P.; Ham, S. Computer Simulations of Intrinsically Disordered Proteins. Annu. Rev. Phys. Chem. 2017, 68, 117–134. [Google Scholar] [CrossRef]
- Rauscher, S.; Gapsys, V.; Gajda, M.J.; Zweckstetter, M.; De Groot, B.L.; Grubmüller, H. Structural Ensembles of Intrinsically Disordered Proteins Depend Strongly on Force Field: A Comparison to Experiment. J. Chem. Theory Comput. 2015, 11, 5513–5524. [Google Scholar] [CrossRef] [Green Version]
- Henriques, J.; Cragnell, C.; Skepo, M. Molecular dynamics simulations of intrinsically disordered proteins: Force field evaluation and comparison with experiment. J. Chem. Theory Comput. 2015, 11, 3420–3431. [Google Scholar] [CrossRef]
- Huang, J.; Rauscher, S.; Nawrocki, G.; Ran, T.; Feig, M.; De Groot, B.L.; Grubmüller, H.; Mackerell, A.D., Jr. CHARMM36m: An improved force field for folded and intrinsically disordered proteins. Nat. Methods 2017, 14, 71–73. [Google Scholar] [CrossRef] [Green Version]
- Piana, S.; Klepeis, J.L.; Shaw, D.E. Assessing the accuracy of physical models used in protein-folding simulations: Quantitative evidence from long molecular dynamics simulations. Curr. Opin. Struct. Biol. 2014, 24, 98–105. [Google Scholar] [CrossRef] [Green Version]
- Best, R.B. Computational and theoretical advances in studies of intrinsically disordered proteins. Curr. Opin. Struct. Biol. 2017, 42, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Piana, S.; Donchev, A.G.; Robustelli, P.; Shaw, D.E. Water Dispersion Interactions Strongly Influence Simulated Structural Properties of Disordered Protein States. J. Phys. Chem. B 2015, 119, 5113–5123. [Google Scholar] [CrossRef] [PubMed]
- Song, D.; Wang, W.; Ye, W.; Ji, D.; Luo, R.; Chen, H.-F. ff14IDPs force field improving the conformation sampling of intrinsically disordered proteins. Chem. Biol. Drug Des. 2017, 89, 5–15. [Google Scholar] [CrossRef] [Green Version]
- Song, D.; Luo, R.; Chen, H. The IDP-Specific Force Field ff14IDPSFF Improves the Conformer Sampling of Intrinsically Disordered Proteins. J. Chem. Inf. Modeling 2017, 57, 1166–1178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Guo, X.; Han, J.; Luo, R.; Chen, H.-F. Order-disorder transition of intrinsically disordered kinase inducible transactivation domain of CREB. J. Chem. Phys. 2018, 148, 225101. [Google Scholar] [CrossRef]
- Mark, P.; Nilsson, L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Henriques, J.; Skepo, M. Molecular Dynamics Simulations of Intrinsically Disordered Proteins: On the Accuracy of the TIP4P-D Water Model and the Representativeness of Protein Disorder Models. J. Chem. Theory Comput. 2016, 12, 3407–3415. [Google Scholar] [CrossRef]
- Onufriev, A.V.; Case, D.A. Generalized Born Implicit Solvent Models for Biomolecules. Annu. Rev. Biophys. 2019, 48, 275–296. [Google Scholar] [CrossRef]
- Sawle, L.; Ghosh, K. Convergence of Molecular Dynamics Simulation of Protein Native States: Feasibility vs Self-Consistency Dilemma. J. Chem. Theory Comput. 2016, 12, 861–869. [Google Scholar] [CrossRef]
- Grossfield, A.; Zuckerman, D.M. Quantifying uncertainty and sampling quality in biomolecular simulations. In Annual Reports in Computational Chemistry; Elsevier Science & Technology: Amsterdam, The Netherlands, 2009; Chapter 2; pp. 23–48. [Google Scholar]
- Smith, L.J.; Daura, X.; van Gunsteren, W.F. Assessing equilibration and convergence in biomolecular simulations. Proteins Struct. Funct. Bioinform. 2002, 48, 487–496. [Google Scholar] [CrossRef]
- Lyman, E.; Zuckerman, D.M. Ensemble-Based Convergence Analysis of Biomolecular Trajectories. Biophys. J. 2006, 91, 164–172. [Google Scholar] [CrossRef] [Green Version]
- Hess, B. Similarities between principal components of protein dynamics and random diffusion. Phys. Rev. E Stat. Phys. Plasmas Fluids Relat. Interdiscip. Top. 2000, 62, 8438–8448. [Google Scholar] [CrossRef] [Green Version]
- Hess, B. Convergence of sampling in protein simulations. Phys. Rev. E Stat. Nonlinear Soft Matter Phys. 2002, 65, 031910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knapp, B.; Frantal, S.; Cibena, M.; Schreiner, W.; Bauer, P. Is an Intuitive Convergence Definition of Molecular Dynamics Simulations Solely Based on the Root Mean Square Deviation Possible? J. Comput. Biol. 2011, 18, 997–1005. [Google Scholar] [CrossRef] [Green Version]
- van Ravenzwaaij, D.; Cassey, P.; Brown, S.D. A simple introduction to Markov Chain Monte-Carlo sampling. Psychon. Bull. Rev. 2018, 25, 143–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boomsma, W.; Frellsen, J.; Harder, T.; Bottaro, S.; Johansson, K.E.; Tian, P.; Stovgaard, K.; Andreetta, C.; Olsson, S.; Valentin, J.B.; et al. PHAISTOS: A framework for Markov chain Monte Carlo simulation and inference of protein structure. J. Comput. Chem. 2013, 34, 1697–1705. [Google Scholar] [CrossRef]
- Case, D.A. AMBER 2016 Reference Manual; University of California: San Francisco, CA, USA, 2016; pp. 1–923. [Google Scholar]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The PyMOL Molecular Graphics System, version 1.8.6.0, Computer Software Reference; Schrodinger, LLC: New York, NY, USA, 2010.
- Xu, D.; Zhang, Y. Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins Struct. Funct. Bioinform. 2012, 80, 1715–1735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.; Zhang, Y. Toward optimal fragment generations for ab initio protein structure assembly. Proteins Struct. Funct. Bioinform. 2013, 81, 229–239. [Google Scholar] [CrossRef] [Green Version]
- Roe, D.R.; Cheatham, T.E. PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput. 2013, 9, 3084–3095. [Google Scholar] [CrossRef]
- Rambo, R. “Scatter”. The SIBYLS Beamline, Version 3.0, Computer Software Reference, 2017. Available online: https://bl1231.als.lbl.gov/scatter/ (accessed on 20 June 2020).
- Petoukhov, M.V.; Franke, D.; Shkumatov, A.V.; Tria, G.; Kikhney, A.G.; Gajda, M.; Gorba, C.; Mertens, H.D.T.; Konarev, P.V.; Svergun, D.I. New developments in the ATSAS program package for small-angle scattering data analysis. J. Appl. Crystallogr. 2012, 45, 342–350. [Google Scholar] [CrossRef] [Green Version]
- Konarev, P.V.; Volkov, V.; Sokolova, A.; Koch, M.H.J.; Svergun, D.I. PRIMUS: A Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 2003, 36, 1277–1282. [Google Scholar] [CrossRef]
- Svergun, D.I. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 1992, 25, 495–503. [Google Scholar] [CrossRef]
- Shen, Y.; Bax, A. SPARTA+: A modest improvement in empirical NMR chemical shift prediction by means of an artificial neural network. J. Biomol. NMR 2010, 48, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Tribello, G.A.; Bonomi, M.; Branduardi, D.; Camilloni, C.; Bussi, G. PLUMED 2: New feathers for an old bird. Comput. Phys. Commun. 2014, 185, 604–613. [Google Scholar] [CrossRef] [Green Version]
- Pietrucci, F.; Laio, A. A Collective Variable for the Efficient Exploration of Protein Beta-Sheet Structures: Application to SH3 and GB1. J. Chem. Theory Comput. 2009, 5, 2197–2201. [Google Scholar] [CrossRef] [PubMed]
- Grant, B.J.; Rodrigues, A.P.C.; Sawy, K.M.E.; Cammon, J.A.M.; Caves, L. Bio3d: An R package for the comparative analysis of protein structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef] [Green Version]
- Scherer, M.K.; Trendelkamp-Schroer, B.; Paul, F.; Pérez-Hernández, G.; Hoffmann, M.; Plattner, N.; Wehmeyer, C.; Prinz, J.-H.; Noé, F. PyEMMA 2: A Software Package for Estimation, Validation, and Analysis of Markov Models. J. Chem. Theory Comput. 2015, 11, 5525–5542. [Google Scholar] [CrossRef]
- Kovacs, J.A.; Wriggers, W. Spatial Heat Maps from Fast Information Matching of Fast and Slow Degrees of Freedom: Application to Molecular Dynamics Simulations. J. Phys. Chem. B 2016, 120, 8473–8484. [Google Scholar] [CrossRef] [Green Version]
- Wriggers, W.R.; Stafford, K.; Shan, Y.; Piana, S.; Maragakis, P.; Lindorff-Larsen, K.; Miller, P.J.; Gullingsrud, J.; Rendleman, C.A.; Eastwood, M.P.; et al. Automated Event Detection and Activity Monitoring in Long Molecular Dynamics Simulations. J. Chem. Theory Comput. 2009, 5, 2595–2605. [Google Scholar] [CrossRef] [PubMed]
- Cragnell, C.; Durand, M.; Cabane, B.; Skep̦ö, M. Coarse-grained modeling of the intrinsically disordered protein Histatin 5 in solution: Monte Carlo simulations in combination with SAXS. Proteins Struct. Funct. Bioinform. 2016, 84, 777–791. [Google Scholar] [CrossRef] [PubMed]
- Raj, P.A.; Marcus, E.; Sukumaran, D.K. Structure of human salivary histatin 5 in aqueous and nonaqueous solutions. Biopolymers 1998, 45, 51–67. [Google Scholar] [CrossRef]
- Andresen, C.; Helander, S.; Lemak, A.; Farès, C.; Csizmok, V.; Carlsson, J.; Penn, L.Z.; Forman-Kay, J.D.; Arrowsmith, C.H.; Lundström, P.; et al. Transient structure and dynamics in the disordered c-Myc transactivation domain affect Bin1 binding. Nucleic Acids Res. 2012, 40, 6353–6366. [Google Scholar] [CrossRef]
- Wahlström, T.; Henriksson, M.A. Impact of MYC in regulation of tumor cell metabolism. BBA Gene Regul. Mech. 2015, 1849, 563–569. [Google Scholar] [CrossRef]
- Chodera, J.D.; Noé, F. Markov state models of biomolecular conformational dynamics. Curr. Opin. Struct. Biol. 2014, 25, 135–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; West-Osterfield, K.; Spears, E.; Li, Z.; Panaccione, A.; Hann, S.R. MB0 and MBI Are Independent and Distinct Transactivation Domains in MYC that Are Essential for Transformation. Genes 2017, 8, 134. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Force Fields | Cluster 1 | Cluster 2 | Experimental |
|---|---|---|---|
| ff14SB | 9.15 Å | 7.71 Å | |
| ff14IDPs | 7.38 Å | 8.15 Å | 13.8 Å |
| ff14IDPSFF | 7.48 Å | 9.87 Å |
| Water Model | Cluster 1 | Cluster 2 | Experimental |
|---|---|---|---|
| TIP3P | 9.15 Å | 7.71 Å | |
| TIP4P-D | 13.47 Å | 12.12 Å | 13.8 Å |
| Implicit GB8 | 10.68 Å | 14.14 Å |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sullivan, S.S.; Weinzierl, R.O.J. Optimization of Molecular Dynamics Simulations of c-MYC1-88—An Intrinsically Disordered System. Life 2020, 10, 109. https://doi.org/10.3390/life10070109
Sullivan SS, Weinzierl ROJ. Optimization of Molecular Dynamics Simulations of c-MYC1-88—An Intrinsically Disordered System. Life. 2020; 10(7):109. https://doi.org/10.3390/life10070109
Chicago/Turabian StyleSullivan, Sandra S., and Robert O.J. Weinzierl. 2020. "Optimization of Molecular Dynamics Simulations of c-MYC1-88—An Intrinsically Disordered System" Life 10, no. 7: 109. https://doi.org/10.3390/life10070109
APA StyleSullivan, S. S., & Weinzierl, R. O. J. (2020). Optimization of Molecular Dynamics Simulations of c-MYC1-88—An Intrinsically Disordered System. Life, 10(7), 109. https://doi.org/10.3390/life10070109