Urea-Assisted Synthesis and Characterization of Saponite with Different Octahedral (Mg, Zn, Ni, Co) and Tetrahedral Metals (Al, Ga, B), a Review


Abstract
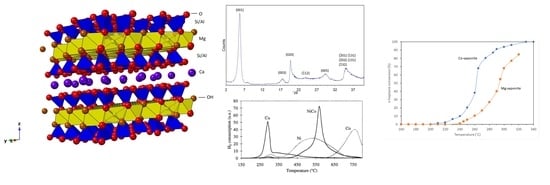
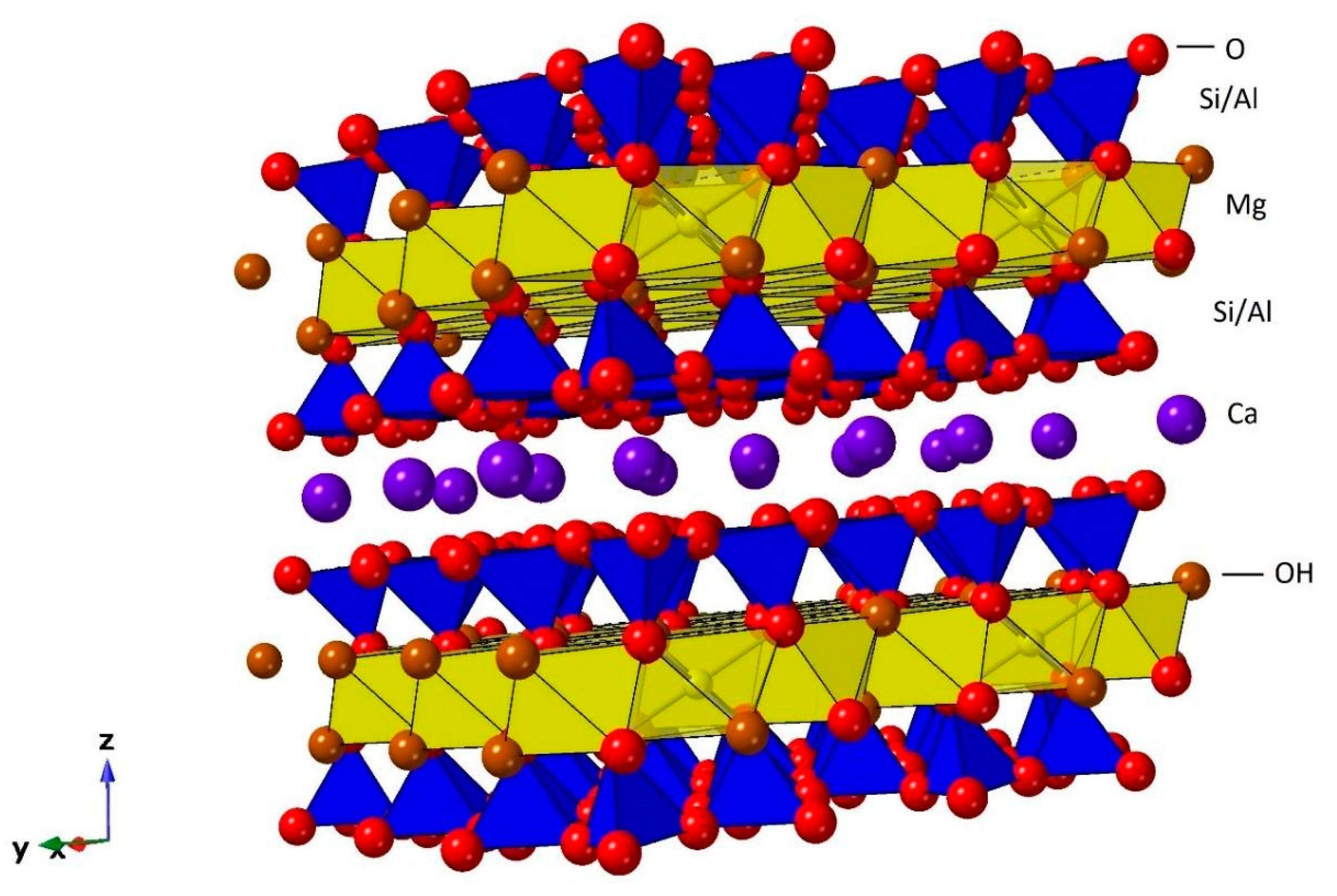
:
1. Introduction
2. Saponite Synthesis and Characterization
2.1. Saponite Composition and Occurrence
2.2. Preparation Methods

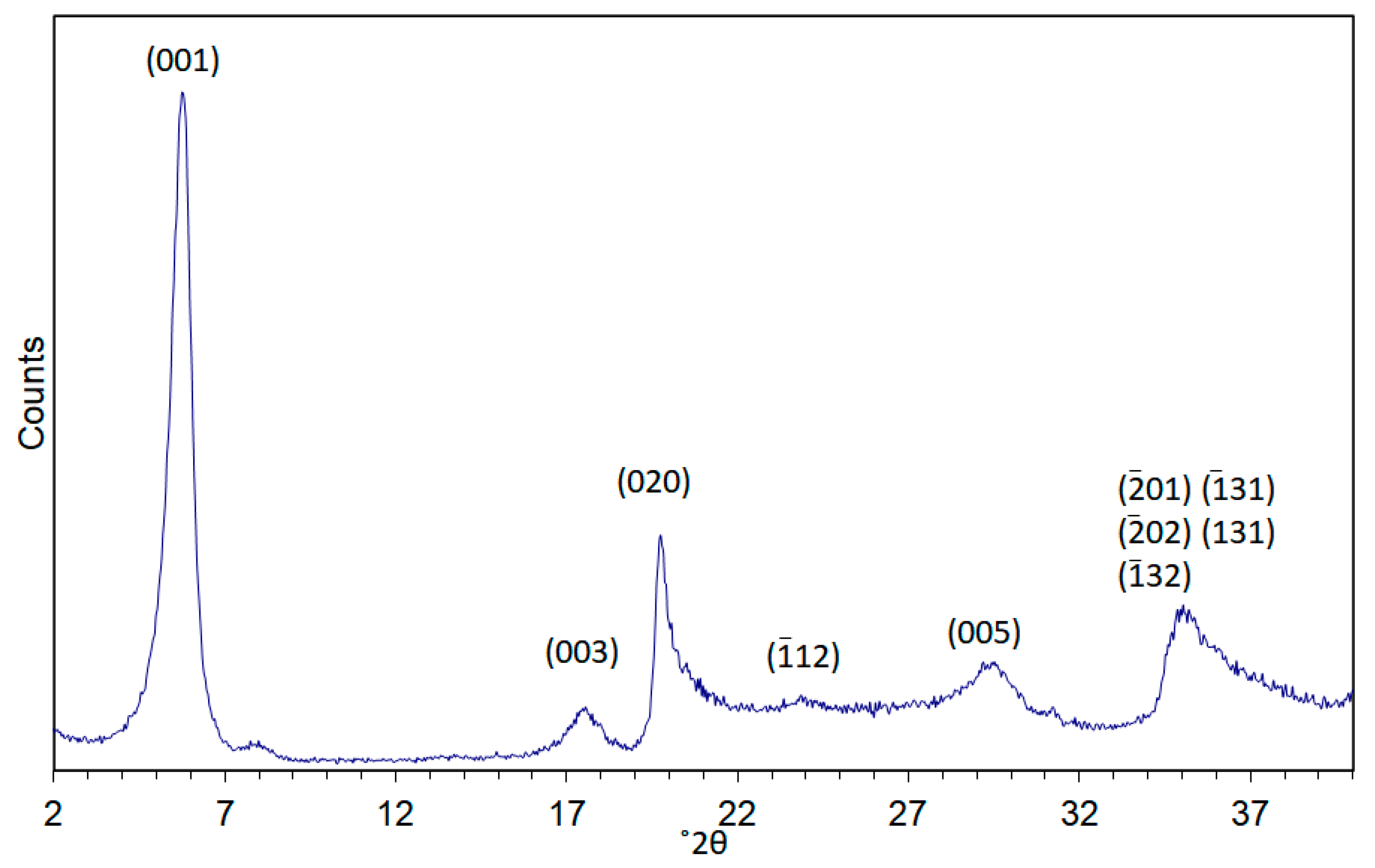
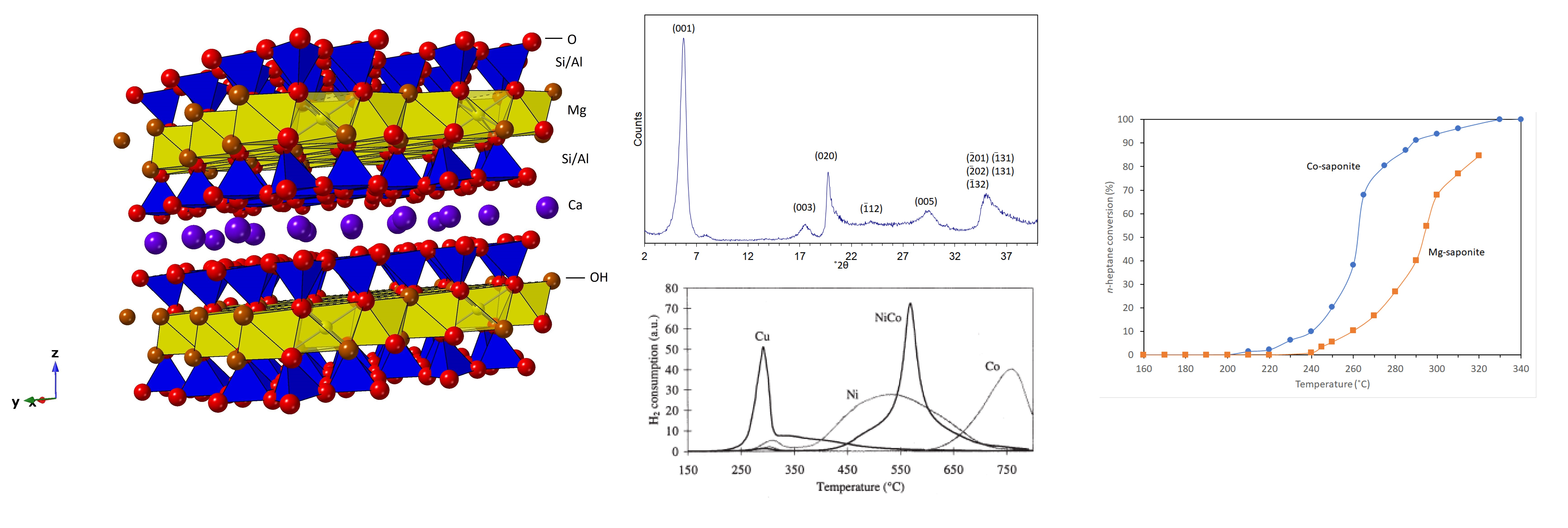{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
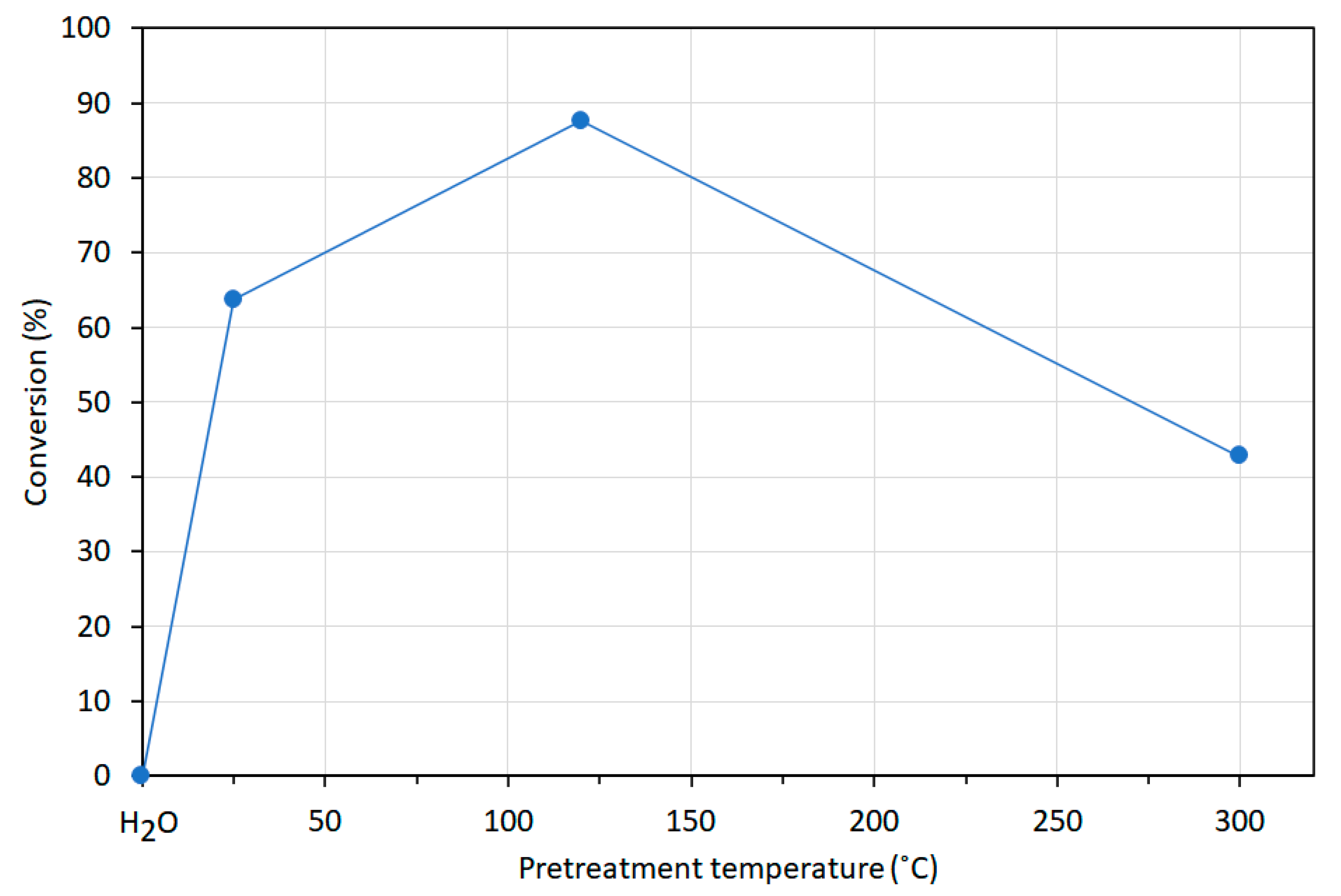{kind=link}
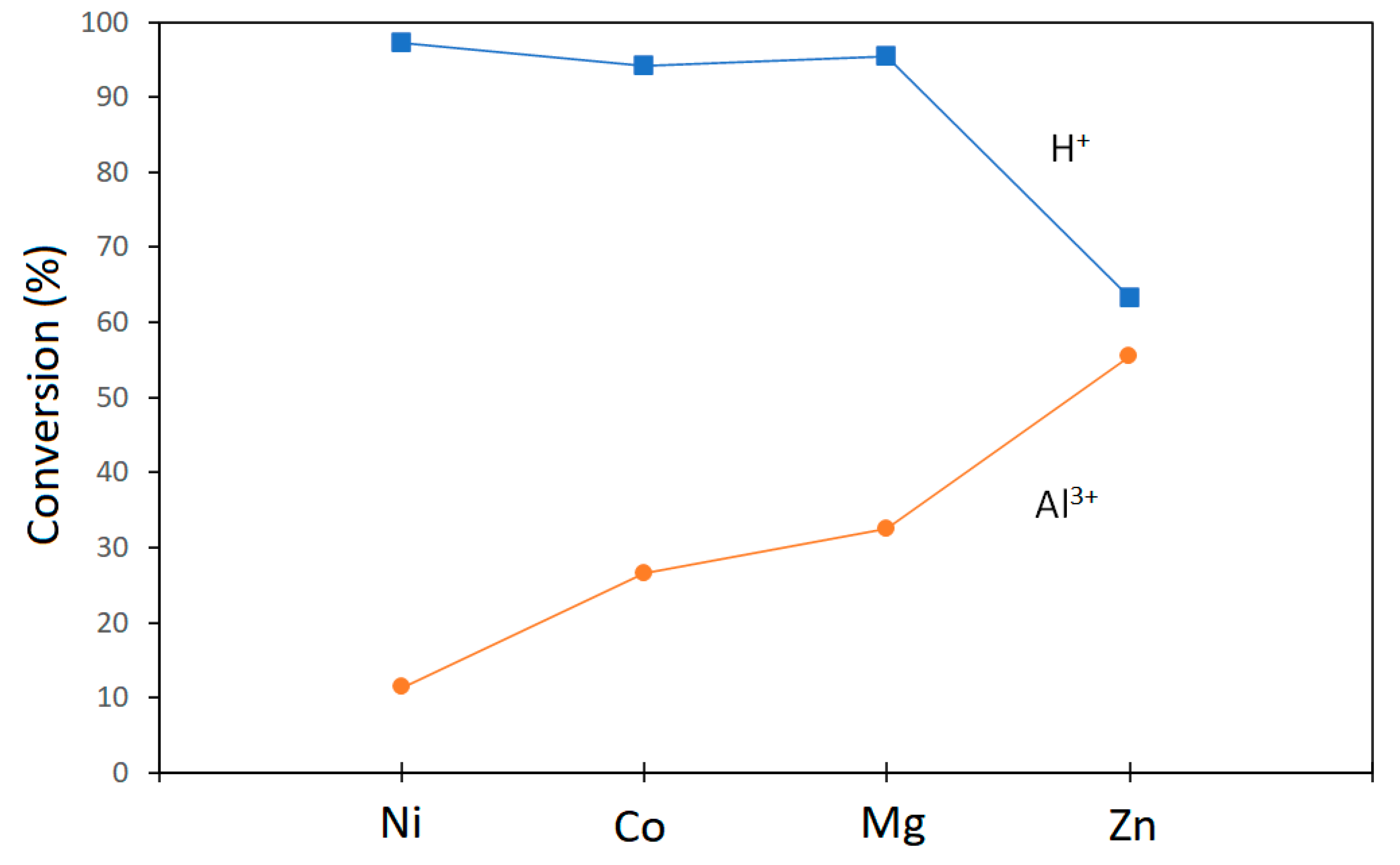{kind=link}
| Author/s, Year | Starting Materials | Conditions | Product/Product Properties | Notes |
|---|---|---|---|---|
| Kloprogge et al., 1993 [28] | SiO2, Al[OCH(CH3)2}3, Mg(CH3COO)2.4H2O, NH4OH; gel composition: (NH4)0.6Mg3Al0.6Si3.4O10(OH)2 | Autoclave at 125 to 280 °C for 72 h | NH4-saponite—high crystallinity, low CEC, high Al content in interlayer spacing or in octahedral sites | uncontrolled intercalation of Al3+ in the interlayer; non-swelling saponites with low amounts of NH4+; deceased catalytic activity |
| Kloprogge et al., 1994 [29] | modified from Kloprogge et al., 1993; solutions containing the desired cation were in the form of a hydroxide or fluoride salt; gel composition: (M)0.6Mg3Al0.6Si3.4O10(OH)2; (M = Na+, K+, Rb+, Ca2+, Ba2+, or Ce4+) | Autoclave at 200 °C and autogenous water pressure for 72 h | Mg-saponite—low CEC, low stacking of saponite sheets; considerable octahedral aluminum substitution; | (1) the presence of interlayer Mg2+ and the absence of interlayer Al3+; (2) a much higher amount of octahedral AI; and (3) that synthesis experiments with fluorine do not result in incorporation of extensive F- into the saponite structure replacing hydroxyl groups, nor in the formation of sellaite, MgF2 |
| Kawi and Yao, 1999 [33] | sodium silicate solution; NaOH/NaHCO3; chlorides of M3+ = Al3+, and M2+ = Mg2+, Ni2+; saponite of theoretical formula [MgxNi6-x](Si7Al)O20(OH)4 | Autoclave at 285 °C for 48 h | saponite with varied Mg/Ni ratio | Mg and Ni were incorporated in octahedral sites; tested as catalyst for dehydration of IPA to propene and dehydrogenation to acetone |
| Vogels et al., 1997 [31] | modified from Kloprogge et al., 1993; stoichiometric powder mix of SiO2 and Mg(CH3COO)2⋅4H2O; NH4Cl or NH4F with the Al[OCH(CH3)2}3 dissolved in the aqueous ammonium solutions before mixing with the powder | Autoclave at 200 °C and autogenous water pressure; varying synthesis time | NH4-saponite | The crystallinity of synthetic ammonium-saponite depends strongly on synthesis time, ammonium concentration and initial constituents of the gel. |
| Higashi, Miki and Komarmeni, 2007 [34] | silicic acid (containing 81.4% SiO2), MnCO3, Al(NO3)3∙9H2O, NaOH solution | Autoclave at 100–250 °C under autogenous pressure for 72–168 h | Mn-saponite with Mn-carbonate impurities | Was not analyzed further due to poor crystallinity. |
| Carniatto et al., 2009 [35] | modified from Kloprogge et al., 1993; SiO2, Al[OCH(CH3)2}3, Mg(CH3COO)2.4H2O, NH4OH, vanadium(IV) oxide sulphate hydrate (VOSO4∙xH2O; tetraethyl orthosilicate (TEOS); acidified ethanol | Autoclave at 240 °C for 72 h | V-saponite, crystalline | Strong potential as additive for polymer materials with flame retardant properties. |
| Bisio et al., 2011 [36] | modified from Kloprogge et al., 1993; the reagents enumerated to form the gel in Kloprogge et al., 1993 were added with hexadecyltrimethylammonium bromide (CTABr) | Autoclave at 200 °C for 72 h | organo-saponite | CTA- ions are essentially confined in the interlayer space of the saponite. |
| Sychev and Prihod’ko, 1998 [37] | Na2SiO3; NaOH; M3+-nitrate where M = Al, Fe or Cr; aqueous M2+-nitrate and urea; Si/M3+ ratio is varied from 3.0–12 | Sol-gel precipitation at 190 °C at 1 atm for 24 h | saponite like materials; poor crystallinity; high CEC | Possess acidic and basic/redox active sites which depends on the chemical composition of both tetrahedral and octahedral sheets of saponite. |
| Vogels et al., 2005 [27] | stoichiometric mixture containing Si/Al3+ gel from Na2SiO3 solution and Al(NO3)3∙9H2O; NaOH solution, M2+-nitrate (M2+ = Mg2+, Zn2+, Ni2+, Co2+, or Cu2+), urea, and water | Sol-gel co-precipitation at 90 °C for 20 h | M2+-saponites where (M2+ = Mg2+, Zn2+, Ni2+, Co2+, or Cu2+) | Thermal stability increases in order Zn2+, Co2+, Mg2+, to Ni2+ from 450 to 800 °C and is determined by the nature of the octahedral cation. |
| Xue and Pinnavaia, 2008 [38] | Modified from Vogels et al., 2005; stoichiometric mixture containing Si/Al3+ gel from water glass solution, Al(NO3)3∙9H2O, Mg(NO3)2∙6H2O, urea and water in a molar ratio of 6.6:0.40:3.0:10 per 400 moles of water | Sol-gel co-precipitation at 90 °C for 24 h | saponite, poor crystallinity, high surface area, BJH pore volume and pore size | Has decent transparency when incorporated in glassy epoxy polymer. |
| Schumann et al., 2012 [39] | 56.25% SiO2, 4.30% Al2O3, 30.88% MgO, and 3.97% K2O; saponite formula: K0.33Mg3(OH)2(Si3.67Al0.33)O10. The gel is added with NaOH and Na oxalate solutions | Sol-gel precipitation at 60 °C and ambient pressure for 3 months! | predominantly saponite with Mg2+ in interlayers and talc byproduct | Gives replicating clay minerals; possibly through template-catalyzed polymerization, charge distribution transmitted from layer to layer. |
| Besselink et al., 2020 [40] | Modified from Vogels et al., 2005 | 2-step sol-gel co-precipitation at 25–95 °C at varying synthesis time from 5 min to 90 days | Mg-saponite - nanocrystals; | Two-step saponite crystallization—(1) amorphous aluminosilicate network formation, (2) crystallization of this amorphous aluminosilicate network towards saponite in the presence of magnesium and urea. |
| Vicente, I. et al., 2010 [41] | slurry (≈9wt.% solids), with a Si4+:Al3+:Mg2+:NH4+ composition ratio of 14.3:2.5:12.5:8 which would result in saponite with the theoretical formula (NH4)1.2[Mg6Al1.2Si6.8O20(OH)4] | Microwave, 180 °C for 6 hrs | NH4-saponite; some have higher crystallinity, higher CEC and higher Al(Td)/Al(Oh) ratio of saponites formed | Physico-chemical properties of resulting saponite is influenced by initial slurry pH |
| Trujillano et al., 2011 [42] | sodium silicate solution (27% wt, d = 1.39g/mL); NaOH and NaHCO3; chlorides M3+ = Al3+ or Fe3+, and M2+ = Mg2+, Ni2+, or Fe2+; saponite theoretical formula [Si7M3+][M2+6O20(OH)4Na.nH2O | Microwave, 180 °C for 8h | saponites containing divalent Mg, Ni or Fe - large surface area | A potentially good oxidation catalyst especially for NiMgAl; possible synergistic effect takes place when two different cations are present in adjacent octahedral positions |
| Gebretsadik et al., 2015 [43] | modified from Trujillano, et al., 2011 and Gebretsadik et al., 2014 | Microwave, 180 °C for 6h | Na-saponite, NH4-saponite | Characterized by higher delamination with smaller lamella and higher BET area; higherincorporation of Al in the tetrahedral sheet (higher Al(T)/Al(O) ratio) |
2.3. Characterization of Synthetic Saponites
2.3.1. Powder X-ray Diffraction (XRD)
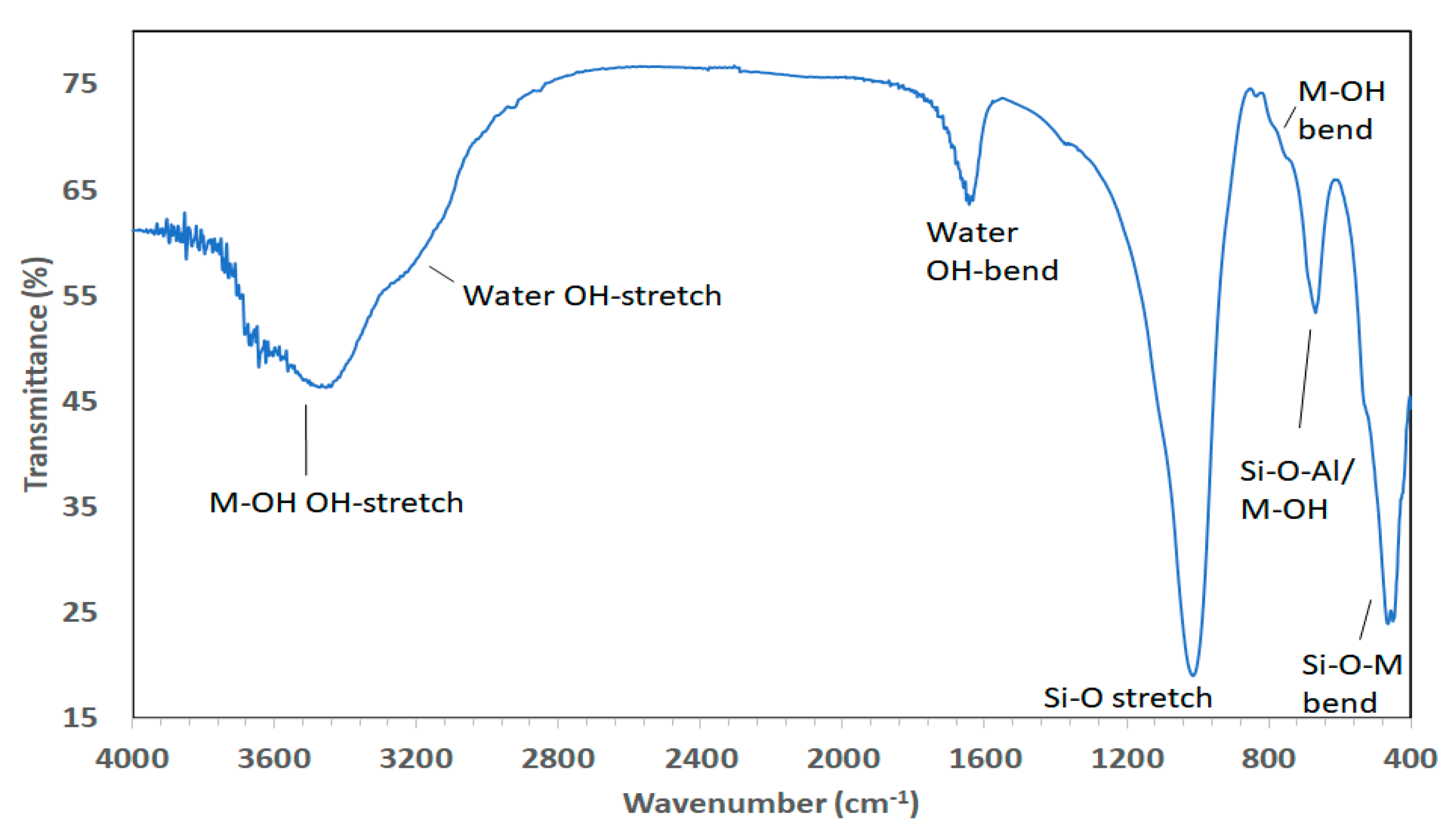
2.3.2. Infrared Spectroscopy (IR)
2.3.3. Transmission Electron Microscopy (TEM)
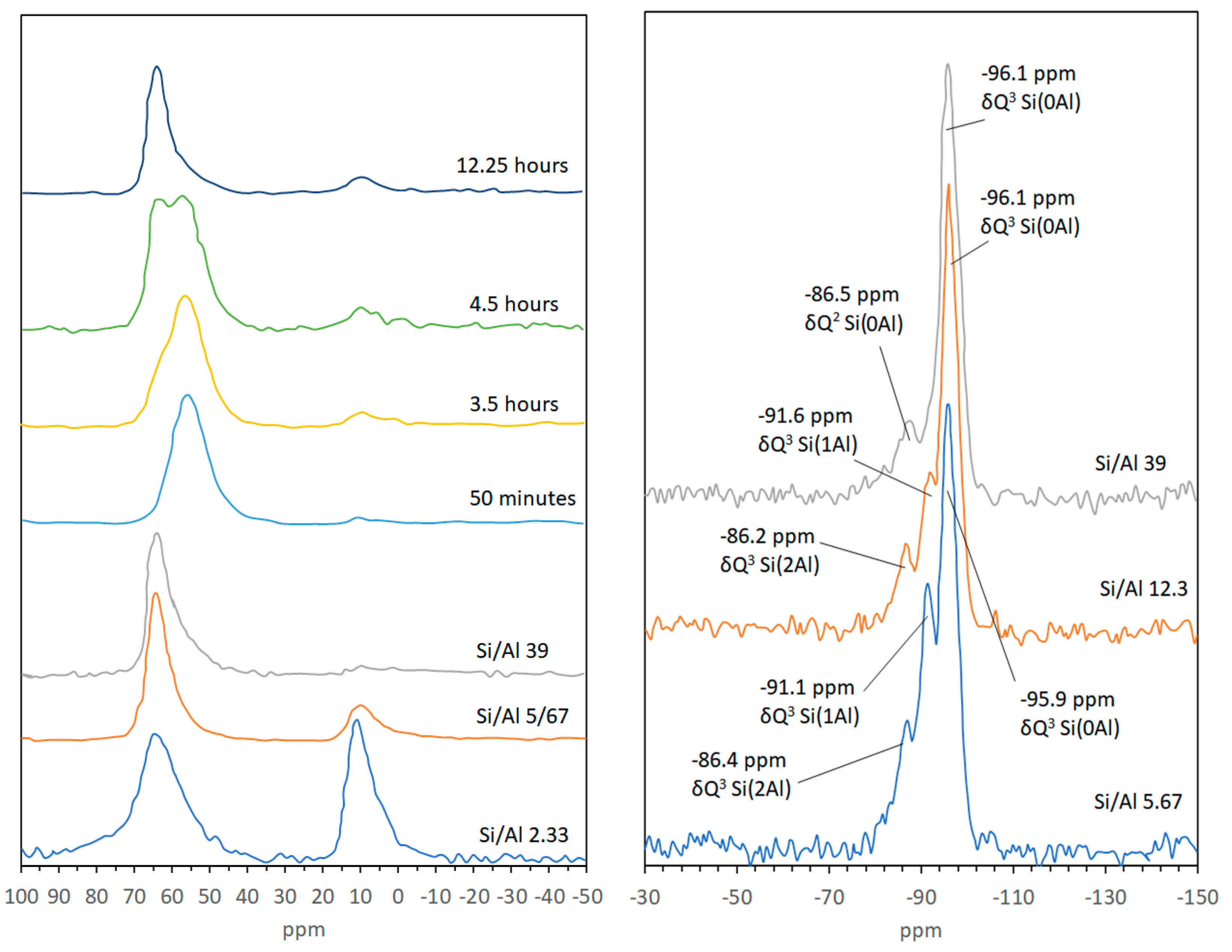
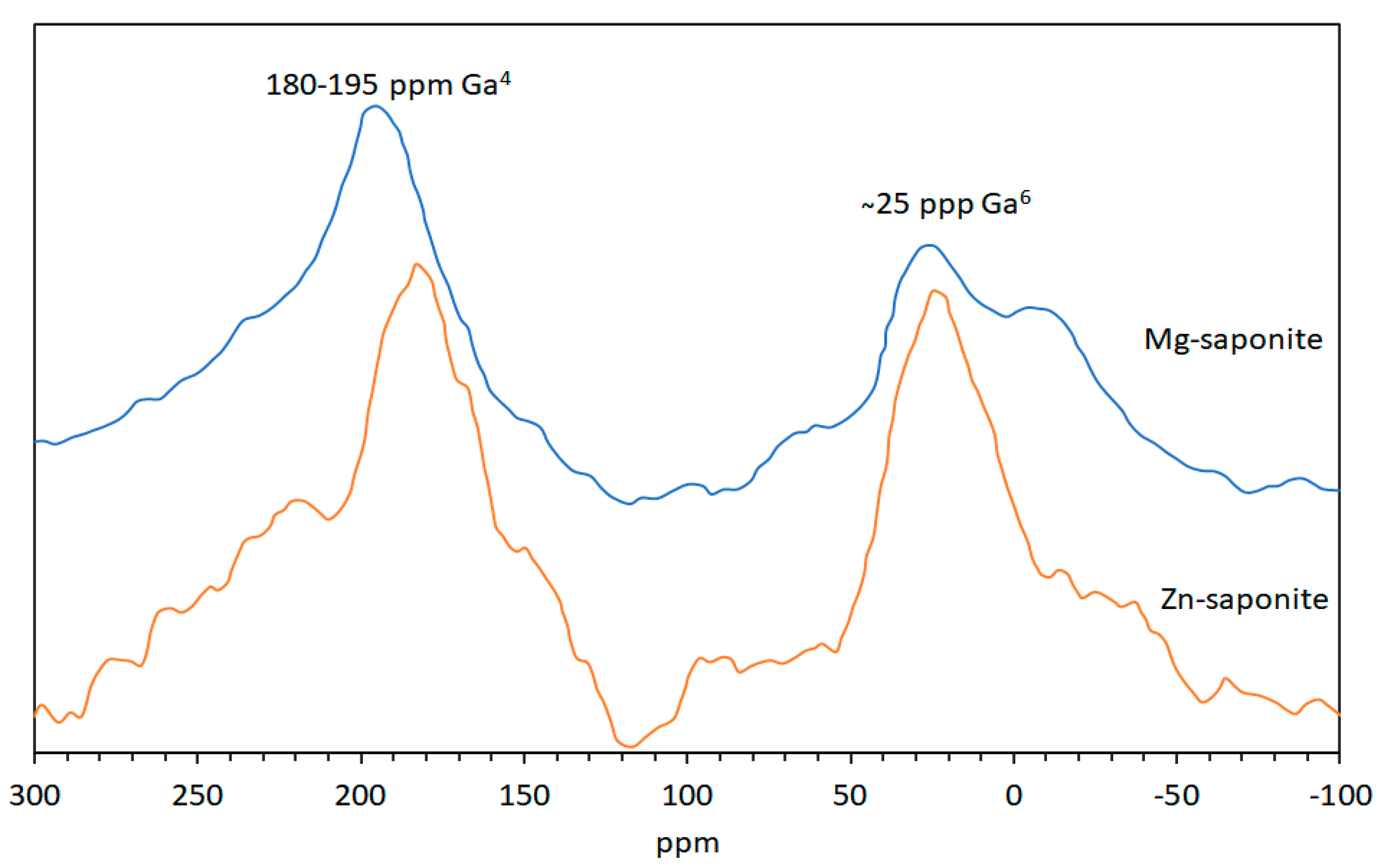
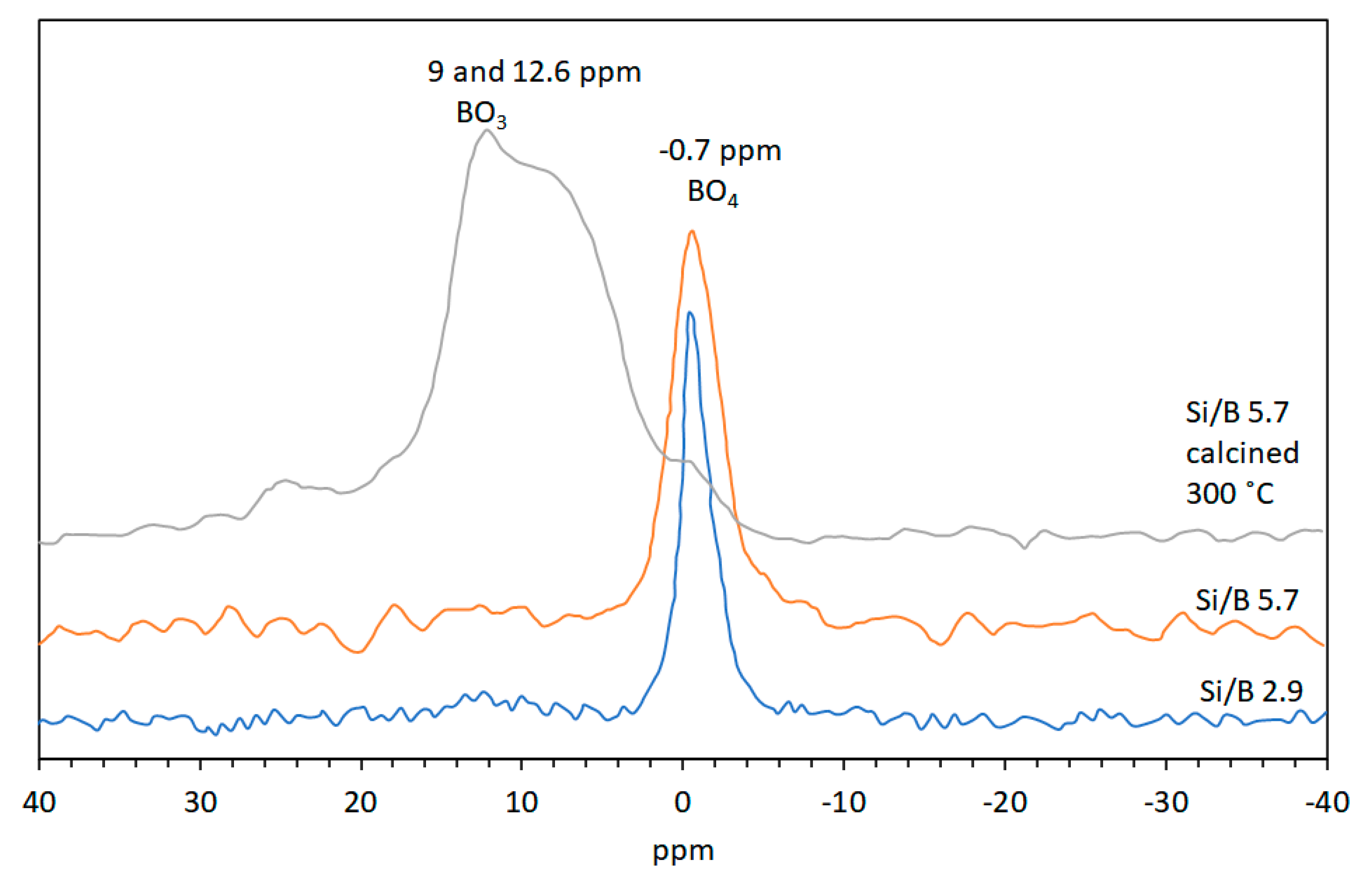
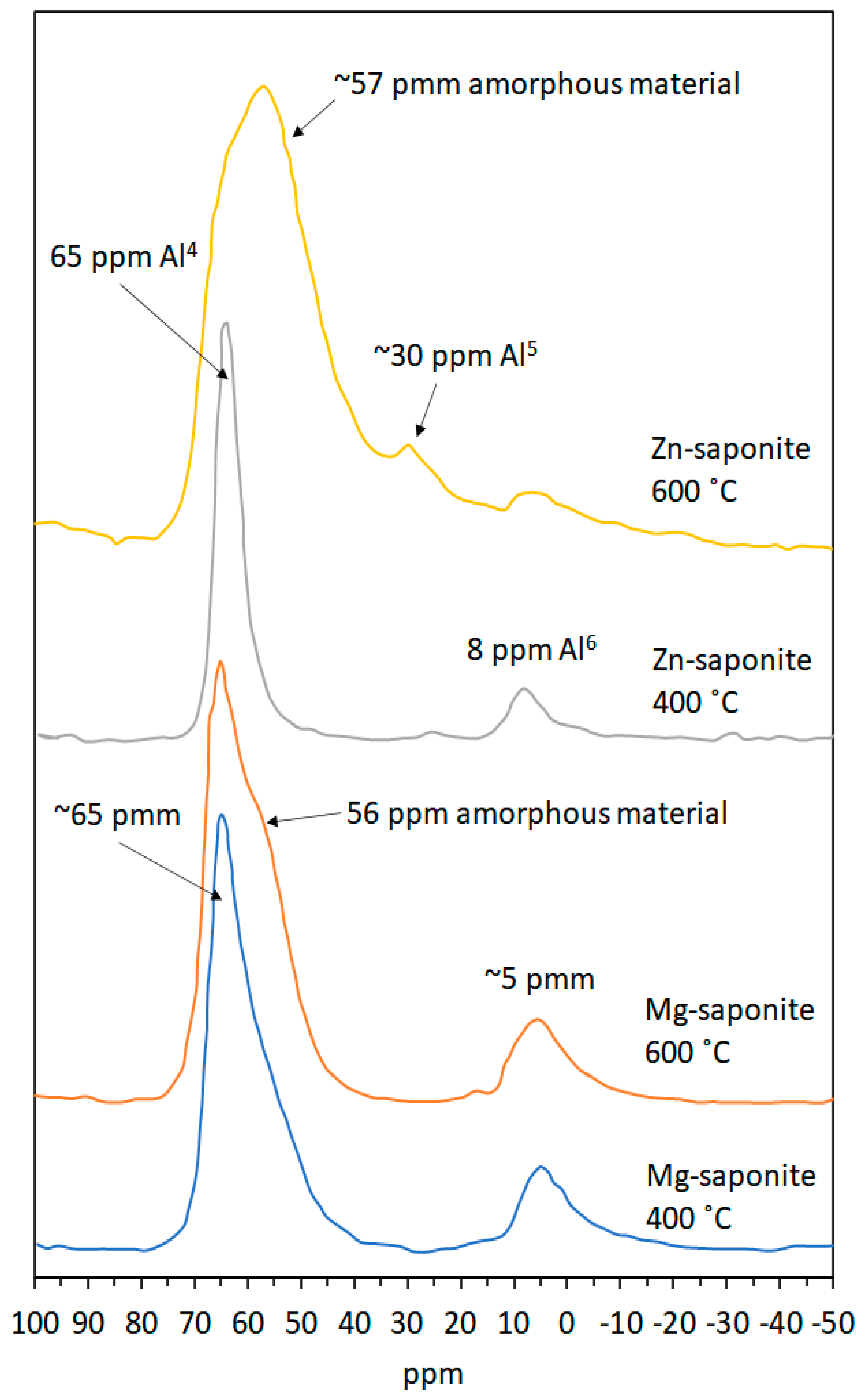
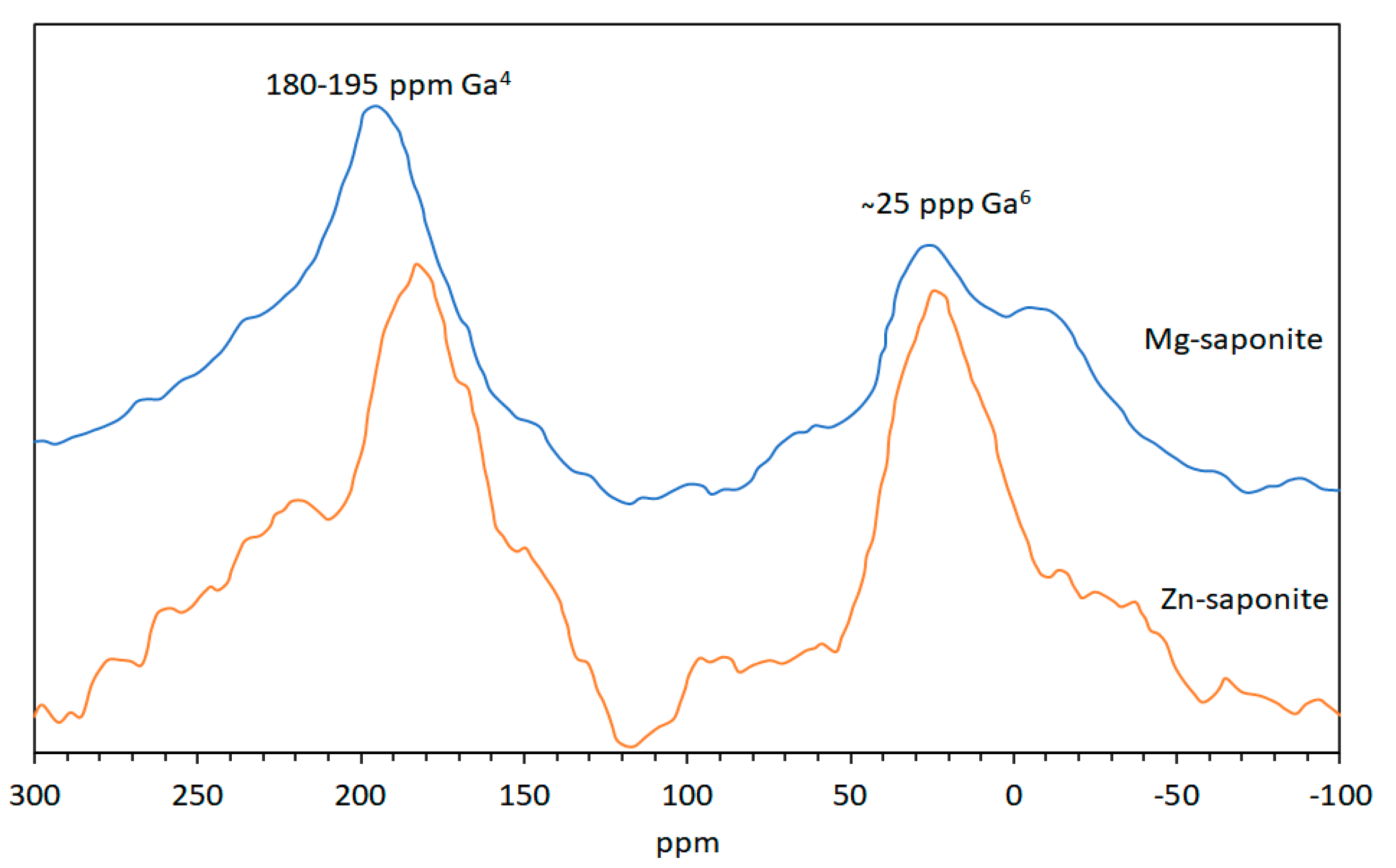
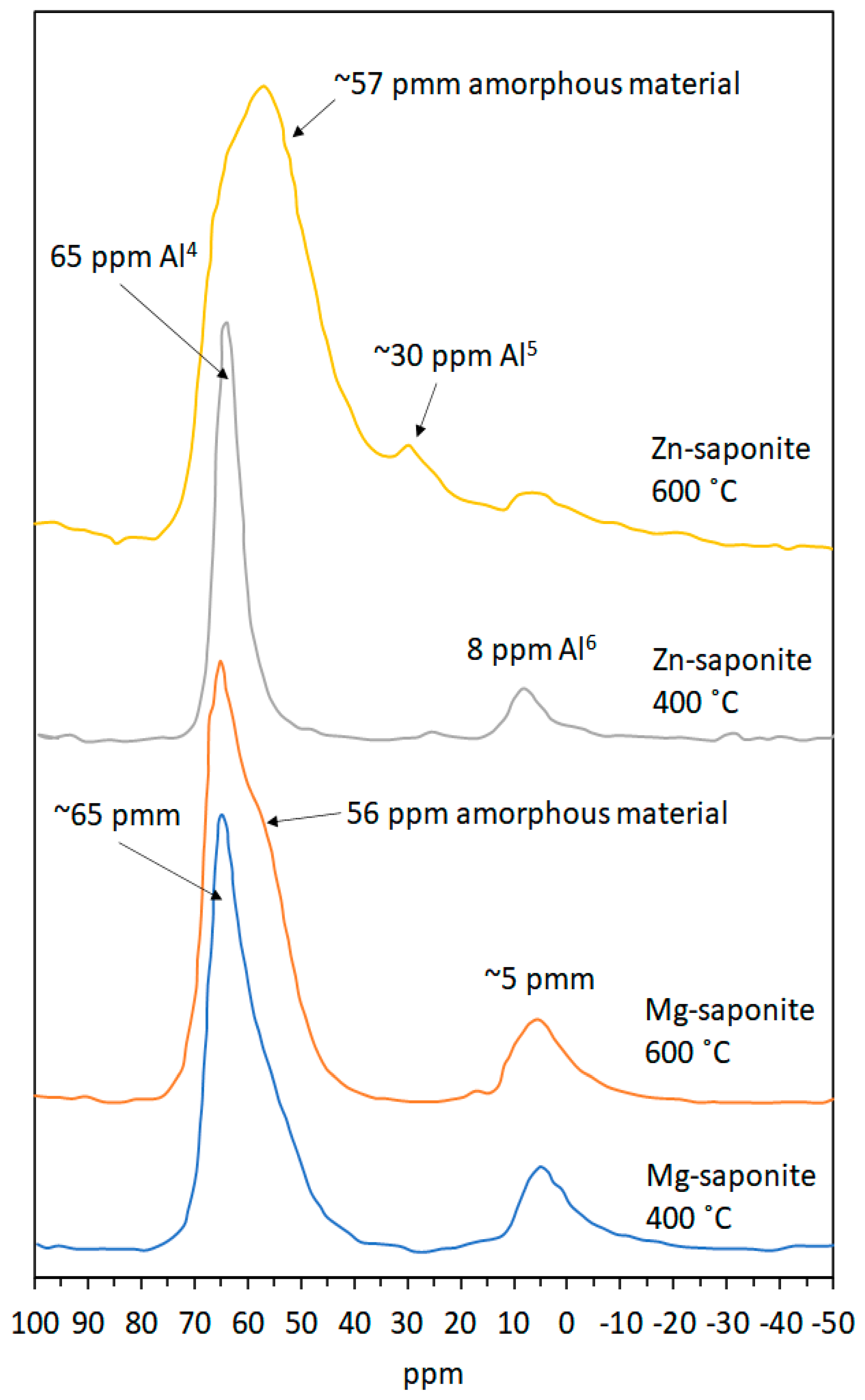
2.3.4. Magic-Angle Spinning-Nuclear Magnetic Resonance (MAS-NMR) Spectroscopy
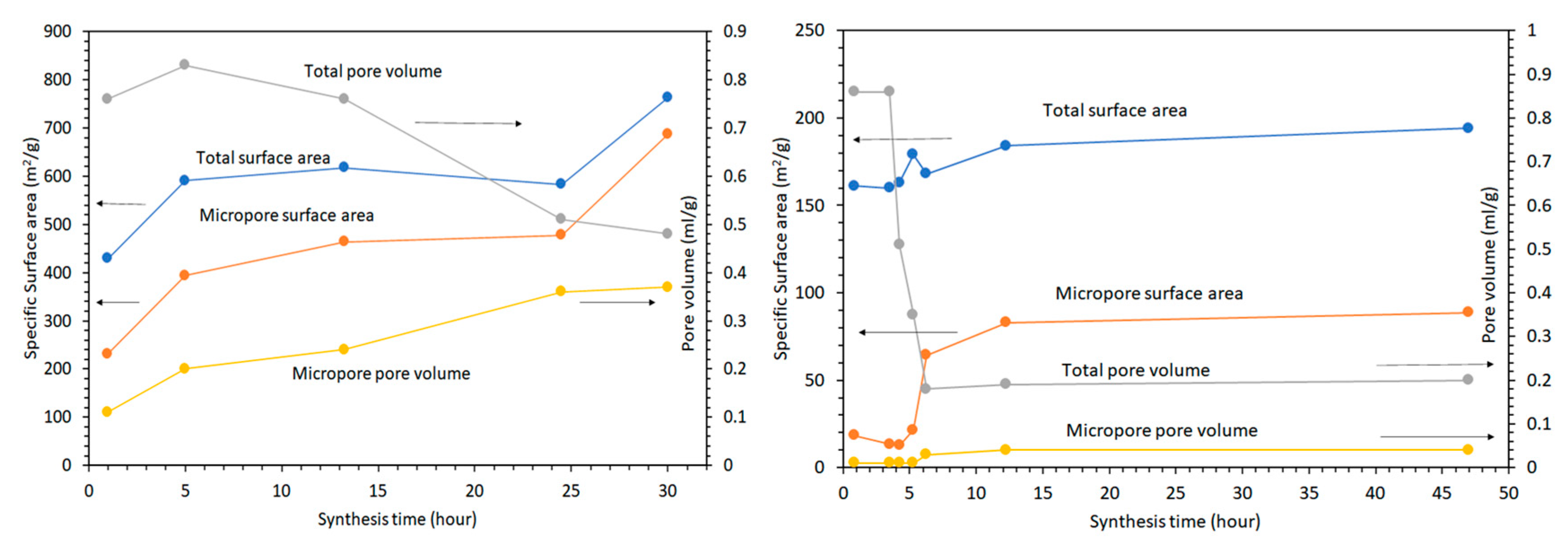
2.3.5. Nitrogen Physisorption Measurements
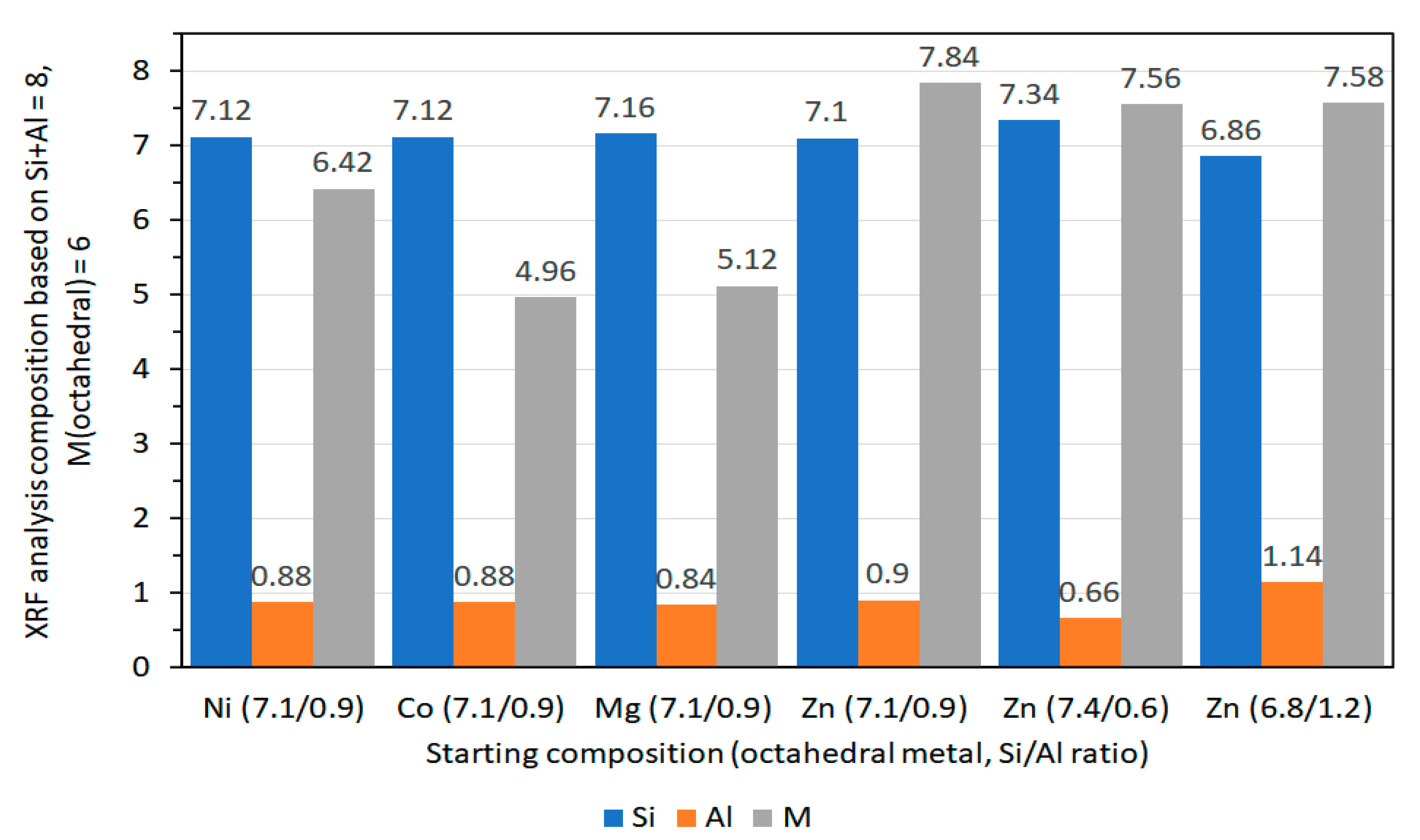
2.3.6. X-Ray Fluorescence Spectroscopy (XRF)
2.3.7. Extended X-Ray Absorption Fine Structure Spectroscopy (EXAFS)
2.3.8. pH Analysis
3. Thermal Properties of Saponites
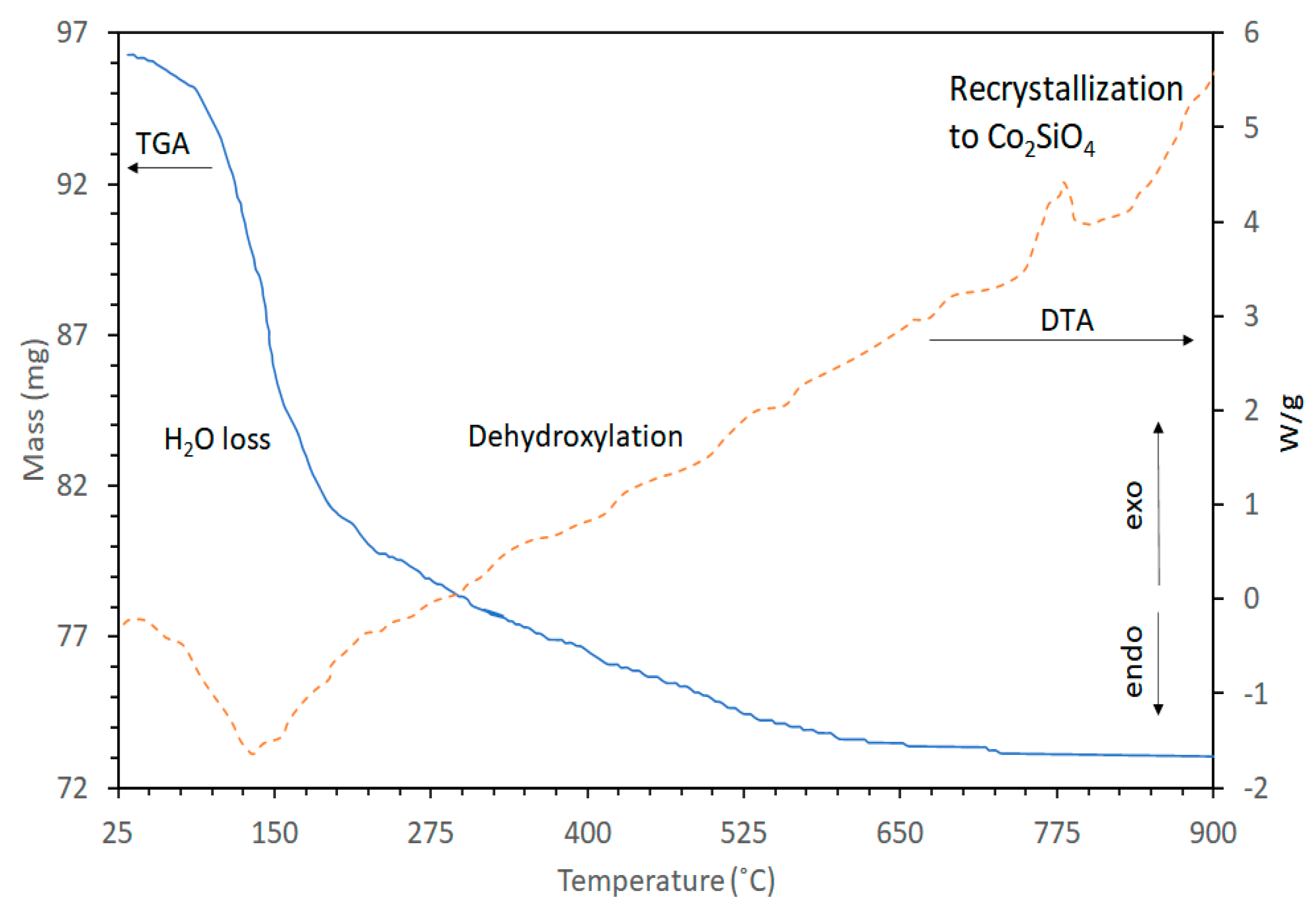
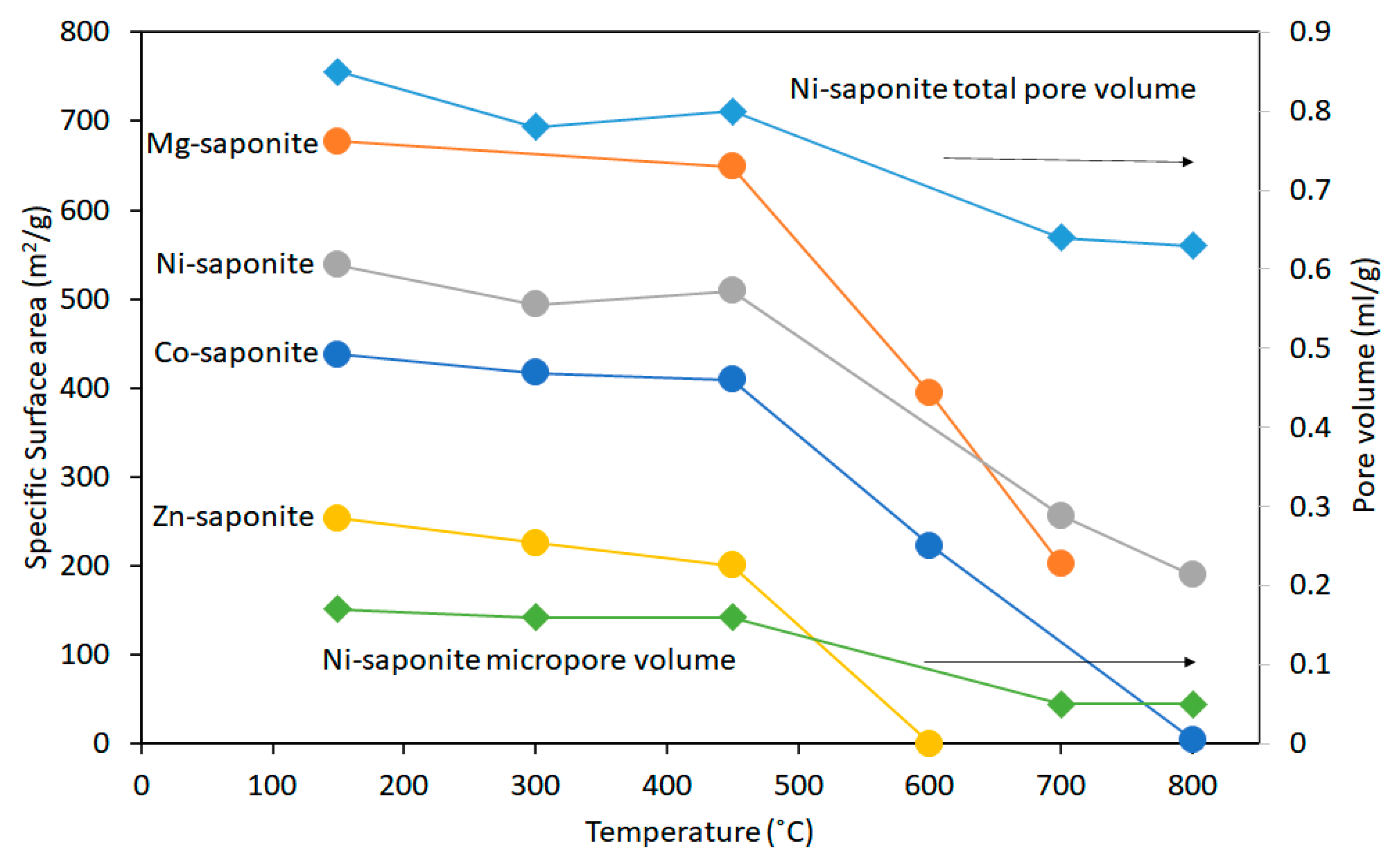
3.1. Thermal Stability
3.2. Hydrothermal Stability
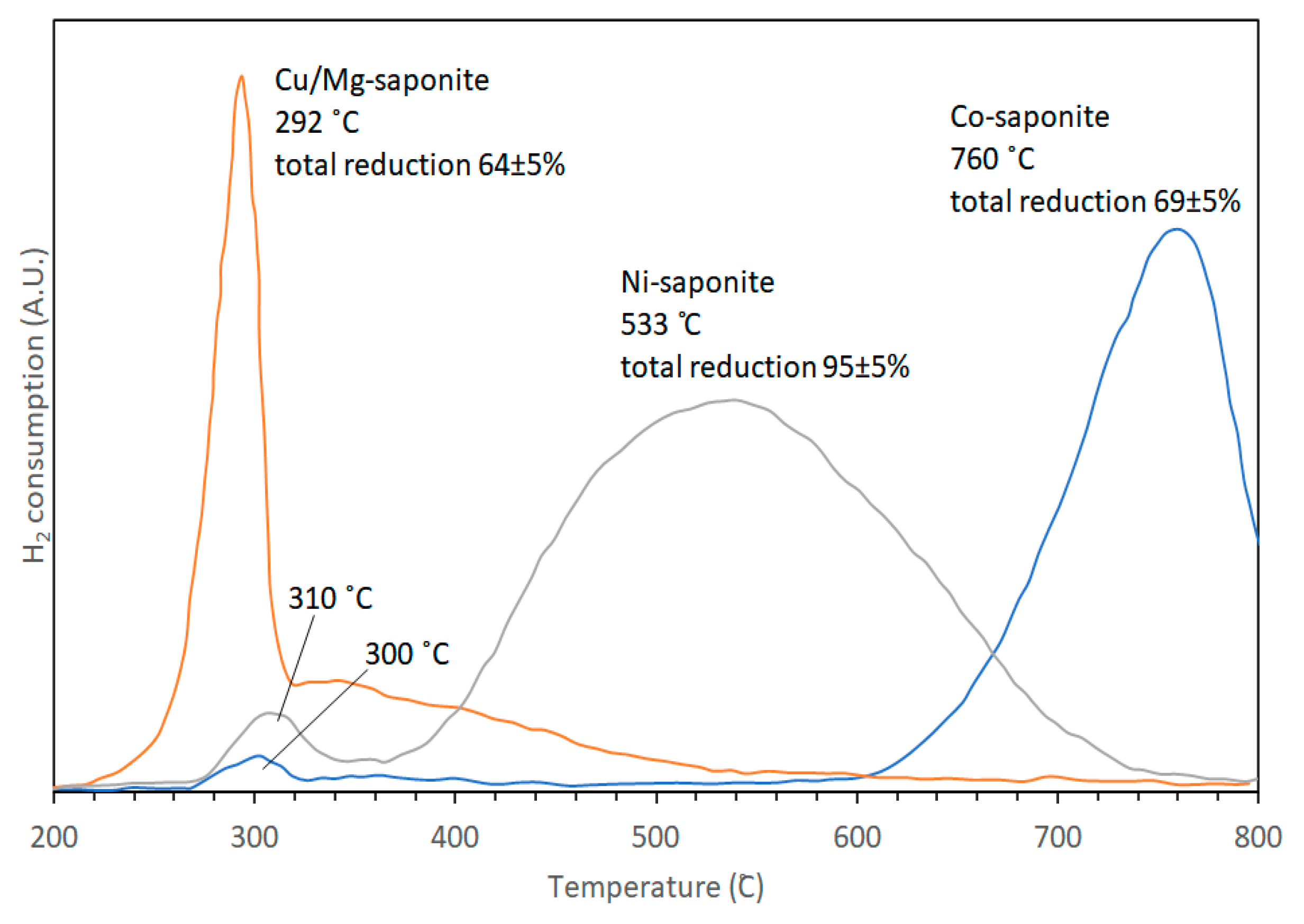
3.3. Thermal Stability in H2
4. Catalytic Properties
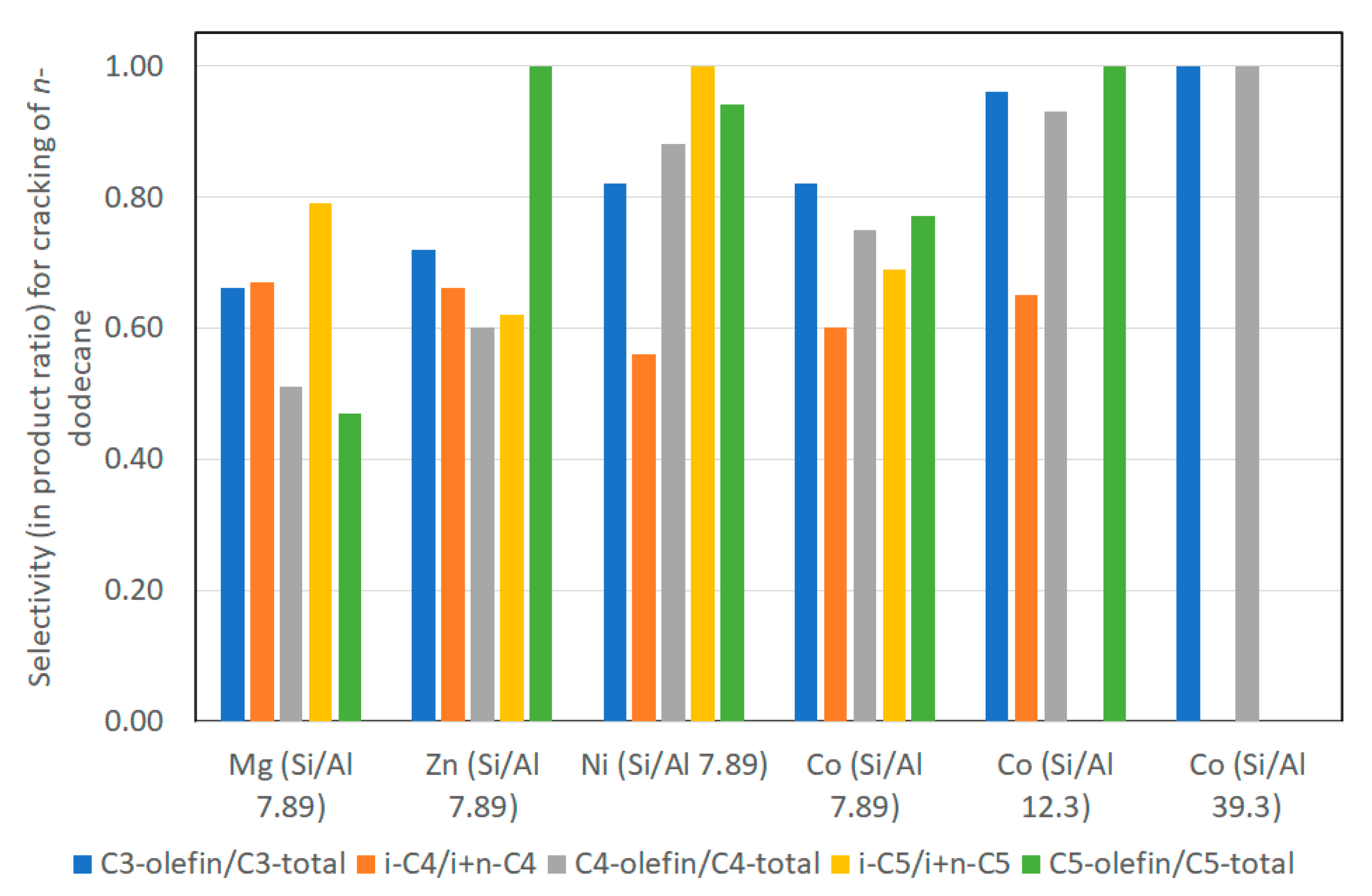
4.1. Cracking of n-Dodecane
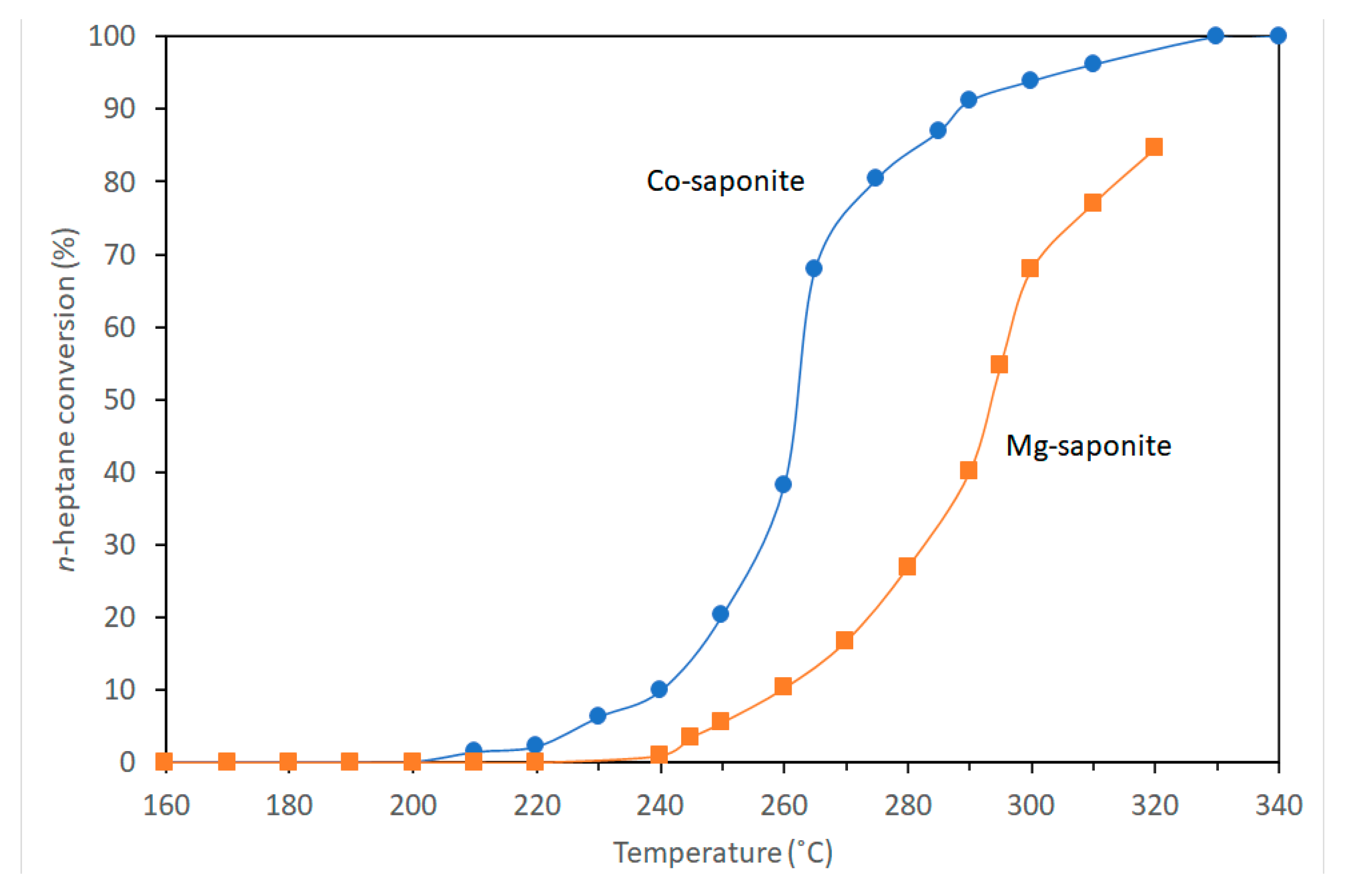
4.2. Hydro-Isomerization of n-Heptane
4.3. Friedel–Crafts Alkylation of Benzene with Propylene to Cumene
5. Summary
Author Contributions
Funding
Conflicts of Interest
References
- Bernal, J.D. The Physical Basis of Life. Proc. Phys. Soc. Sect. B 1949, 62, 597–618. [Google Scholar] [CrossRef]
- Cairns-Smith, A.G.; Hartman, H. Clay Minerals and the Origin of Life; Cambridge University Press: Cambridge, UK, 1986. [Google Scholar]
- Balogh, M.; Laszlo, P. Organic Chemistry Using Clays; Springer: Berlin/Heidelberg, Germany, 1993. [Google Scholar]
- Aldersley, M.F.; Joshi, P.C.; Price, J.D.; Ferris, J.P. The role of montmorillonite in its catalysis of RNA synthesis. Appl. Clay Sci. 2011, 54, 1–14. [Google Scholar] [CrossRef]
- Ertem, G.; Ferris, J.P. Synthesis of RNA oligomers on heterogeneous templates. Nature 1996, 379, 238–240. [Google Scholar] [CrossRef] [PubMed]
- Ferris, J.P.; Joshi, P.C.; Wang, K.J.; Miyakawa, S.; Huang, W. Catalysis in prebiotic chemistry: Application to the synthesis of RNA oligomers. Adv. Space Res. 2004, 33, 100–105. [Google Scholar] [CrossRef]
- Joshi, P.C.; Aldersley, M.F.; Ferris, J.P. Homochiral Selectivity in RNA Synthesis: Montmorillonite-catalyzed Quaternary Reactions of D, L-Purine with D, L- Pyrimidine Nucleotides. Orig. Life Evol. Biosph. 2011, 41, 213–236. [Google Scholar] [CrossRef]
- Huang, W.; Ferris, J.P. Synthesis of 35–40 mers of RNA oligomers from unblocked monomers. A simple approach to the RNA world. Chem. Commun. 2003, 1458–1459. [Google Scholar] [CrossRef]
- Negron-Mendoza, A.; Ramos-Bernal, S.; Mosqueira, F.G. Synthesis in prebiotic clay environments induced by radiation. Int. J. Astrobiol. 2004, 3, 295–300. [Google Scholar] [CrossRef]
- Moorbath, S. Age of the oldest rocks with biogenic components. J. Biol. Phys. 1995, 20, 85–94. [Google Scholar] [CrossRef]
- Deamer, D.; Singaram, S.; Rajamani, S.; Kompanichenko, V.; Guggenheim, S. Self-assembly processes in the prebiotic environment. Philos. Trans. R Soc. Lond. B Biol. Sci. 2006, 361, 1809–1818. [Google Scholar] [CrossRef] [Green Version]
- Williams, L.B.; Canfield, B.; Voglesonger, K.M.; Holloway, J.R. Organic molecules formed in a “primordial womb”. Geology 2005, 33, 913–916. [Google Scholar] [CrossRef]
- Aldersley, M.F.; Joshi, P.C. RNA dimer synthesis using montmorillonite as a catalyst: The role of surface layer charge. Appl. Clay Sci. 2013, 83–84, 77–82. [Google Scholar] [CrossRef]
- Bu, H.; Yuan, P.; Liu, H.; Liu, D.; Qin, Z.; Zhong, X.; Song, H.; Li, Y. Formation of macromolecules with peptide bonds via the thermal evolution of amino acids in the presence of montmorillonite: Insight into prebiotic geochemistry on the early Earth. Chem. Geol. 2019, 510, 72–83. [Google Scholar] [CrossRef]
- Scappini, F.; Casadei, F.; Zamboni, R.; Franchi, M.; Gallori, E.; Monti, S. Protective effect of clay minerals on adsorbed nucleic acid against UV radiation: Possible role in the origin of life. Int. J. Astrobiol. 2004, 3, 17–19. [Google Scholar] [CrossRef]
- Ferris, J.P.; Ertem, G. Montmorillonite catalysis of RNA oligomer formation in aqueous solution. A model for the prebiotic formation of RNA. J. Am. Chem. Soc. 1993, 115, 12270–12275. [Google Scholar] [CrossRef] [PubMed]
- Ménez, B.; Pisapia, C.; Andreani, M.; Jamme, F.; Vanbellingen, Q.P.; Brunelle, A.; Richard, L.; Dumas, P.; Réfrégiers, M. Abiotic synthesis of amino acids in the recesses of the oceanic lithosphere. Nature 2018, 564, 59–63. [Google Scholar] [CrossRef]
- Bernhardt, H.S. Making Molecules with Clay: Layered Double Hydroxides, Pentopyranose Nucleic Acids and the Origin of Life. Life 2019, 9, 19. [Google Scholar] [CrossRef] [Green Version]
- Erastova, V.; Degiacomi, M.T.; Fraser, D.G.; Greenwell, H.C. Mineral surface chemistry control for origin of prebiotic peptides. Nat. Commun. 2017, 8, 2033. [Google Scholar] [CrossRef] [Green Version]
- Ponnamperuma, C.; Shimoyama, A.; Friebele, E. Clay and the origin of life. Orig. Life 1982, 12, 9–40. [Google Scholar] [CrossRef]
- Ferris, J.P. Mineral Catalysis and Prebiotic Synthesis: Montmorillonite-Catalyzed Formation of RNA. Elements 2005, 1, 145–149. [Google Scholar] [CrossRef]
- Brack, A. Chapter 10.4—Clay Minerals and the Origin of Life. In Developments in Clay Science; Bergaya, F., Lagaly, G., Eds.; Elsevier: Amsterdam, The Netherlands, 2013; Volume 5, pp. 507–521. [Google Scholar]
- Sueoka, Y.; Yamashita, S.; Kouduka, M.; Suzuki, Y. Deep Microbial Colonization in Saponite-Bearing Fractures in Aged Basaltic Crust: Implications for Subsurface Life on Mars. Front. Microbiol. 2019, 10, 2793. [Google Scholar] [CrossRef] [Green Version]
- dos Santos, R.; Patel, M.; Cuadros, J.; Martins, Z. Influence of mineralogy on the preservation of amino acids under simulated Mars conditions. Icarus 2016, 277, 342–353. [Google Scholar] [CrossRef]
- Fialho, D.M.; Roche, T.P.; Hud, N.V. Prebiotic Syntheses of Noncanonical Nucleosides and Nucleotides. Chem. Rev. 2020, 120, 4806–4830. [Google Scholar] [CrossRef] [PubMed]
- Menor-Salván, C. From the Dawn of Organic Chemistry to Astrobiology: Urea as a Foundational Component in the Origin of Nucleobases and Nucleotides. In Prebiotic Chemistry and Chemical Evolution of Nucleic Acids; Menor-Salván, C., Ed.; Springer International Publishing: Cham, Switzerland, 2018; pp. 85–142. [Google Scholar] [CrossRef]
- Vogels, R.J.M.J.; Kloprogge, J.T.; Geus, J.W. Synthesis and characterization of saponite clays. Am. Mineral. 2005, 90, 931–944. [Google Scholar] [CrossRef]
- Kloprogge, J.T.; Breukelaar, J.; Jansen, J.B.H.; Geus, J.W. Development of Ammonium-Saponites from Gels with Variable Ammonium Concentration and Water Content at Low Temperatures. Clays Clay Miner. 1993, 41, 103–110. [Google Scholar] [CrossRef]
- Kloprogge, J.T.; Breukelaar, J.; Geus, J.W.; Jansen, J.B.H. Characterization of Mg-Saponites Synthesized from Gels Containing Amounts of Na+, K+, Rb+, Ca2+, Ba2+, or Ce4+ Equivalent to The CEC of the Saponite. Clays Clay Miner. 1994, 42, 18–22. [Google Scholar] [CrossRef]
- Booij, E.; Kloprogge, J.T.; Van Veen, J.A.R. Preparation, Structural Characteristics and Catalytic Properties of Large-Pore Rare Earth Element (Ce, La)/Al-Pillared Smectites. Clays Clay Miner. 1996, 44, 774–782. [Google Scholar] [CrossRef]
- Vogels, R.J.M.J.; Breukelaar, J.; Kloprogge, J.T.; Jansen, J.B.H.; Geus, J.W. Hydrothermal Crystallization of Ammonium-Saponite at 200 °C and Autogenous Water Pressure. Clays Clay Miner. 1997, 45, 1–7. [Google Scholar] [CrossRef]
- Kloprogge, J.T.; Komarneni, S.; Amonette, J.E. Synthesis of Smectite Clay Minerals: A Critical Review. Clays Clay Miner. 1999, 47, 529–554. [Google Scholar] [CrossRef]
- Kawi, S.; Yao, Y.Z. Saponite catalysts with systematically varied Mg/Ni ratio: Synthesis, characterization, and catalysis. Microporous Mesoporous Mater. 1999, 33, 49–59. [Google Scholar] [CrossRef]
- Higashi, S.; Miki, H.; Komarneni, S. Mn-smectites: Hydrothermal synthesis and characterization. Appl. Clay Sci. 2007, 38, 104–112. [Google Scholar] [CrossRef] [Green Version]
- Carniato, F.; Bisio, C.; Gatti, G.; Roncoroni, S.; Recchia, S.; Marchese, L. On the Properties of a Novel V-Containing Saponite Catalyst for Propene Oxidative Dehydrogenation. Catal. Lett. 2009, 131, 42–48. [Google Scholar] [CrossRef]
- Bisio, C.; Carniato, F.; Paul, G.; Gatti, G.; Boccaleri, E.; Marchese, L. One-Pot Synthesis and Physicochemical Properties of an Organo-Modified Saponite Clay. Langmuir 2011, 27, 7250–7257. [Google Scholar] [CrossRef] [PubMed]
- Sychev, M.; Prihod’ko, R. Characterization and catalytic activity of non-hydrothermally synthesized saponite-like materials. Stud. Surf. Sci. Catal. 1998, 118, 967–974. [Google Scholar]
- Xue, S.; Pinnavaia, T.J. Porous synthetic smectic clay for the reinforcement of epoxy polymers. Microporous Mesoporous Mater. 2008, 107, 134–140. [Google Scholar] [CrossRef]
- Schumann, D.; Hartman, H.; Eberl, D.D.; Sears, S.K.; Hesse, R.; Vali, H. Formation of replicating saponite from a gel in the presence of oxalate: Implications for the formation of clay minerals in carbonaceous chondrites and the origin of life. Astrobiology 2012, 12, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Besselink, R.; Stawski, T.M.; Freeman, H.M.; Hövelmann, J.; Tobler, D.J.; Benning, L.G. Mechanism of Saponite Crystallization from a Rapidly Formed Amorphous Intermediate. Cryst. Growth Des. 2020, 20, 3365–3373. [Google Scholar] [CrossRef] [Green Version]
- Vicente, I.; Salagre, P.; Cesteros, Y.; Medina, F.; Sueiras, J.E. Microwave-assisted synthesis of saponite. Appl. Clay Sci. 2010, 48, 26–31. [Google Scholar] [CrossRef]
- Trujillano, R.; Rico, E.; Vicente, M.A.; Rives, V.; Ciuffi, K.J.; Cestari, A.; Gil, A.; Korili, S.A. Rapid microwave-assisted synthesis of saponites and their use as oxidation catalysts. Appl. Clay Sci. 2011, 53, 326–330. [Google Scholar] [CrossRef]
- Gebretsadik, F.B.; Mance, D.; Baldus, M.; Salagre, P.; Cesteros, Y. Microwave synthesis of delaminated acid saponites using quaternary ammonium salt or polymer as template. Study of pH influence. Appl. Clay Sci. 2015, 114, 20–30. [Google Scholar] [CrossRef]
- Vogels, R.J.M.J.; Kloprogge, J.T.; Geus, J.W.; Beers, A.W.F. Synthesis and characterization of saponite clays: Part 2. Thermal stability. Am. Mineral. 2005, 90, 945–953. [Google Scholar] [CrossRef]
- Vogels, R.J.M.J.; Kloprogge, J.T.; Geus, J.W. Synthesis and characterisation of boron and gallium substituted saponite clays below 100°C at one atmosphere. Microporous Mesoporous Mater. 2005, 77, 159–165. [Google Scholar] [CrossRef] [Green Version]
- Vogels, R.J.M.J.; Kloprogge, J.T.; Geus, J.W. Homogeneous forced hydrolysis of aluminum through the thermal decomposition of urea. J. Colloid Interface Sci. 2005, 285, 86–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prihod’ko, R.; Sychev, M.; Hensen, E.J.M.; van Veen, J.A.R.; van Santen, R.A. Preparation, characterization and catalytic activity of non-hydrothermally synthesized saponite-like materials. Stud. Surf. Sci. Catal. 2002, 142A, 271–278. [Google Scholar]
- Yu, B.-S.; Hung, W.-H.; Fang, J.-N.; Yu, Y.-T. Synthesis of Zn-Saponite Using a Microwave Circulating Reflux Method under Atmospheric Pressure. Minerals 2020, 10, 45. [Google Scholar] [CrossRef] [Green Version]
- Gull, M.; Omran, A.; Feng, T.; Pasek, M.A. Silicate-, Magnesium Ion-, and Urea-Induced Prebiotic Phosphorylation of Uridine via Pyrophosphate; Revisiting the Hot Drying Water Pool Scenario. Life 2020, 10, 122. [Google Scholar] [CrossRef]
- Vogels, R.J.M.J.; Kloprogge, J.T.; Geus, J.W. Catalytic activity of synthetic saponite clays: Effects of tetrahedral and octahedral composition. J. Catal. 2005, 231, 443–452. [Google Scholar] [CrossRef] [Green Version]
- Kahle, M.; Kleber, M.; Jahn, R. Review of XRD-based quantitative analyses of clay minerals in soils: The suitability of mineral intensity factors. Geoderma 2002, 109, 191–205. [Google Scholar] [CrossRef]
- Decarreau, A. Étude expérimentale de la cristallogenèse des smectites. Mesures des coefficients de partage smectite trioctahédrique -solution aqueuse pour les métaux M2+ de la première série de transition. Sci. Geol. Mémoire 1983, 74, 1–191. [Google Scholar]
- Gates, W.P.; Kloprogge, J.T.; Madejova, J.; Bergaya, F. Infrared and Raman Spectroscopies of Clay Minerals; Elsevier: Amsterdam, The Netherlands, 2017; Volume 8, p. 604. [Google Scholar]
- Kloprogge, J.T. Application of vibrational spectroscopy in clay mineral synthesis. In Infrared and Raman Spectroscopies of Clay Minerals; Gates, W.P., Kloprogge, J.T., Madejova, J., Bergaya, F., Eds.; Elsevier: Amsterdam, The Netherlands, 2017; Volume 8, pp. 222–287. [Google Scholar]
- van der Marel, H.W.; Beutelspacher, H. Atlas of Infrared Spectroscopy of Clay Minerals and Their Admixtures; Elsevier: Amsterdam, The Netherlands, 1976. [Google Scholar]
- Kloprogge, J.T.; Frost, R.L. The effect of synthesis temperature on the FT-Raman and FT-IR spectra of saponites. Vib. Spectrosc. 2000, 23, 119–127. [Google Scholar] [CrossRef]
- Vogels, R.J.M.J.; Kerkhoffs, M.J.H.V.; Geus, J.W. Non-hydrothermal synthesis, characterization and catalytic properties of saponite clays. Stud. Surf. Sci. Catal. 1995, 91, 1153–1161. [Google Scholar]
- Bradley, S.M.; Howe, R.F.; Kydd, R.A. Correlation between 27Al and 71Ga NMR chemical shifts. Magn. Reson. Chem. 1993, 31, 883–886. [Google Scholar] [CrossRef]
- Sanz, J.; Serratosa, J.M. Silicon-29 and aluminum-27 high-resolution MAS-NMR spectra of phyllosilicates. J. Am. Chem. Soc. 1984, 106, 4790–4793. [Google Scholar] [CrossRef]
- Turner, G.L.; Smith, K.A.; Kirkpatrick, R.J.; Oldfield, E. Boron-11 nuclear magnetic resonance spectroscopic study of borate and borosilicate minerals and a borosilicate glass. J. Magn. Reson. (1969) 1986, 67, 544–550. [Google Scholar] [CrossRef]
- Kim, Y.; Kirkpatrick, R.J. 11B NMR investigation of boron interaction with mineral surfaces: Results for boehmite, silica gel and illite. Geochim. Cosmochim. Acta 2006, 70, 3231–3238. [Google Scholar] [CrossRef]
- de Ruiter, R.; Pamin, K.; Kentgens, A.P.M.; Jansen, J.C.; van Bekkum, H. Synthesis of molecular sieve [B]-BEA and modification of the boron site. Zeolites 1993, 13, 611–621. [Google Scholar] [CrossRef]
- Scholle, K.F.M.G.J.; Veeman, W.S. The influence of hydration on the coordination state of boron in H-Boralite studied by 11B magic angle spinning n.m.r. Zeolites 1985, 5, 118–122. [Google Scholar] [CrossRef]
- Michot, L.J.; Villiéras, F. Chapter 2.10—Surface Area and Porosity. In Developments in Clay Science; Bergaya, F., Lagaly, G., Eds.; Elsevier: Amsterdam, The Netherlands, 2013; Volume 5, pp. 319–332. [Google Scholar]
- Matthias, T.; Katsumi, K.; Alexander, V.N.; James, P.O.; Francisco, R.-R.; Jean, R.; Kenneth, S.W.S. Physisorption of gases, with special reference to the evaluation of surface area and pore size distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef] [Green Version]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of Gases in Multimolecular Layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Galarneau, A.; Villemot, F.; Rodriguez, J.; Fajula, F.; Coasne, B. Validity of the t-plot Method to Assess Microporosity in Hierarchical Micro/Mesoporous Materials. Langmuir 2014, 30, 13266–13274. [Google Scholar] [CrossRef]
- Morishige, K.; Tateishi, M. Accurate Relations between Pore Size and the Pressure of Capillary Condensation and the Evaporation of Nitrogen in Cylindrical Pores. Langmuir 2006, 22, 4165–4169. [Google Scholar] [CrossRef]
- Sing, K.S.W. Reporting physisorption data for gas/solid systems with special reference to the determination of surface area and porosity (Recommendations 1984). J. Pure Appl. Chem. 1985, 57, 603. [Google Scholar] [CrossRef]
- Cychosz, K.A.; Guillet-Nicolas, R.; García-Martínez, J.; Thommes, M. Recent advances in the textural characterization of hierarchically structured nanoporous materials. Chem. Soc. Rev. 2017, 46, 389–414. [Google Scholar] [CrossRef] [PubMed]
- Zou, S.; Zhang, M.; Mo, S.; Cheng, H.; Fu, M.; Chen, P.; Chen, L.; Shi, W.; Ye, D. Catalytic Performance of Toluene Combustion over Pt Nanoparticles Supported on Pore-Modified Macro-Meso-Microporous Zeolite Foam. Nanomaterials 2020, 10, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Üner, O.; Bayrak, Y. The effect of carbonization temperature, carbonization time and impregnation ratio on the properties of activated carbon produced from Arundo donax. Microporous Mesoporous Mater. 2018, 268, 225–234. [Google Scholar] [CrossRef]
- Pedreira-Segade, U.; Hao, J.; Razafitianamaharavo, A.; Pelletier, M.; Marry, V.; Le Crom, S.; Michot, L.J.; Daniel, I. How do Nucleotides Adsorb Onto Clays? Life 2018, 8, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- West, M.; Ellis, A.T.; Potts, P.J.; Streli, C.; Vanhoof, C.; Wegrzynek, D.; Wobrauschek, P. Atomic spectrometry update-X-ray fluorescence spectrometry. J. Anal. At. Spectrom. 2011, 26, 1919–1963. [Google Scholar] [CrossRef]
- Nicholls, D. Cobalt. In Complexes and First-Row Transition Elements; Macmillan Education UK: London, UK, 1974; pp. 185–193. [Google Scholar] [CrossRef]
- He, H.; Li, T.; Tao, Q.; Chen, T.; Zhang, D.; Zhu, J.; Yuan1, P.; Zhu, R. Aluminum ion occupancy in the structure of synthetic saponites: Effect on crystallinity. J. Am. Mineral. 2014, 99, 109. [Google Scholar] [CrossRef]
- Koningsberger, D.C.; Mojet, B.L.; van Dorssen, G.E.; Ramaker, D.E. XAFS spectroscopy; fundamental principles and data analysis. Top. Catal. 2000, 10, 143–155. [Google Scholar] [CrossRef]
- Koningsberger, D.C.; Miller, J.T. Local structure determination of aluminum in Y zeolite: Application of low energy X-ray absorption fine structure spectroscopy. Catal. Lett. 1994, 29, 77–90. [Google Scholar] [CrossRef]
- Koningsberger, D.C.; Miller, J.T. Withdrawal of electron density by cations from framework aluminium in Y zeolite determined by Al XAFS spectroscopy. Stud. Surf. Sci. Catal. 1995, 97, 125–131. [Google Scholar]
- Koningsberger, D.C.; Miller, J.T. The development of strong acidity by non-framework aluminium in H-USY determined by Al EXAFS. In Proceedings of the 11th International Conference on Catalysis—40th Anniversary, Part B, Baltimore, MD, USA, 30 June–5 July 1996; pp. 841–850. [Google Scholar]
- Nemecz, E. Clay Minerals (English Translation); Akadémiai Hiadó: Budapest, Hungary, 1981; p. 547. [Google Scholar]
- Reinholdt, M.; Miehé-Brendlé, J.; Delmotte, L.; Tuilier, M.-H.; le Dred, R.; Cortès, R.; Flank, A.-M. Fluorine Route Synthesis of Montmorillonites Containing Mg or Zn and Characterization by XRD, Thermal Analysis, MAS NMR, and EXAFS Spectroscopy. Eur. J. Inorg. Chem. 2001, 2001, 2831–2841. [Google Scholar] [CrossRef]
- Bisio, C.; Gatti, G.; Boccaleri, E.; Marchese, L.; Superti, G.B.; Pastore, H.O.; Thommes, M. Understanding physico–chemical properties of saponite synthetic clays. Microporous Mesoporous Mater. 2008, 107, 90–101. [Google Scholar] [CrossRef]
- Baldermann, A.; Dohrmann, R.; Kaufhold, S.; Nickel, C.; Letofsky-Papst, I.; Dietzel, M. The Fe-Mg-saponite solid solution series—A hydrothermal synthesis study. Clay Miner. 2014, 49, 391–415. [Google Scholar] [CrossRef]
- Marshall, W.L. Hydrothermal synthesis of amino acids. Geochim. Cosmochim. Acta 1994, 58, 2099–2106. [Google Scholar] [CrossRef]
- Ohara, S.; Kakegawa, T.; Nakazawa, H. Pressure Effects on the Abiotic Polymerization of Glycine. Orig. Life Evol. Biosph. 2007, 37, 215–223. [Google Scholar] [CrossRef]
- Woessner, D.E. Characterization of Clay Minerals by 27Al Nuclear Magnetic Resonance Spectroscopy. Am. Mineral. 1989, 74, 203–215. [Google Scholar]
- Engelhardt, G.; Michel, D. High Resolution Solid State NMR of Silicates and Zeolites; Wiley: New York, NY, USA, 1987; p. 300. [Google Scholar]
- Hazen, R.M.; Wones, D.R. The effect of cation substitutions on the physical properties of trioctahedral micas. Am. Mineral. 1972, 57, 103–129. [Google Scholar]
- Gillams, R.J.; Jia, T.Z. Mineral Surface-Templated Self-Assembling Systems: Case Studies from Nanoscience and Surface Science towards Origins of Life Research. Life 2018, 8, 10. [Google Scholar] [CrossRef] [Green Version]
- Van der Grift, C.J.G.; Mulder, A.; Geus, J.W. Characterization of silica supported copper catalysts by means of temperature-programmed reduction. Appl. Catal. 1990, 60, 181–192. [Google Scholar] [CrossRef]
- Carriat, J.Y.; Che, M.; Kermarec, M.; Decarreau, A. Influence of orderdisorder parameters on the reducibility of Ni- and Cu-containing silicates: Application to talc and chrysocolla. Catal. Lett. 1994, 25, 127–140. [Google Scholar] [CrossRef]
- Gérard, P.; Lemaitre, J. Characterization of hydrous nickel containing silicates by temperature programmed reduction. Bull. De Minéralogie 1981, 104, 655–660. [Google Scholar]
- Ferris, J.P.; Hill, A.R.; Liu, R.; Orgel, L.E. Synthesis of long prebiotic oligomers on mineral surfaces. Nature 1996, 381, 59–61. [Google Scholar] [CrossRef]
- Choudary, B.M.; Kantam, M.L.; Sateesh, M.; Rao, K.K.; Santhi, P.L. Iron pillared clays—Efficient catalysts for Friedel–Crafts reactions. Appl. Catal. A Gen. 1997, 149, 257–264. [Google Scholar] [CrossRef]
- Corma, A.; Miguel, P.J.; Orchille´s, A.V. Influence of hydrocarbon chain length and zeolite structure on the catalyst activity and deactivation for n-alkanes cracking. Appl. Catal. A Gen. 1994, 117, 29–40. [Google Scholar] [CrossRef]
- Rimola, A.; Ugliengo, P.; Sodupe, M. Formation versus hydrolysis of the peptide bond from a quantum-mechanical viewpoint: The role of mineral surfaces and implications for the origin of life. Int. J. Mol. Sci. 2009, 10, 746–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, W.; Baross, J.; Kelley, D.; Russell, M.J. Hydrothermal vents and the origin of life. Nat. Rev. Microbiol. 2008, 6, 805–814. [Google Scholar] [CrossRef]
- Toxvaerd, S. Origin of homochirality in biosystems. Int. J. Mol. Sci. 2009, 10, 1290–1299. [Google Scholar] [CrossRef]
- Leliveld, B.R.G.; Kerkhoffs, M.J.H.V.; Broersma, F.A.; van Dillen, J.A.J.; Geus, J.W.; Koningsberger, D.C. Acidic properties of synthetic saponites studied by pyridine IR and TPD-TG of n-propylamine. J. Chem. Soc. Faraday Trans. 1998, 94, 315–321. [Google Scholar] [CrossRef]
- Emeis, C.A. Determination of Integrated Molar Extinction Coefficients for Infrared Absorption Bands of Pyridine Adsorbed on Solid Acid Catalysts. J. Catal. 1993, 141, 347–354. [Google Scholar] [CrossRef]
- Biaglow, A.I.; Gittleman, C.; Gorte, R.J.; Madon, R.J. 2-Propanamine adsorption on a fluid catalytic cracking catalyst. J. Catal. 1991, 129, 88–93. [Google Scholar] [CrossRef]
- Meima, G.R.; van der Aalst, M.J.M.; Samson, M.S.U.; Garces, J.M.; Lee, J.G. Cumene production based on modified mordenite catalysts. In Proceedings from the Ninth International Zeolite Conference; von Ballmoos, R., Higgins, J.B., Treacy, M.M.J., Eds.; Butterworth-Heinemann: Oxford, UK, 1993; pp. 327–334. [Google Scholar] [CrossRef]
- Kaeding, W.W. Shape-selective reactions with zeolite catalysts: VII. Alkylation and disproportionation of cumene to produce diisopropylbenzene. J. Catal. 1989, 120, 409–412. [Google Scholar] [CrossRef]
- Urabe, K.; Sakurai, H.; Izumi, Y. Pillared synthetic saponite as an efficient alkylation catalyst. J. Chem. Soc. Chem. Commun. 1986, 1074–1076. [Google Scholar] [CrossRef]








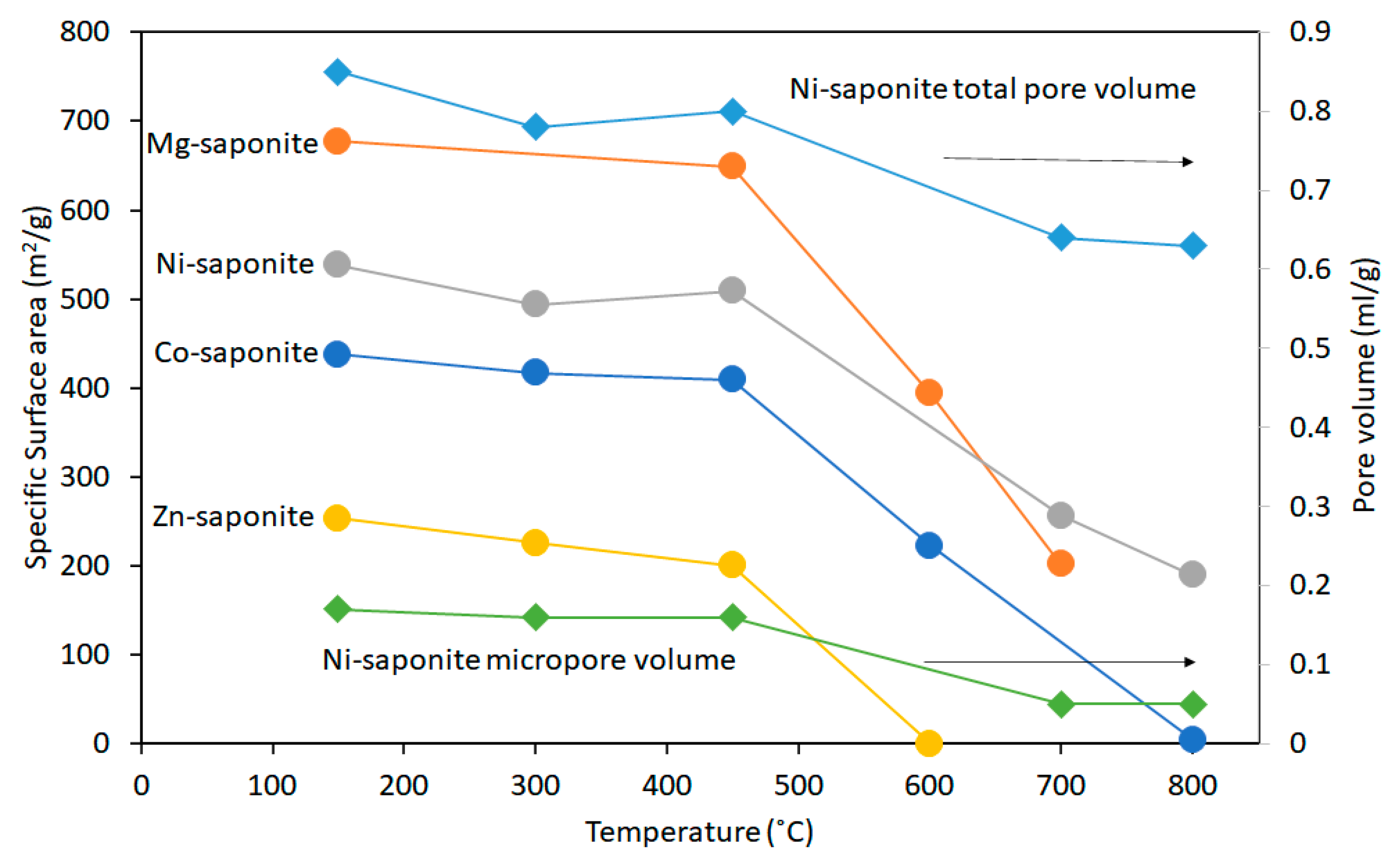
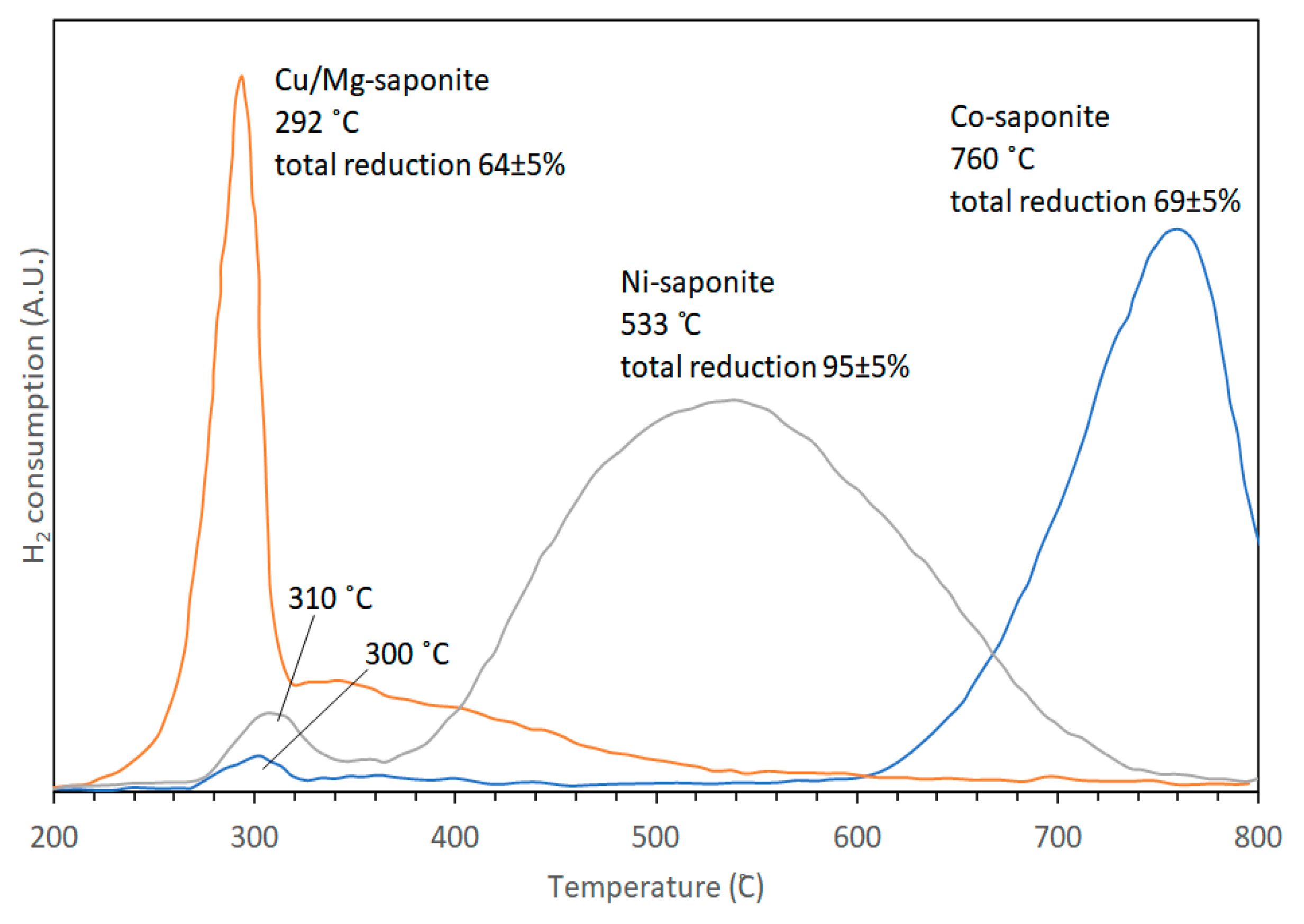

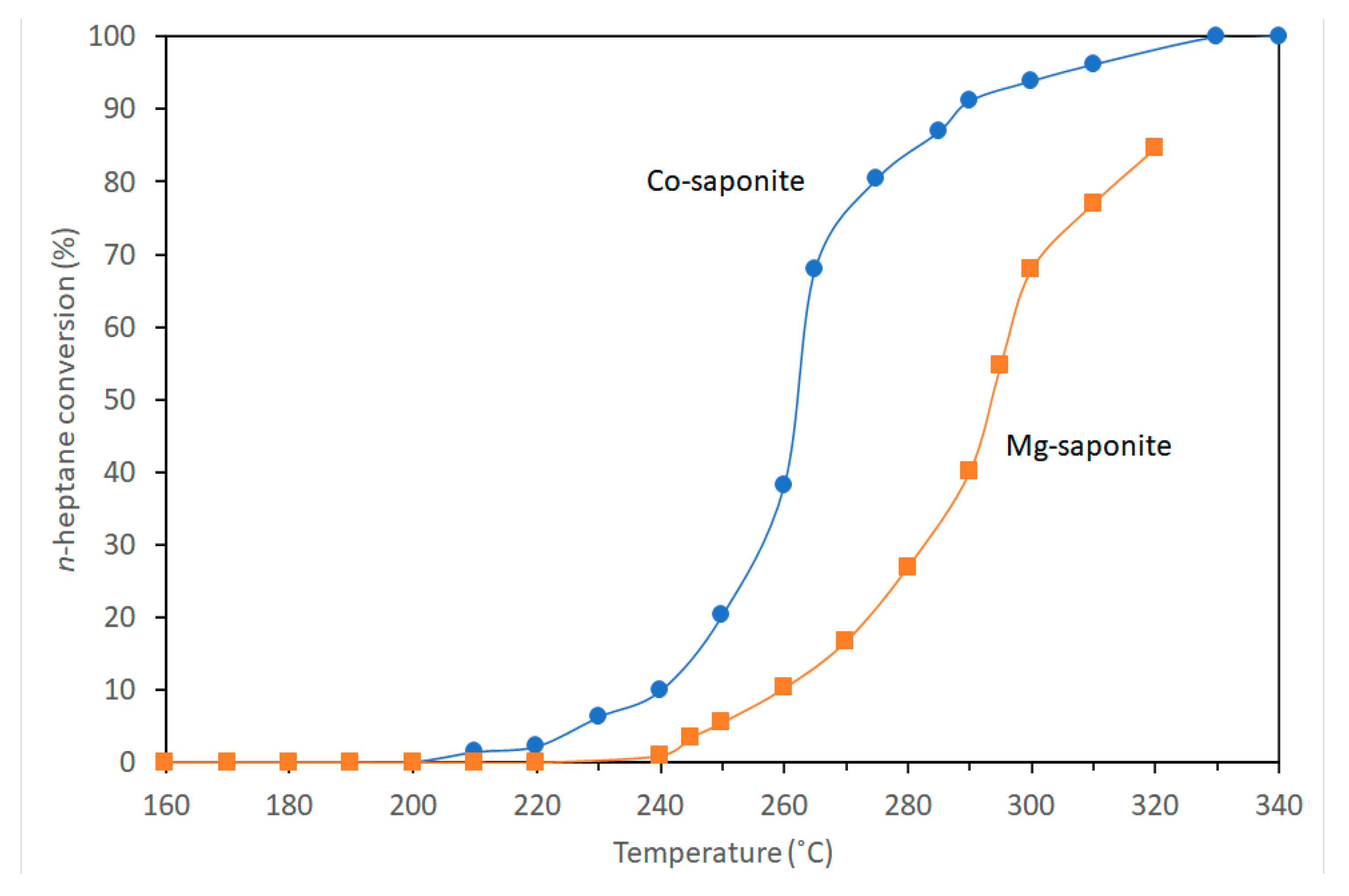


| Conditions (Catalysts wt%, Reaction Temperature and Reaction Time) | Conversion (%) | p-DIPB (%) | o-DIPB (%) | m-DIPB (%) |
|---|---|---|---|---|
| 1.5 wt% catalyst, 190 °C, 2 h | 99 | 47 | 50 | 3 |
| 1.5 wt% catalyst, 160 °C, 0.25 h | 98 | 47 | 36 | 17 |
| 0.2 wt% catalyst, 160 °C, 0.25 h | 87 | 49 | 35 | 16 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ponce, C.P.; Kloprogge, J.T. Urea-Assisted Synthesis and Characterization of Saponite with Different Octahedral (Mg, Zn, Ni, Co) and Tetrahedral Metals (Al, Ga, B), a Review. Life 2020, 10, 168. https://doi.org/10.3390/life10090168
Ponce CP, Kloprogge JT. Urea-Assisted Synthesis and Characterization of Saponite with Different Octahedral (Mg, Zn, Ni, Co) and Tetrahedral Metals (Al, Ga, B), a Review. Life. 2020; 10(9):168. https://doi.org/10.3390/life10090168
Chicago/Turabian StylePonce, Concepcion P., and J. Theo Kloprogge. 2020. "Urea-Assisted Synthesis and Characterization of Saponite with Different Octahedral (Mg, Zn, Ni, Co) and Tetrahedral Metals (Al, Ga, B), a Review" Life 10, no. 9: 168. https://doi.org/10.3390/life10090168
APA StylePonce, C. P., & Kloprogge, J. T. (2020). Urea-Assisted Synthesis and Characterization of Saponite with Different Octahedral (Mg, Zn, Ni, Co) and Tetrahedral Metals (Al, Ga, B), a Review. Life, 10(9), 168. https://doi.org/10.3390/life10090168