Rapid and Efficient Colony-PCR for High Throughput Screening of Genetically Transformed Chlamydomonas reinhardtii

 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
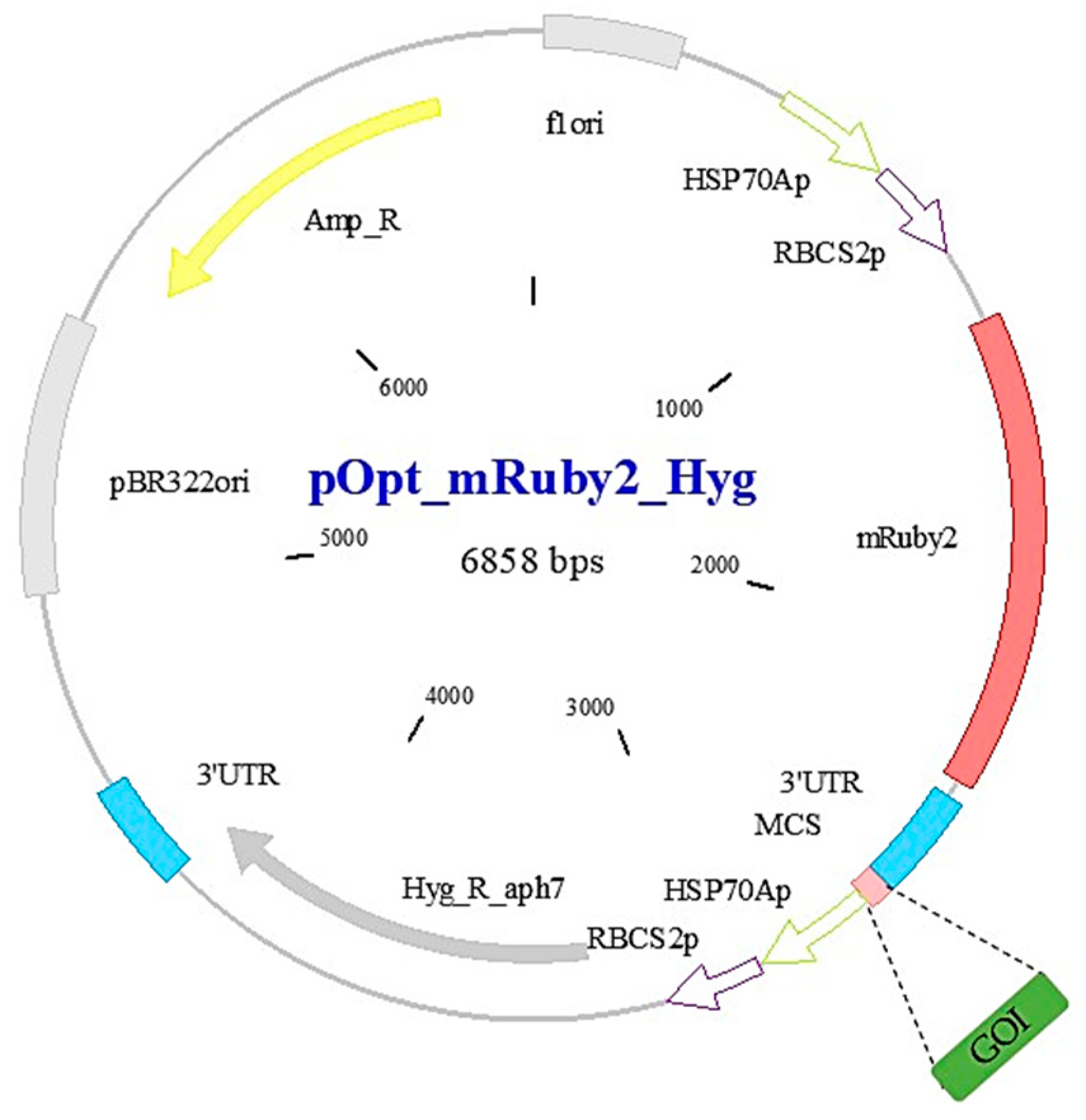
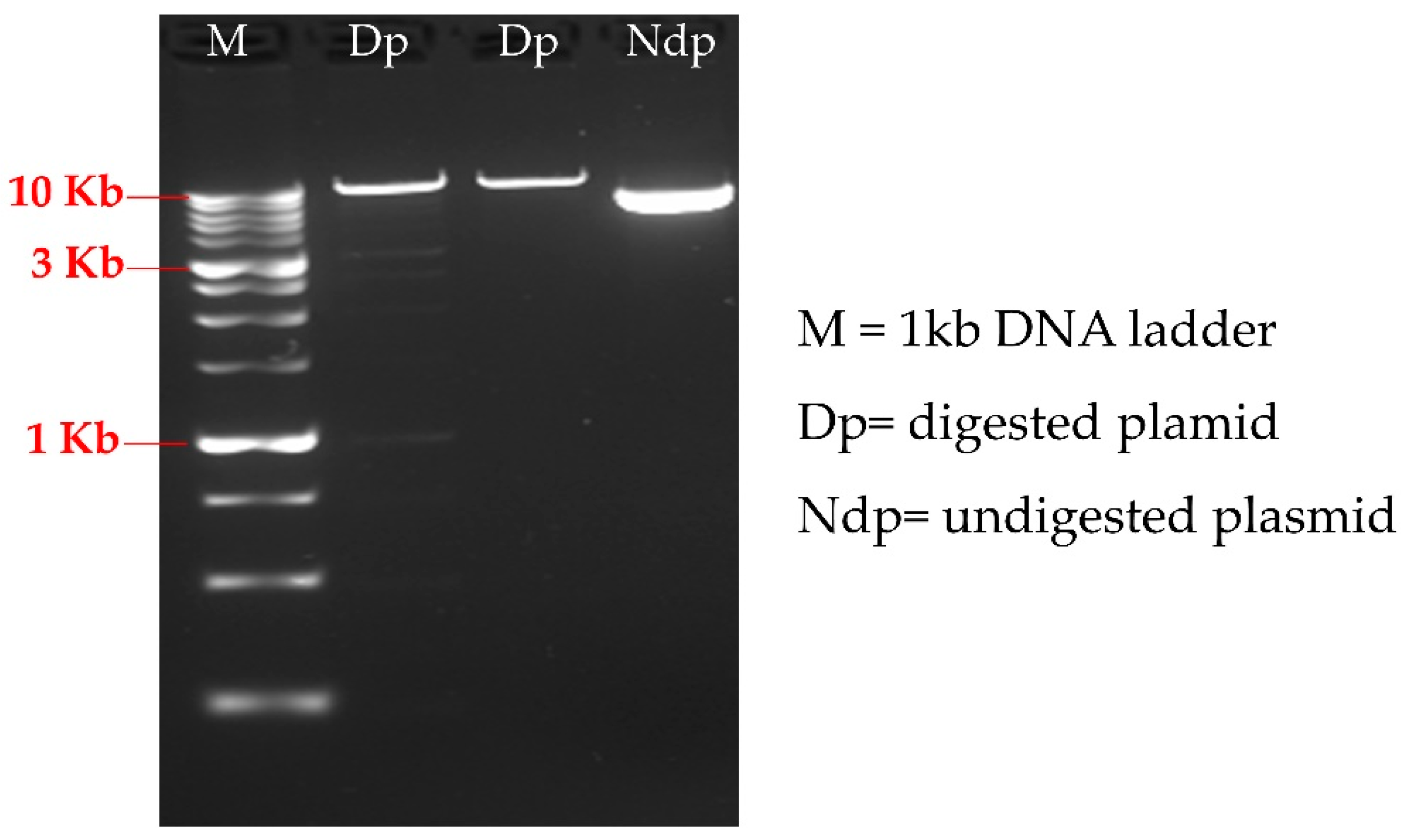
2.1. C. reinhardtii Strain, Plasmid and Primers Design
- Nuc_PGK_F: 5′AGTCCGAACAACCCACTTAC 3′;
- Nuc_PGK_R: 5′ CAGAGCTCAGGAGGTGAAATAG 3′;
- GOI_F: 5′ GCTGGGCATCAAGAAGACCG 3′;
- GOI_R: 5′ CCTGCGTGTAGTTGTTCGGG 3.’
2.2. C. reinhardtii Growth and Transformation Conditions
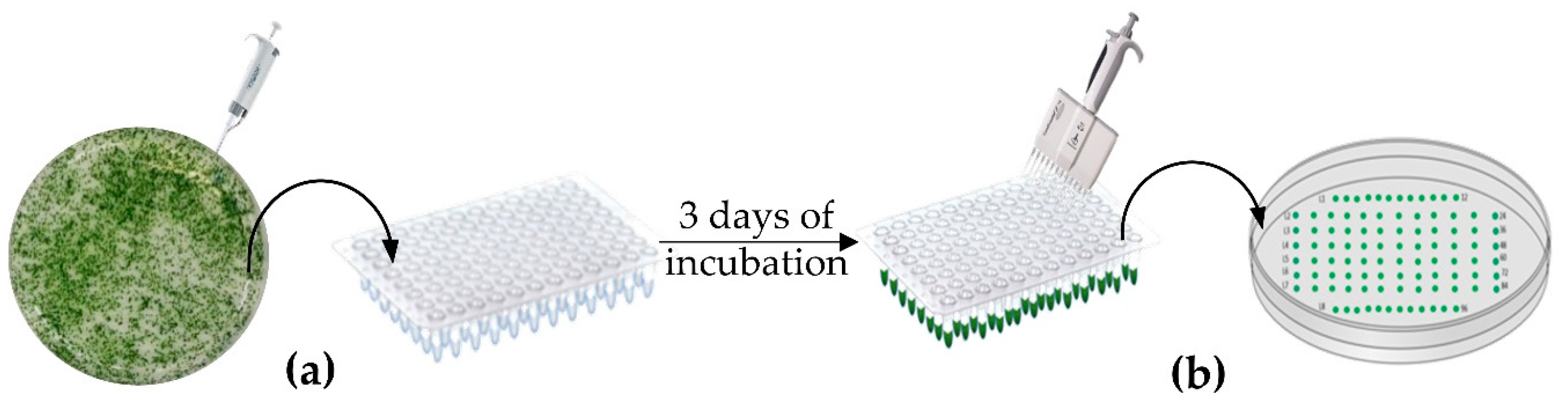
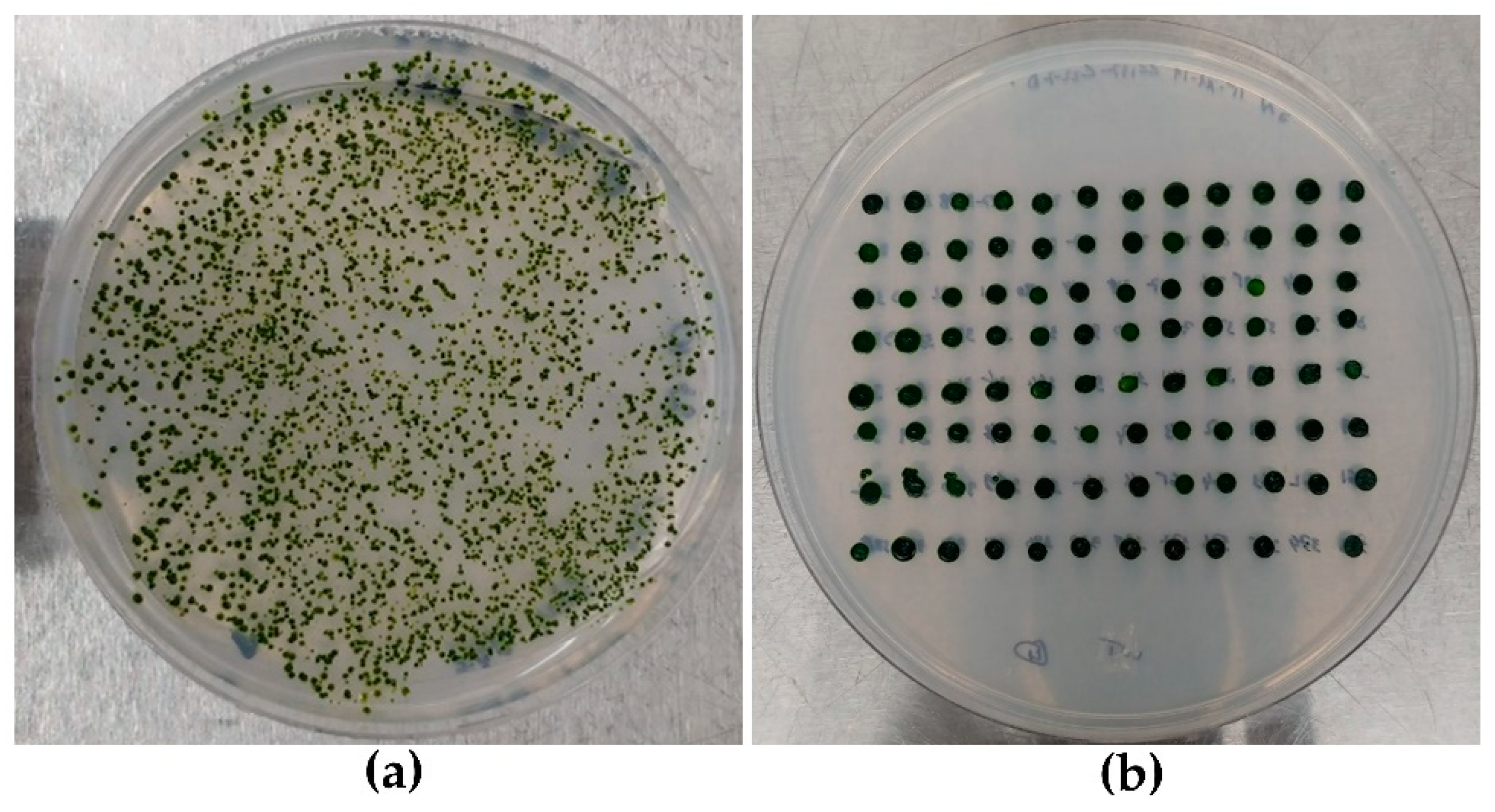
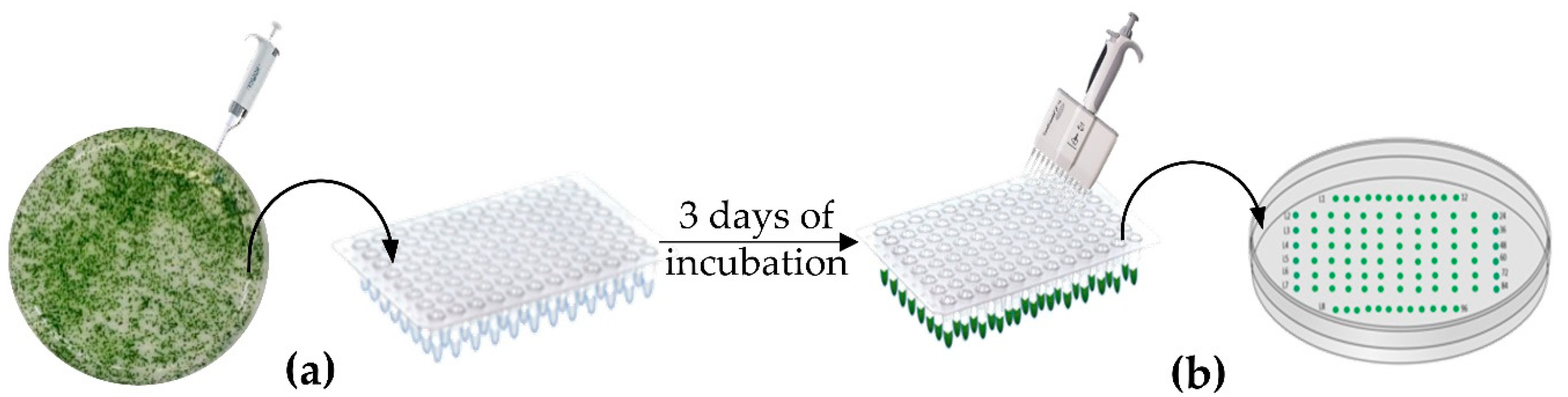
2.3. Strains Layout and Selection Method
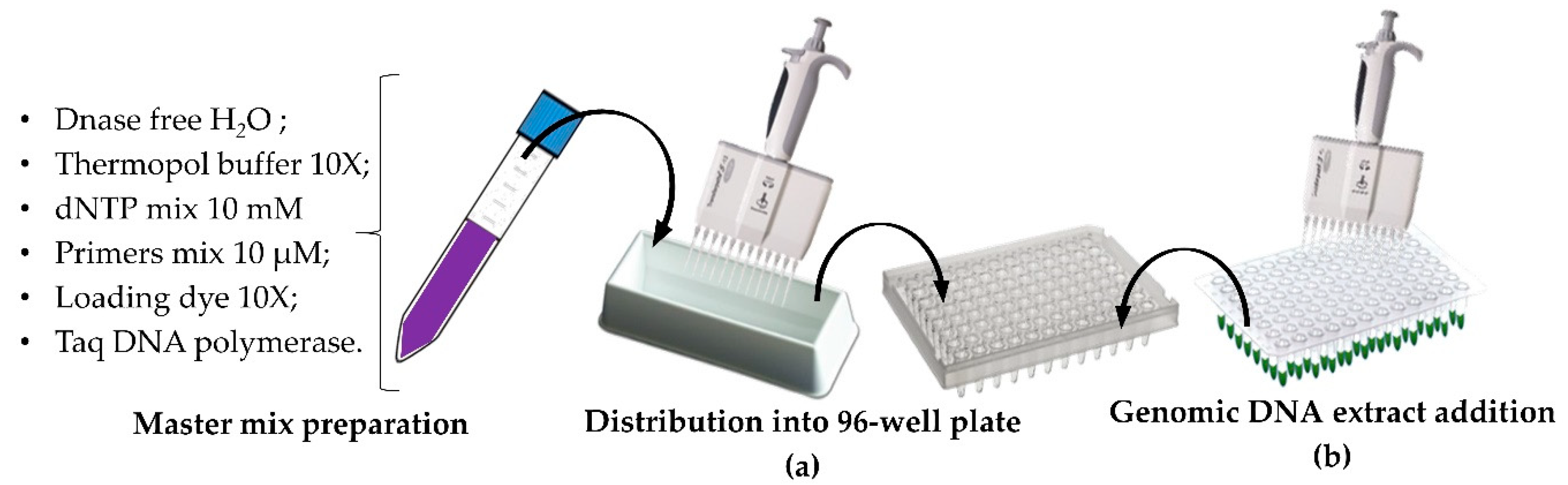
2.4. High Throughput Genomic DNA Extraction
2.5. HT PCR and HT Sample Loading onto Adapted Agarose Gel
3. Results
3.1. Strains Layout and HTS DNA Extraction
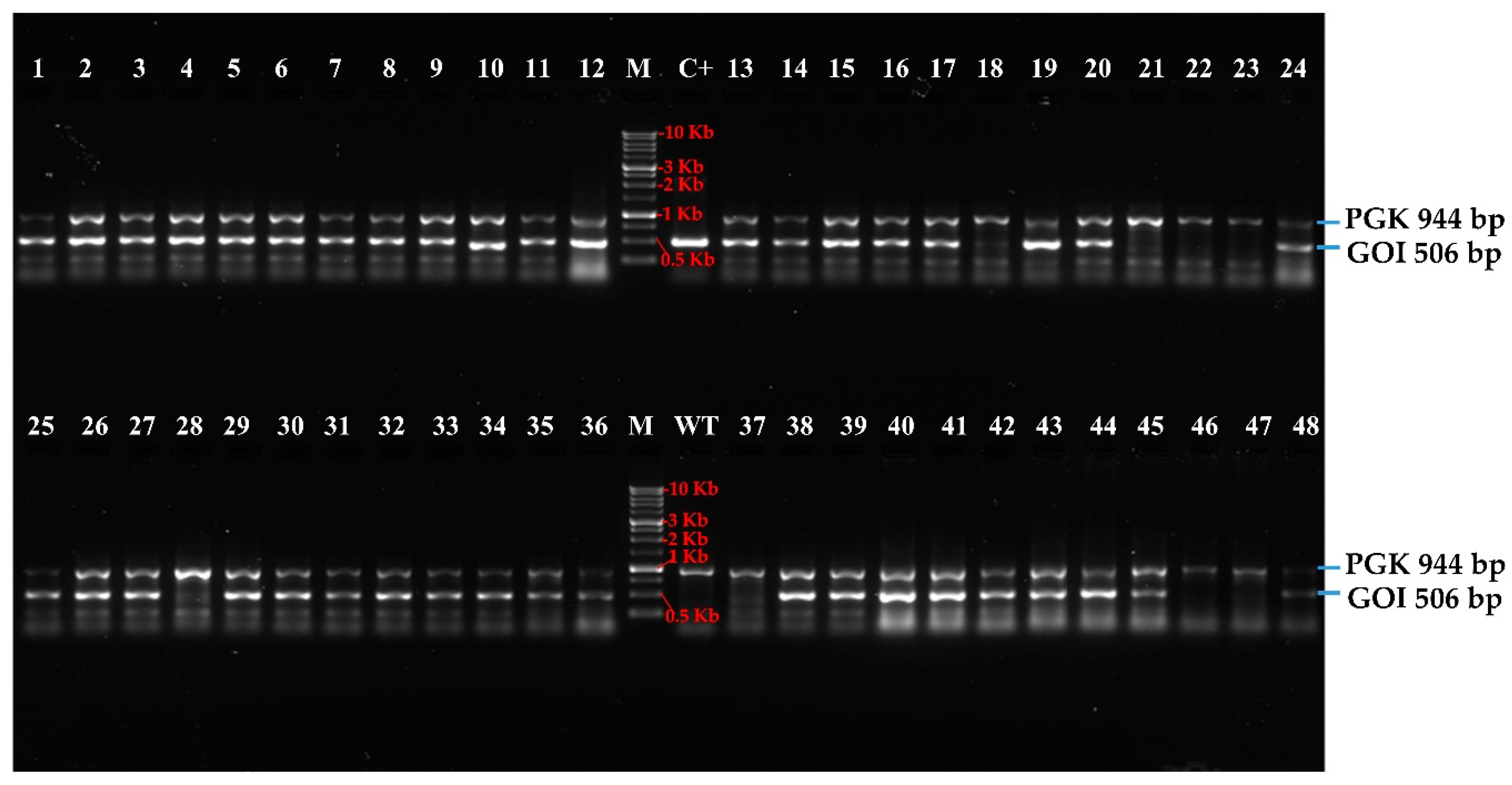
3.2. High-Troughput Screening cPCR and Revelation
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Read# | Well ID | Location | 260 Raw | 280 Raw | 320 Raw | 260 | 280 | 260/280 | ng/µL |
|---|---|---|---|---|---|---|---|---|---|
| 1 | SPL1 | A2 | 0.167 | 0.134 | 0.074 | 0.09 | 0.057 | 1.562 | 89.714 |
| 2 | SPL2 | A3 | 0.167 | 0.124 | 0.056 | 0.107 | 0.065 | 1.639 | 106.894 |
| 3 | SPL3 | B2 | 0.158 | 0.131 | 0.084 | 0.07 | 0.044 | 1.569 | 69.743 |
| 4 | SPL4 | B3 | 0.19 | 0.143 | 0.06 | 0.126 | 0.08 | 1.569 | 126.219 |
| 5 | SPL5 | C2 | 0.129 | 0.101 | 0.057 | 0.067 | 0.041 | 1.633 | 67.486 |
| 6 | SPL6 | C3 | 0.17 | 0.129 | 0.063 | 0.102 | 0.063 | 1.629 | 102.019 |
| 7 | SPL7 | D2 | 0.121 | 0.092 | 0.05 | 0.067 | 0.04 | 1.675 | 66.743 |
| 8 | SPL8 | D3 | 0.117 | 0.094 | 0.059 | 0.054 | 0.033 | 1.653 | 53.816 |
| 9 | SPL9 | E2 | 0.112 | 0.088 | 0.051 | 0.056 | 0.034 | 1.647 | 56.21 |
| 10 | SPL10 | E3 | 0.141 | 0.105 | 0.051 | 0.087 | 0.052 | 1.684 | 86.768 |
| 11 | SPL11 | F2 | 0.101 | 0.081 | 0.049 | 0.048 | 0.029 | 1.622 | 47.791 |
| 12 | SPL12 | F3 | 0.114 | 0.092 | 0.049 | 0.061 | 0.04 | 1.526 | 60.933 |
| B | SPL13 | G2 | 0.042 | 0.043 | 0.036 | 0 | 0 | 0 | 0 |
| 13 | SPL1 | A2 | 0.156 | 0.117 | 0.053 | 0.1 | 0.061 | 1.631 | 99.51 |
| 14 | SPL2 | A3 | 0.09 | 0.074 | 0.051 | 0.034 | 0.02 | 1.758 | 34.371 |
| 15 | SPL3 | B2 | 0.24 | 0.173 | 0.06 | 0.175 | 0.11 | 1.598 | 175.279 |
| 16 | SPL4 | B3 | 0.117 | 0.092 | 0.054 | 0.059 | 0.035 | 1.682 | 58.657 |
| 17 | SPL5 | C2 | 0.114 | 0.091 | 0.055 | 0.055 | 0.033 | 1.651 | 55.252 |
| 18 | SPL6 | C3 | 0.12 | 0.092 | 0.05 | 0.066 | 0.04 | 1.667 | 65.907 |
| 19 | SPL7 | D2 | 0.393 | 0.279 | 0.07 | 0.317 | 0.205 | 1.549 | 317.437 |
| 20 | SPL8 | D3 | 0.103 | 0.082 | 0.049 | 0.05 | 0.03 | 1.63 | 49.642 |
| 21 | SPL9 | E2 | 0.1 | 0.081 | 0.051 | 0.044 | 0.027 | 1.66 | 44.324 |
| 22 | SPL10 | E3 | 0.104 | 0.082 | 0.047 | 0.053 | 0.032 | 1.677 | 53.017 |
| 23 | SPL11 | F2 | 0.145 | 0.111 | 0.057 | 0.083 | 0.051 | 1.624 | 82.756 |
| 24 | SPL12 | F3 | 0.164 | 0.143 | 0.106 | 0.054 | 0.034 | 1.604 | 54.168 |
| B | SPL13 | G2 | 0.045 | 0.042 | 0.039 | 0 | 0 | 0 | 0 |
| 25 | SPL1 | A2 | 0.118 | 0.093 | 0.049 | 0.066 | 0.041 | 1.593 | 65.524 |
| 26 | SPL2 | A3 | 0.122 | 0.094 | 0.051 | 0.067 | 0.04 | 1.666 | 67.201 |
| 27 | SPL3 | B2 | 0.174 | 0.126 | 0.05 | 0.12 | 0.074 | 1.63 | 119.823 |
| 28 | SPL4 | B3 | 0.259 | 0.191 | 0.068 | 0.186 | 0.119 | 1.562 | 186.418 |
| 29 | SPL5 | C2 | 0.233 | 0.169 | 0.054 | 0.174 | 0.111 | 1.566 | 174.01 |
| 30 | SPL6 | C3 | 0.132 | 0.102 | 0.051 | 0.077 | 0.048 | 1.587 | 76.95 |
| 31 | SPL7 | D2 | 0.071 | 0.061 | 0.046 | 0.022 | 0.013 | 1.751 | 21.983 |
| 32 | SPL8 | D3 | 0.114 | 0.09 | 0.055 | 0.055 | 0.033 | 1.672 | 54.91 |
| 33 | SPL9 | E2 | 0.072 | 0.061 | 0.044 | 0.024 | 0.014 | 1.723 | 24.317 |
| 34 | SPL10 | E3 | 0.083 | 0.067 | 0.044 | 0.034 | 0.02 | 1.689 | 34.399 |
| 35 | SPL11 | F2 | 0.145 | 0.114 | 0.065 | 0.076 | 0.046 | 1.637 | 75.822 |
| 36 | SPL12 | F3 | 0.105 | 0.084 | 0.049 | 0.053 | 0.033 | 1.618 | 52.875 |
| B | SPL13 | G2 | 0.042 | 0.041 | 0.038 | 0 | 0 | 0 | 0 |
| 37 | SPL1 | A2 | 0.116 | 0.093 | 0.05 | 0.063 | 0.041 | 1.547 | 63.025 |
| 38 | SPL2 | A3 | 0.277 | 0.201 | 0.062 | 0.21 | 0.136 | 1.545 | 209.66 |
| 39 | SPL3 | B2 | 0.15 | 0.114 | 0.051 | 0.095 | 0.06 | 1.577 | 95.231 |
| 40 | SPL4 | B3 | 0.354 | 0.259 | 0.064 | 0.286 | 0.192 | 1.49 | 285.522 |
| 41 | SPL5 | C2 | 0.216 | 0.163 | 0.061 | 0.151 | 0.098 | 1.532 | 150.736 |
| 42 | SPL6 | C3 | 0.113 | 0.088 | 0.045 | 0.064 | 0.04 | 1.602 | 64.316 |
| 43 | SPL7 | D2 | 0.206 | 0.155 | 0.057 | 0.145 | 0.096 | 1.517 | 144.948 |
| 44 | SPL8 | D3 | 0.327 | 0.241 | 0.074 | 0.248 | 0.164 | 1.514 | 247.814 |
| 45 | SPL9 | E2 | 0.177 | 0.135 | 0.058 | 0.114 | 0.074 | 1.547 | 113.956 |
| 46 | SPL10 | E3 | 0.154 | 0.118 | 0.051 | 0.099 | 0.065 | 1.53 | 98.815 |
| 47 | SPL11 | F2 | 0.381 | 0.282 | 0.097 | 0.277 | 0.18 | 1.538 | 276.991 |
| 48 | SPL12 | F3 | 0.157 | 0.118 | 0.052 | 0.101 | 0.063 | 1.609 | 100.925 |
| Component | 25 μL Reaction | Final Concentration |
|---|---|---|
| 10× ThermoPol Reaction Buffer | 2.5 µL | 1X |
| 10 mM dNTPs | 0.5 µL | 200 µM |
| 10 µM Forward Primer mix (PGK/GOI) | 0.5 µL | 0.2 µM (0.05–1 µM) |
| 10 µM Reverse Primer mix (PGK/GOI) | 0.5 µL | 0.2 µM (0.05–1 µM) |
| 10× Loading Dye Buffer | 1 µL | 0.4X |
| Template DNA | 2 µL | <1000 ng |
| Taq DNA Polymerase | 0.125 µL | 1.25 units/50 µL PCR |
| Nuclease-free water | To 25 µL |
References
- Malcata, F.X.; Pinto, I.S.; Guedes, A.C. Marine Macro-and Microalgae: An Overview; CRC Press: Boca Raton, FL, USA, 2018. [Google Scholar]
- Salomé, P.A.; Merchant, S.S. A Series of Fortunate Events: Introducing Chlamydomonas as a Reference Organism. Plant Cell 2019, 31, 1682–1707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merchant, S.S.; Prochnik, S.E.; Vallon, O.; Harris, E.H.; Karpowicz, S.J.; Witman, G.B.; Terry, A.; Salamov, A.; Fritz-Laylin, L.K.; Maréchal-Drouard, L.; et al. The Chlamydomonas Genome Reveals the Evolution of Key Animal and Plant Functions. Science 2007, 318, 245–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scranton, M.A.; Ostrand, J.T.; Fields, F.J.; Mayfield, S.P. Chlamydomonas as a model for biofuels and bio-products production. Plant J. 2015, 82, 523–531. [Google Scholar] [CrossRef] [Green Version]
- Scaife, M.A.; Nguyen, G.T.; Rico, J.; Lambert, D.; Helliwell, K.E.; Smith, A.G. Establishing Chlamydomonas reinhardtii as an industrial biotechnology host. Plant J. 2015, 82, 532–546. [Google Scholar] [CrossRef]
- Kong, F.; Yamaoka, Y.; Ohama, T.; Lee, Y.; Li-Beisson, Y. Molecular Genetic Tools and Emerging Synthetic Biology Strategies to Increase Cellular Oil Content in Chlamydomonas reinhardtii. Plant Cell Physiol. 2019, 60, 1184–1196. [Google Scholar] [CrossRef] [PubMed]
- Leon, R. Transgenic microalgae as green cell-factories. Trends Biotechnol. 2004, 22, 45–52. [Google Scholar] [CrossRef]
- Leon, R.; Fernandez, E. Nuclear transformation of eukaryotic microalgae. In Transgenic Microalgae as Green Cell Factories; Springer: Berlin/Heidelberg, Germany, 2007; pp. 1–11. [Google Scholar]
- Kindle, K.L. High-frequency nuclear transformation of Chlamydomonas reinhardtii. Proc. Natl. Acad. Sci. USA 1990, 87, 1228–1232. [Google Scholar] [CrossRef] [Green Version]
- Shimogawara, K.; Fujiwara, S.; Grossman, A.; Usuda, H. High-efficiency transformation of Chlamydomonas reinhardtii by electroporation. GenetICS 1998, 148, 1821–1828. [Google Scholar]
- Kumar, S.V.; Misquitta, R.W.; Reddy, V.S.; Rao, B.J.; Rajam, M.V. Genetic transformation of the green alga—Chlamydomonas reinhardtii by Agrobacterium tumefaciens. Plant Sci. 2004, 166, 731–738. [Google Scholar] [CrossRef]
- Zhang, Y.; Yan, Y.-P.; Wu, Y.-C.; Hua, W.; Chen, C.; Ge, Q.; Wang, Z.-Z. Pathway engineering for phenolic acid accumulations in Salvia miltiorrhiza by combinational genetic manipulation. Metab. Eng. 2014, 21, 71–80. [Google Scholar] [CrossRef]
- Jinkerson, R.E.; Jonikas, M.C. Molecular techniques to interrogate and edit the Chlamydomonas nuclear genome. Plant J. 2015, 82, 393–412. [Google Scholar] [CrossRef] [PubMed]
- Nour-Eldin, H.H.; Specht, E.A.; Ostrand, J.; Hoang, K.T.; Karunanithi, P.S.; Mayfield, S.P. High-throughput system for quantifying and characterizing homologous recombination in Chlamydomonas reinhardtii. Algal Res. 2018, 31, 167–172. [Google Scholar] [CrossRef]
- Yamano, T.; Iguchi, H.; Fukuzawa, H. Rapid transformation of Chlamydomonas reinhardtii without cell-wall removal. J. Biosci. Bioeng. 2013, 115, 691–694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shahar, N.; Landman, S.; Weiner, I.; Elman, T.; Dafni, E.; Feldman, Y.; Tuller, T.; Yacoby, I. The Integration of Multiple Nuclear-Encoded Transgenes in the Green Alga Chlamydomonas reinhardtii Results in Higher Transcription Levels. Front. Plant Sci. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Weiner, I.; Atar, S.; Schweitzer, S.; Eilenberg, H.; Feldman, Y.; Avitan, M.; Blau, M.; Danon, A.; Tuller, T.; Yacoby, I. Enhancing heterologous expression in Chlamydomonas reinhardtii by transcript sequence optimization. Plant J. 2018, 94, 22–31. [Google Scholar] [CrossRef] [Green Version]
- Mussgnug, J.H. Nuclear Transformation and Toolbox Development. In Chlamydomonas: Molecular Genetics and Physiology; Springer: Berlin/Heidelberg, Germany, 2017; pp. 27–58. [Google Scholar]
- Rasala, B.A.; Lee, P.A.; Shen, Z.; Briggs, S.P.; Mendez, M.; Mayfield, S.P. Robust Expression and Secretion of Xylanase1 in Chlamydomonas reinhardtii by Fusion to a Selection Gene and Processing with the FMDV 2A Peptide. PLoS ONE 2012, 7, e43349. [Google Scholar] [CrossRef] [Green Version]
- Lauersen, K.; Berger, H.; Mussgnug, J.H.; Kruse, O. Efficient recombinant protein production and secretion from nuclear transgenes in Chlamydomonas reinhardtii. J. Biotechnol. 2013, 167, 101–110. [Google Scholar] [CrossRef]
- Lauersen, K.; Kruse, O.; Mussgnug, J.H. Targeted expression of nuclear transgenes in Chlamydomonas reinhardtii with a versatile, modular vector toolkit. Appl. Microbiol. Biotechnol. 2015, 99, 3491–3503. [Google Scholar] [CrossRef]
- Kong, F.; Yamasaki, T.; Kurniasih, S.D.; Hou, L.; Li, X.; Ivanova, N.; Okada, S.; Ohama, T. Robust expression of heterologous genes by selection marker fusion system in improved Chlamydomonas strains. J. Biosci. Bioeng. 2015, 120, 239–245. [Google Scholar] [CrossRef] [Green Version]
- Plucinak, T.M.; Horken, K.M.; Jiang, W.; Fostvedt, J.; Nguyen, S.T.; Weeks, D.P. Improved and versatile viral 2A platforms for dependable and inducible high-level expression of dicistronic nuclear genes in Chlamydomonas reinhardtii. Plant J. 2015, 82, 717–729. [Google Scholar] [CrossRef]
- Rasala, B.A.; Mayfield, S.P. Photosynthetic biomanufacturing in green algae; production of recombinant proteins for industrial, nutritional, and medical uses. Photosynth. Res. 2014, 123, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Onishi, M.; Pringle, J.R. Robust Transgene Expression from Bicistronic mRNA in the Green Alga Chlamydomonas reinhardtii. G3: Genes Genomes Genet. 2016, 6, 4115–4125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molino, J.V.D.; De Carvalho, J.C.M.; Mayfield, S.P. Comparison of secretory signal peptides for heterologous protein expression in microalgae: Expanding the secretion portfolio for Chlamydomonas reinhardtii. PLoS ONE 2018, 13, e0192433. [Google Scholar] [CrossRef]
- Lauersen, K.; Baier, T.; Wichmann, J.; Wördenweber, R.; Mussgnug, J.H.; Hübner, W.; Huser, T.R.; Kruse, O. Efficient phototrophic production of a high-value sesquiterpenoid from the eukaryotic microalga Chlamydomonas reinhardtii. Metab. Eng. 2016, 38, 331–343. [Google Scholar] [CrossRef] [PubMed]
- López-Paz, C.; Liu, D.; Geng, S.; Umen, J.G. Identification of Chlamydomonas reinhardtii endogenous genic flanking sequences for improved transgene expression. Plant J. 2017, 92, 1232–1244. [Google Scholar] [CrossRef] [Green Version]
- Baier, T.; Wichmann, J.; Kruse, O.; Lauersen, K. Intron-containing algal transgenes mediate efficient recombinant gene expression in the green microalga Chlamydomonas reinhardtii. Nucleic Acids Res. 2018, 46, 6909–6919. [Google Scholar] [CrossRef]
- Wichmann, J.; Baier, T.; Wentnagel, E.; Lauersen, K.; Kruse, O. Tailored carbon partitioning for phototrophic production of (E)-α-bisabolene from the green microalga Chlamydomonas reinhardtii. Metab. Eng. 2018, 45, 211–222. [Google Scholar] [CrossRef]
- Cao, M.; Fu, Y.; Guo, Y.; Pan, J. Chlamydomonas (Chlorophyceae) colony PCR. Protoplasma 2009, 235, 107–110. [Google Scholar] [CrossRef]
- Sheu, D.-S.; Wang, Y.-T.; Lee, C.-Y. Rapid detection of polyhydroxyalkanoate-accumulating bacteria isolated from the environment by colony PCR. Microbiology 2000, 146, 2019–2025. [Google Scholar] [CrossRef] [Green Version]
- Chlamydomonas Resource Center. Available online: http://chlamycollection.org/ (accessed on 10 September 2020).
- Liu, C.; Wu, G.; Huang, X.; Liu, S.; Cong, B. Validation of housekeeping genes for gene expression studies in an ice alga Chlamydomonas during freezing acclimation. Extremophiles 2012, 16, 419–425. [Google Scholar] [CrossRef]
- Integrated DNA Techonologies. Available online: https://www.idtdna.com/ (accessed on 10 September 2020).
- Wittkopp, T. Nuclear Transformation of Chlamydomonas reinhardtii by Electroporation. BIO-PROTOCOL 2018, 8. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Yang, L.; Wen, X.; Chen, Z.; Liang, Q.; Li, J.; Wang, W. Rapid and high efficiency transformation of Chlamydomonas reinhardtii by square-wave electroporation. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [Green Version]
- Geissmann, Q. OpenCFU, a New Free and Open-Source Software to Count Cell Colonies and Other Circular Objects. PLoS ONE 2013, 8, e54072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Gouil, C.; Déry, C. A rapid procedure for the screening of recombinant plasmids. Nucleic Acids Res. 1991, 19, 6655. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Fahs, A.; Feldman, C.; Shah, S.; Gu, Y.; Wang, Y.; Machado, R.F.; Wunderink, R.G.; Chen, J. A reliable and effective method of DNA isolation from old human blood paper cards. SpringerPlus 2013, 2, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Bi, C.; Tong, S.; Gong, Z.; Hou, H. An improved and reliable method for microalgae direct PCR. Environ. Boil. Fishes 2019, 31, 2411–2421. [Google Scholar] [CrossRef]
- Lai, M.-Y.; Abdul-Majid, N.; Lau, Y.-L. Identification of Host Proteins Interacting with Toxoplasma gondii SAG1 by Yeast Two-Hybrid Assay. Acta Parasitol. 2019, 64, 575–581. [Google Scholar] [CrossRef]
- Kindo, A.J.; Vijayaraman, R.S.; Ramaraj, V. Direct colony polymerase chain reaction for rapid identification of yeasts isolated from blood specimen. J. Acad. Clin. Microbiol. 2016, 18, 91. [Google Scholar] [CrossRef]
- Walch, G.; Knapp, M.; Rainer, G.; Peintner, U. Colony-PCR Is a Rapid Method for DNA Amplification of Hyphomycetes. J. Fungi 2016, 2, 12. [Google Scholar] [CrossRef] [Green Version]
- Azevedo, F.; Pereira, H.; Johansson, B. Colony PCR. In PCR.; Springer: Berlin/Heidelberg, Germany, 2017; pp. 129–139. [Google Scholar]
- Run, C.; Fang, L.; Fan, J.; Fan, C.-M.; Luo, Y.; Hu, Z.-M.; Li, Y. Stable nuclear transformation of the industrial alga Chlorella pyrenoidosa. Algal Res. 2016, 17, 196–201. [Google Scholar] [CrossRef]
- New England Biolabs Inc. Available online: https://international.neb.com/protocols/0001/01/01/taq-dna-polymerase-with-thermopol-buffer-m0267 (accessed on 10 September 2020).




| Number of Colonies Counted After Electroporation | Number of Clones Selected Randomly | Viable Clones After Six Round Sub-Culturing (% of Viable Cells) | Positive Clones After HTS-Colony-PCR (% of Positive Colonies) |
|---|---|---|---|
| 7000 | 1440 | 1336 (93) | 646 (48, 3) |
| Steps | STS | HTS | Difference |
|---|---|---|---|
| Time (min) | |||
| DNA extraction | 360 | 40 | 320 |
| Mix PCR preparation and distribution | 150 | 35 | 115 |
| Gel preparation and sample loading | 150 | 20 | 130 |
| Total | 660 | 95 | 565 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nouemssi, S.B.; Ghribi, M.; Beauchemin, R.; Meddeb-Mouelhi, F.; Germain, H.; Desgagné-Penix, I. Rapid and Efficient Colony-PCR for High Throughput Screening of Genetically Transformed Chlamydomonas reinhardtii. Life 2020, 10, 186. https://doi.org/10.3390/life10090186
Nouemssi SB, Ghribi M, Beauchemin R, Meddeb-Mouelhi F, Germain H, Desgagné-Penix I. Rapid and Efficient Colony-PCR for High Throughput Screening of Genetically Transformed Chlamydomonas reinhardtii. Life. 2020; 10(9):186. https://doi.org/10.3390/life10090186
Chicago/Turabian StyleNouemssi, Serge Basile, Manel Ghribi, Rémy Beauchemin, Fatma Meddeb-Mouelhi, Hugo Germain, and Isabel Desgagné-Penix. 2020. "Rapid and Efficient Colony-PCR for High Throughput Screening of Genetically Transformed Chlamydomonas reinhardtii" Life 10, no. 9: 186. https://doi.org/10.3390/life10090186
APA StyleNouemssi, S. B., Ghribi, M., Beauchemin, R., Meddeb-Mouelhi, F., Germain, H., & Desgagné-Penix, I. (2020). Rapid and Efficient Colony-PCR for High Throughput Screening of Genetically Transformed Chlamydomonas reinhardtii. Life, 10(9), 186. https://doi.org/10.3390/life10090186