Association of Genes Related to Oxidative Stress with the Extent of Coronary Atherosclerosis

 , , , ,
, , , ,  and
and
Abstract
:1. Introduction
2. Results
2.1. Individual Oxidative Stress-Related Polymorphisms and Coronary Atherosclerosis
2.2. The Association of Eight Oxidative Stress-Related SNPs and Coronary Atherosclerosis
3. Discussion
4. Materials and Methods
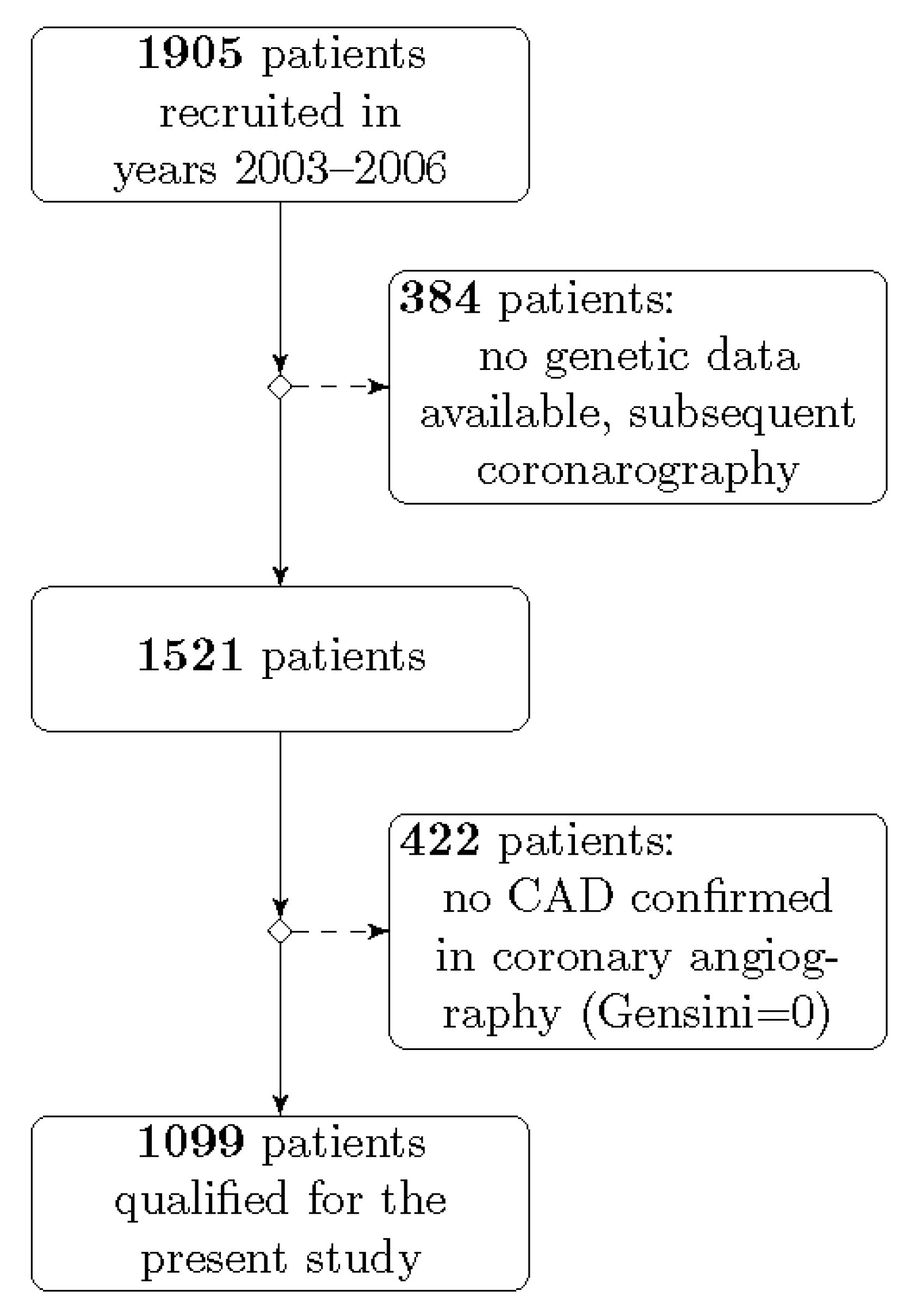
4.1. Study Population
4.2. Coronary Atherosclerosis Evaluation
4.3. Genetic Analyses
4.4. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| SNP | single nucleotide polymorphism |
| GRS | genetic risk score |
| CAD | coronary artery disease |
| ROS | reactive oxygen species |
| BMI | body mass index |
| LDL | low density lipoprotein |
| HDL | high density lipoprotein |
| SD | standard deviation |
| MACE | major adverse cardiovascular events |
References
- Herrington, W.; Lacey, B.; Sherliker, P.; Armitage, J.; Lewington, S. Epidemiology of Atherosclerosis and the Potential to Reduce the Global Burden of Atherothrombotic Disease. Circ. Res. 2016, 118, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Cervantes Gracia, K.; Llanas-Cornejo, D.; Husi, H. CVD and Oxidative Stress. J. Clin. Med. 2017, 6, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative Stress in Atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 42. [Google Scholar] [CrossRef] [PubMed]
- Peluso, I.; Morabito, G.; Urban, L.; Ioannone, F.; Serafini, M. Oxidative stress in atherosclerosis development: The central role of LDL and oxidative burst. Endocr. Metab. Immune Disord. Drug Targets 2012, 12, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Vujic, N.; Schlager, S.; Eichmann, T.O.; Madreiter-Sokolowski, C.T.; Goeritzer, M.; Rainer, S.; Schauer, S.; Rosenberger, A.; Woelfler, A.; Doddapattar, P.; et al. Monoglyceride lipase deficiency modulates endocannabinoid signaling and improves plaque stability in ApoE-knockout mice. Atherosclerosis 2016, 244, 9–21. [Google Scholar] [CrossRef] [Green Version]
- Katare, C.; Saxena, S.; Agrawal, S.; Joseph, A.Z.; Subramani, S.K.; Yadav, D.; Singh, N.; Bisen, P.S.; Prasad, G.B. Lipid-lowering and antioxidant functions of bottle gourd (Lagenaria siceraria) extract in human dyslipidemia. J. Evid. Based Complement. Altern. Med. 2014, 19, 112–118. [Google Scholar] [CrossRef]
- Doddapattar, P.; Radović, B.; Patankar, J.V.; Obrowsky, S.; Jandl, K.; Nusshold, C.; Kolb, D.; Vujić, N.; Doshi, L.; Chandak, P.G.; et al. Xanthohumol ameliorates atherosclerotic plaque formation, hypercholesterolemia, and hepatic steatosis in ApoE-deficient mice. Mol. Nutr. Food Res. 2013, 57, 1718–1728. [Google Scholar] [CrossRef]
- De Rosa, S.; Cirillo, P.; Paglia, A.; Sasso, L.; Di Palma, V.; Chiariello, M. Reactive oxygen species and antioxidants in the pathophysiology of cardiovascular disease: Does the actual knowledge justify a clinical approach? Curr. Vasc. Pharmacol. 2010, 8, 259–275. [Google Scholar] [CrossRef]
- Jadaun, P.; Yadav, D.; Bisen, P.S. Spirulina platensis prevents high glucose-induced oxidative stress mitochondrial damage mediated apoptosis in cardiomyoblasts. Cytotechnology 2018, 70, 523–536. [Google Scholar] [CrossRef]
- Martin-Ventura, J.L.; Rodrigues-Diez, R.; Martinez-Lopez, D.; Salaices, M.; Blanco-Colio, L.M.; Briones, A.M. Oxidative Stress in Human Atherothrombosis: Sources, Markers and Therapeutic Targets. Int. J. Mol. Sci. 2017, 18, 2315. [Google Scholar] [CrossRef] [Green Version]
- Shunmoogam, N.; Naidoo, P.; Chilton, R. Paraoxonase (PON)-1: A brief overview on genetics, structure, polymorphisms and clinical relevance. Vasc. Health Risk Manag. 2018, 14, 137–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camps, J.; Marsillach, J.; Joven, J. The paraoxonases: Role in human diseases and methodological difficulties in measurement. Crit. Rev. Clin. Lab. Sci. 2009, 46, 83–106. [Google Scholar] [CrossRef] [PubMed]
- Khersonsky, O.; Tawfik, D.S. Structure-reactivity studies of serum paraoxonase PON1 suggest that its native activity is lactonase. Biochemistry 2005, 44, 6371–6382. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, X.; Zhang, J.; Liang, Z.; Cai, W.; Huang, M.; Yan, C.; Zhu, Z.; Han, Y. Association between PON1 rs662 polymorphism and coronary artery disease. Eur. J. Clin. Nutr. 2014, 68, 1029–1035. [Google Scholar] [CrossRef]
- Humbert, R.; Adler, D.A.; Disteche, C.M.; Hassett, C.; Omiecinski, C.J.; Furlong, C.E. The molecular basis of the human serum paraoxonase activity polymorphism. Nat. Genet. 1993, 3, 73–76. [Google Scholar] [CrossRef]
- Davies, H.G.; Richter, R.J.; Keifer, M.; Broomfield, C.A.; Sowalla, J.; Furlong, C.E. The effect of the human serum paraoxonase polymorphism is reversed with diazoxon, soman and sarin. Nat. Genet. 1996, 14, 334–336. [Google Scholar] [CrossRef]
- Paolisso, G.; Manzella, D.; Tagliamonte, M.R.; Barbieri, M.; Marfella, R.; Zito, G.; Bonafè, M.; Giugliano, D.; Franceschi, C.; Varricchio, M. The BB-paraoxonase genotype is associated with impaired brachial reactivity after acute hypertriglyceridemia in healthy subjects. J. Clin. Endocrinol. Metab. 2001, 86, 1078–1082. [Google Scholar] [CrossRef]
- Serrato, M.; Marian, A.J. A variant of human paraoxonase/arylesterase (HUMPONA) gene is a risk factor for coronary artery disease. J. Clin. Investig. 1995, 96, 3005–3008. [Google Scholar] [CrossRef] [Green Version]
- Roest, M.; van Himbergen, T.M.; Barendrecht, A.B.; Peeters, P.H.; van der Schouw, Y.T.; Voorbij, H.A. Genetic and environmental determinants of the PON-1 phenotype. Eur. J. Clin. Investig. 2007, 37, 187–196. [Google Scholar] [CrossRef]
- Khan, A.A.; Alsahli, M.A.; Rahmani, A.H. Myeloperoxidase as an Active Disease Biomarker: Recent Biochemical and Pathological Perspectives. Med. Sci. 2018, 6, 33. [Google Scholar] [CrossRef] [Green Version]
- Daugherty, A.; Dunn, J.L.; Rateri, D.L.; Heinecke, J.W. Myeloperoxidase, a catalyst for lipoprotein oxidation, is expressed in human atherosclerotic lesions. J. Clin. Investig. 1994, 94, 437–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goeritzer, M.; Bernhart, E.; Plastira, I.; Reicher, H.; Leopold, C.; Eichmann, T.O.; Rechberger, G.; Madreiter-Sokolowski, C.T.; Prasch, J.; Eller, P.; et al. Myeloperoxidase and Septic Conditions Disrupt Sphingolipid Homeostasis in Murine Brain Capillaries In Vivo and Immortalized Human Brain Endothelial Cells In Vitro. Int. J. Mol. Sci. 2020, 21, 1143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piedrafita, F.J.; Molander, R.B.; Vansant, G.; Orlova, E.A.; Pfahl, M.; Reynolds, W.F. An Alu element in the myeloperoxidase promoter contains a composite SP1-thyroid hormone-retinoic acid response element. J. Biol. Chem. 1996, 271, 14412–14420. [Google Scholar] [CrossRef] [Green Version]
- Ergen, A.; İsbir, S.; Timirci, Ö.; Tekeli, A.; İsbir, T. Effects of myeloperoxidase -463 G/A gene polymorphism and plasma levels on coronary artery disease. Mol. Biol. Rep. 2011, 38, 887–891. [Google Scholar] [CrossRef] [PubMed]
- Lian, S.; Zhao, L.; Xun, X.; Lou, J.; Li, M.; Li, X.; Wang, S.; Zhang, L.; Hu, X.; Bao, Z. Genome-Wide Identification and Characterization of SODs in Zhikong Scallop Reveals Gene Expansion and Regulation Divergence after Toxic Dinoflagellate Exposure. Mar. Drugs 2019, 17, 700. [Google Scholar] [CrossRef] [Green Version]
- Bresciani, G.; Cruz, I.B.; de Paz, J.A.; Cuevas, M.J.; González-Gallego, J. The MnSOD Ala16Val SNP: Relevance to human diseases and interaction with environmental factors. Free Radic. Res. 2013, 47, 781–792. [Google Scholar] [CrossRef]
- Xu, M.; Xu, M.; Han, L.; Yuan, C.; Mei, Y.; Zhang, H.; Chen, S.; Sun, K.; Zhu, B. Role for Functional SOD2 Polymorphism in Pulmonary Arterial Hypertension in a Chinese Population. Int. J. Environ. Res. Public Health 2017, 14, 266. [Google Scholar] [CrossRef] [Green Version]
- Dalton, T.P.; Chen, Y.; Schneider, S.N.; Nebert, D.W.; Shertzer, H.G. Genetically altered mice to evaluate glutathione homeostasis in health and disease. Free Radic. Biol. Med. 2004, 37, 1511–1526. [Google Scholar] [CrossRef]
- Lu, S.C. Regulation of glutathione synthesis. Mol. Asp. Med. 2009, 30, 42–59. [Google Scholar] [CrossRef] [Green Version]
- Franklin, C.C.; Backos, D.S.; Mohar, I.; White, C.C.; Forman, H.J.; Kavanagh, T.J. Structure, function, and post-translational regulation of the catalytic and modifier subunits of glutamate cysteine ligase. Mol. Asp. Med. 2009, 30, 86–98. [Google Scholar] [CrossRef] [Green Version]
- Katakami, N.; Sakamoto, K.; Kaneto, H.; Matsuhisa, M.; Shimizu, I.; Ishibashi, F.; Osonoi, T.; Kashiwagi, A.; Kawamori, R.; Hori, M.; et al. Combined effect of oxidative stress-related gene polymorphisms on atherosclerosis. Biochem. Biophys. Res. Commun. 2009, 379, 861–865. [Google Scholar] [CrossRef] [PubMed]
- Cockcroft, J.R. Exploring vascular benefits of endothelium-derived nitric oxide. Am. J. Hypertens. 2005, 18, 177s–183s. [Google Scholar] [CrossRef] [PubMed]
- Yetik-Anacak, G.; Catravas, J.D. Nitric oxide and the endothelium: History and impact on cardiovascular disease. Vasc. Pharmacol. 2006, 45, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Ragia, G.; Nikolaidis, E.; Tavridou, A.; Arvanitidis, K.I.; Kanoni, S.; Dedoussis, G.V.; Bougioukas, G.; Manolopoulos, V.G. Endothelial nitric oxide synthase gene polymorphisms -786T > C and 894G > T in coronary artery bypass graft surgery patients. Hum. Genom. 2010, 4, 375–383. [Google Scholar] [CrossRef] [Green Version]
- Luo, Z.; Jia, A.; Lu, Z.; Muhammad, I.; Adenrele, A.; Song, Y. Associations of the NOS3 rs1799983 polymorphism with circulating nitric oxide and lipid levels: A systematic review and meta-analysis. Postgrad. Med. J. 2019, 95, 361–371. [Google Scholar] [CrossRef]
- Oliveira-Paula, G.H.; Lacchini, R.; Tanus-Santos, J.E. Endothelial nitric oxide synthase: From biochemistry and gene structure to clinical implications of NOS3 polymorphisms. Gene 2016, 575, 584–599. [Google Scholar] [CrossRef]
- Miyamoto, Y.; Saito, Y.; Kajiyama, N.; Yoshimura, M.; Shimasaki, Y.; Nakayama, M.; Kamitani, S.; Harada, M.; Ishikawa, M.; Kuwahara, K.; et al. Endothelial nitric oxide synthase gene is positively associated with essential hypertension. Hypertension 1998, 32, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Rai, H.; Parveen, F.; Kumar, S.; Kapoor, A.; Sinha, N. Association of endothelial nitric oxide synthase gene polymorphisms with coronary artery disease: An updated meta-analysis and systematic review. PLoS ONE 2014, 9, e113363. [Google Scholar] [CrossRef] [Green Version]
- Dworakowski, R.; Anilkumar, N.; Zhang, M.; Shah, A.M. Redox signalling involving NADPH oxidase-derived reactive oxygen species. Biochem. Soc. Trans. 2006, 34, 960–964. [Google Scholar] [CrossRef]
- Manea, A. NADPH oxidase-derived reactive oxygen species: Involvement in vascular physiology and pathology. Cell Tissue Res. 2010, 342, 325–339. [Google Scholar] [CrossRef]
- Inoue, N.; Kawashima, S.; Kanazawa, K.; Yamada, S.; Akita, H.; Yokoyama, M. Polymorphism of the NADH/NADPH oxidase p22 phox gene in patients with coronary artery disease. Circulation 1998, 97, 135–137. [Google Scholar] [CrossRef] [Green Version]
- Racis, M.; Sobiczewski, W.; Stanisławska-Sachadyn, A.; Wirtwein, M.; Bluj, E.; Nedoszytko, M.; Borzyszkowska, J.; Limon, J.; Rynkiewicz, A.; Gruchała, M. NADPH Oxidase Gene Polymorphism is Associated with Mortality and Cardiovascular Events in 7-Year Follow-Up. J. Clin. Med. 2020, 9, 1475. [Google Scholar] [CrossRef]
- San José, G.; Fortuño, A.; Beloqui, O.; Díez, J.; Zalba, G. NADPH oxidase CYBA polymorphisms, oxidative stress and cardiovascular diseases. Clin. Sci. 2008, 114, 173–182. [Google Scholar] [CrossRef] [Green Version]
- San José, G.; Moreno, M.U.; Oliván, S.; Beloqui, O.; Fortuño, A.; Díez, J.; Zalba, G. Functional effect of the p22phox -930A/G polymorphism on p22phox expression and NADPH oxidase activity in hypertension. Hypertension 2004, 44, 163–169. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, S.; Kugiyama, K.; Sugiyama, S.; Miyamoto, S.; Koide, S.; Fukushima, H.; Honda, O.; Yoshimura, M.; Ogawa, H. Polymorphism in the 5’-flanking region of human glutamate-cysteine ligase modifier subunit gene is associated with myocardial infarction. Circulation 2002, 105, 2968–2973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bresciani, G.; da Cruz, I.B.; González-Gallego, J. Manganese superoxide dismutase and oxidative stress modulation. Adv. Clin. Chem. 2015, 68, 87–130. [Google Scholar] [CrossRef]
- Deng, Z.; Xiang, H.; Gao, W. Significant association between paraoxonase 1 rs662 polymorphism and coronary heart disease: A meta-analysis in the Chinese population. Herz 2020, 45, 347–355. [Google Scholar] [CrossRef]
- Li, P.; Qiu, T.; Qin, C. NADPH oxidase p22phox C242T polymorphism and ischemic cerebrovascular disease: An updated meta-analysis. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2015, 21, 231–238. [Google Scholar] [CrossRef] [Green Version]
- Aviram, M.; Rosenblat, M.; Billecke, S.; Erogul, J.; Sorenson, R.; Bisgaier, C.L.; Newton, R.S.; La Du, B. Human serum paraoxonase (PON 1) is inactivated by oxidized low density lipoprotein and preserved by antioxidants. Free Radic. Biol. Med. 1999, 26, 892–904. [Google Scholar] [CrossRef]
- Gaidukov, L.; Rosenblat, M.; Aviram, M.; Tawfik, D.S. The 192R/Q polymorphs of serum paraoxonase PON1 differ in HDL binding, lipolactonase stimulation, and cholesterol efflux. J. Lipid Res. 2006, 47, 2492–2502. [Google Scholar] [CrossRef] [Green Version]
- Gupta, N.; Gill, K.; Singh, S. Paraoxonases: Structure, gene polymorphism and role in coronary artery disease. Indian J. Med. Res. 2009, 130, 361–368. [Google Scholar] [PubMed]
- Kuchta, A.; Strzelecki, A.; Ćwiklińska, A.; Totoń, M.; Gruchała, M.; Zdrojewski, Z.; Kortas-Stempak, B.; Gliwińska, A.; Dąbkowski, K.; Jankowski, M. PON-1 Activity and Plasma 8-Isoprostane Concentration in Patients with Angiographically Proven Coronary Artery Disease. Oxidative Med. Cell. Longev. 2015, 2015, 5136937. [Google Scholar] [CrossRef] [PubMed]
- Zargari, M.; Sharafeddin, F.; Mahrooz, A.; Alizadeh, A.; Masoumi, P. The common variant Q192R at the paraoxonase 1 (PON1) gene and its activity are responsible for a portion of the altered antioxidant status in type 2 diabetes. Exp. Biol. Med. 2016, 241, 1489–1496. [Google Scholar] [CrossRef] [PubMed]
- Bayrak, A.; Bayrak, T.; Tokgözoglu, S.L.; Volkan-Salanci, B.; Deniz, A.; Yavuz, B.; Alikasifoglu, M.; Demirpençe, E. Serum PON-1 activity but not Q192R polymorphism is related to the extent of atherosclerosis. J. Atheroscler. Thromb. 2012, 19, 376–384. [Google Scholar] [CrossRef] [Green Version]
- Antikainen, M.; Murtomäki, S.; Syvänne, M.; Pahlman, R.; Tahvanainen, E.; Jauhiainen, M.; Frick, M.H.; Ehnholm, C. The Gln-Arg191 polymorphism of the human paraoxonase gene (HUMPONA) is not associated with the risk of coronary artery disease in Finns. J. Clin. Investig. 1996, 98, 883–885. [Google Scholar] [CrossRef]
- Kaman, D.; Ilhan, N.; Metin, K.; Akbulut, M.; Ustündağ, B. A preliminary study of human paraoxonase and PON 1 L/M55-PON 1 Q/R 192 polymorphisms in Turkish patients with coronary artery disease. Cell Biochem. Funct. 2009, 27, 88–92. [Google Scholar] [CrossRef]
- Sanghera, D.K.; Saha, N.; Aston, C.E.; Kamboh, M.I. Genetic polymorphism of paraoxonase and the risk of coronary heart disease. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 1067–1073. [Google Scholar] [CrossRef]
- Zeng, Q.; Zeng, J. A meta-analysis on relationship between paraoxonase 1 polymorphisms and atherosclerotic cardiovascular diseases. Life Sci. 2019, 232, 116646. [Google Scholar] [CrossRef]
- Huo, X.; Guo, Y.; Zhang, Y.; Li, J.; Wen, X.; Liu, J. Paraoxonase 1 gene (Q192R) polymorphism confers susceptibility to coronary artery disease in type 2 diabetes patients: Evidence from case-control studies. Drug Discov. Ther. 2019, 13, 80–88. [Google Scholar] [CrossRef] [Green Version]
- Hernández-Díaz, Y.; Tovilla-Zárate, C.A.; Juárez-Rojop, I.E.; González-Castro, T.B.; Rodríguez-Pérez, C.; López-Narváez, M.L.; Rodríguez-Pérez, J.M.; Cámara-Álvarez, J.F. Effects of paraoxonase 1 gene polymorphisms on heart diseases: Systematic review and meta-analysis of 64 case-control studies. Medicine 2016, 95, e5298. [Google Scholar] [CrossRef]
- Paynter, N.P.; Chasman, D.I.; Paré, G.; Buring, J.E.; Cook, N.R.; Miletich, J.P.; Ridker, P.M. Association between a literature-based genetic risk score and cardiovascular events in women. JAMA 2010, 303, 631–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.; Zheng, Q.; Han, Y.; Qiao, S.; Chen, J.; Yuan, Z.; Yu, B.; Ge, L.; Jia, J.; Gong, Y.; et al. Genetic predisposition to coronary artery disease is predictive of recurrent events: A Chinese prospective cohort study. Hum. Mol. Genet. 2020, 29, 1044–1053. [Google Scholar] [CrossRef] [PubMed]
- Katakami, N.; Kaneto, H.; Matsuoka, T.A.; Takahara, M.; Imamura, K.; Ishibashi, F.; Kanda, T.; Kawai, K.; Osonoi, T.; Kashiwagi, A.; et al. Accumulation of gene polymorphisms related to oxidative stress is associated with myocardial infarction in Japanese type 2 diabetic patients. Atherosclerosis 2010, 212, 534–538. [Google Scholar] [CrossRef] [PubMed]
- Katakami, N.; Kaneto, H.; Matsuoka, T.A.; Takahara, M.; Osonoi, T.; Saitou, M.; Kawai, K.; Ishibashi, F.; Kashiwagi, A.; Kawamori, R.; et al. Accumulation of oxidative stress-related gene polymorphisms and the risk of coronary heart disease events in patients with type 2 diabetes—An 8-year prospective study. Atherosclerosis 2014, 235, 408–414. [Google Scholar] [CrossRef]
- Wirtwein, M.; Melander, O.; Sjogren, M.; Hoffmann, M.; Narkiewicz, K.; Gruchala, M.; Sobiczewski, W. Relationship between selected DNA polymorphisms and coronary artery disease complications. Int. J. Cardiol. 2017, 228, 814–820. [Google Scholar] [CrossRef]
- Wirtwein, M.; Melander, O.; Sjőgren, M.; Hoffmann, M.; Narkiewicz, K.; Gruchala, M.; Sobiczewski, W. Genetic risk factors influence nighttime blood pressure and related cardiovascular complications in patients with coronary heart disease. Hypertens. Res. Off. J. Jpn. Soc. Hypertens. 2018, 41, 53–59. [Google Scholar] [CrossRef]
- Wirtwein, M.; Melander, O.; Sjőgren, M.; Hoffmann, M.; Narkiewicz, K.; Gruchala, M.; Sobiczewski, W. Elevated ambulatory systolic-diastolic pressure regression index is genetically determined in hypertensive patients with coronary heart disease. Blood Press. 2017, 26, 174–180. [Google Scholar] [CrossRef]
- Gensini, G.G. A more meaningful scoring system for determining the severity of coronary heart disease. Am. J. Cardiol. 1983, 51, 606. [Google Scholar] [CrossRef]
- Neeland, I.J.; Patel, R.S.; Eshtehardi, P.; Dhawan, S.; McDaniel, M.C.; Rab, S.T.; Vaccarino, V.; Zafari, A.M.; Samady, H.; Quyyumi, A.A. Coronary angiographic scoring systems: An evaluation of their equivalence and validity. Am. Heart J. 2012, 164, 547–552. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Parameters | Total | Gensini <40 | Gensini ≥40 | p Value | |
|---|---|---|---|---|---|
| n = 1099 | n = 540 (49.1%) | n = 559 (50.9%) | |||
| Age (years): mean ± SD | 64.2 ± 9.4 | 63.9 ± 9.5 | 64.5 ± 9.4 | ns a | |
| median (min, max), n | 65 (36, 87), 1094 | 64.5 (36, 87), 538 | 65.5 (36, 84), 556 | ||
| Male: n (%) | 726 (66.1%) | 327 (60.6%) | 399 (71.5%) | 0.0001 b | |
| BMI (kg/m2): mean ± SD | 28.0 ± 4.1 | 27.9 ± 4.1 | 28.1 ± 4.0 | ns a | |
| median (min, max), n | 27.7 (14.9, 42.8), 1047 | 27.7 (14.9, 42.8), 516 | 27.7 (16.6, 40.9), 531 | ||
| Hypertension: n (%) | 856 (81.1%) | 419 (80.7%) | 438 (81.4%) | ns b | |
| Diabetes: n (%) | 265 (25.0%) | 132 (25.3%) | 133 (24.7%) | ns b | |
| Total cholesterol (mg/dL): mean ± SD | 207.1 ± 51.6 | 206.8 ± 50.1 | 207.3 ± 53.0 | ns a | |
| median (min, max), n | 198 (92, 552), 983 | 198 (98, 417), 486 | 198 (92, 552), 497 | ||
| LDL cholesterol (mg/dL): mean ± SD | 123.3 ± 43.8 | 122.4 ± 43.4 | 124.2 ± 44.3 | ns a | |
| median (min, max), n | 116 (7, 452), 905 | 115 (7, 317), 447 | 117 (37, 452), 458 | ||
| HDL cholesterol (mg/dL): mean ± SD | 54.8 ± 14.5 | 56.6 ± 16.1 | 52.9 ± 12.4 | 0.001 a | |
| median (min, max), n | 52 (26, 175), 956 | 53 (26, 175), 480 | 51 (26, 141), 489 | ||
| Triglycerides (mg/dL): mean ± SD | 147.2 ±94.8 | 144.8 ± 98.4 | 149.5 ± 91.1 | ns a | |
| median (min, max), n | 124 (36, 897), 975 | 122.5 (42, 897), 482 | 128 (36, 878), 493 | ||
| History of smoking: n (%) | 678 (67.0%) | 316 (63.2%) | 362 (70.7%) | 0.011 b | |
| CAD in family: n (%) | 546 (54%) | 267 (53.9%) | 279 (54.0%) | ns b | |
| Coronary narrowings ≥50% n (%) | 1070 (97.4%) | 511 (94.6%) | 559 (100%) | <0.0001 b | |
| Coronary narrowings ≥70% n (%) | 990 (90.1%) | 431 (79.8%) | 559 (100%) | <0.0001 b | |
| SNP | Risk allele | Risk allele frequency | |||
| PON1 c.575 A>G | G | 0.270 | 0.250 | 0.291 | 0.035 b |
| MPO c.−463 A>G | A | 0.164 | 0.157 | 0.170 | ns b |
| SOD2 c.47C>T | T | 0.476 | 0.462 | 0.490 | ns b |
| GCLM c.588 C>T | T | 0.165 | 0.166 | 0.165 | ns b |
| eNOS c.894 G>T | T | 0.277 | 0.269 | 0.284 | ns b |
| eNOS c.−786 C>T | C | 0.364 | 0.352 | 0.377 | ns b |
| CYBA c.214 T>C | T | 0.346 | 0.342 | 0.351 | ns b |
| CYBA c.−930 G>A | G | 0.596 | 0.590 | 0.602 | ns b |
| SNPs/GRS | Genotype/GRS Group | Gensini Score <40 (% of Genotype Carriers) | Gensini Score ≥40 (% of Genotype Carriers) | OR (95% CI), p |
|---|---|---|---|---|
| rs662 PON1 c.575A>G | AA | 299 (55.7) | 275 (49.6) | 1 (Ref) |
| AG | 208 (38.7) | 236 (42.6) | 1.23 (0.96–1.58), p = 0.097 | |
| GG | 30 (5.6) | 43 (7.8) | 1.55 (0.95–2.55), p = 0.076 | |
| p for trend = 0.028 | ||||
| A allele (ref) vs. GG | 1.42 (0.88–2.30), p = 0.151 | |||
| AA (ref) vs. G allele | 1.27 (1.004–1.617), p = 0.046 | |||
| rs2333227 MPO c.−463A>G | GG | 375 (69.8) | 385 (69.5) | 1 (Ref) |
| GA | 155 (28.9) | 150 (27.1) | 0.94 (0.72–1.22), p = 0.663 | |
| AA | 7 (1.3) | 19 (3.4) | 2.64 (1.10–6.36), p = 0.025 | |
| p for trend = 0.432 | ||||
| A allel vs. GG (ref) | 1.02 (0.78–1.31), p = 0.903 | |||
| AA vs. G allel (ref) | 2.69 (1.12–6.45), p = 0.021 | |||
| rs4880 SOD2 c.47C>T | CC | 148 (27.6) | 147 (26.5) | 1 (Ref) |
| CT | 282 (52.5) | 271 (48.9) | 0.97 (0.73–1.28), p = 0.828 | |
| TT | 107 (19.9) | 136 (24.6) | 1.28 (0.91–1.79), p = 0.156 | |
| p for trend = 0.183 | ||||
| T allele vs. CC (ref) | 1.05 (0.81–1.37), p = 0.703 | |||
| TT vs. C allele (ref) | 1.31 (0.98–1.74), p = 0.067 | |||
| rs41303970 GCLM c.588C>T | CC | 376 (70.0) | 387 (69.9) | 1 (Ref) |
| CT | 144 (26.8) | 151 (27.2) | 0.99 (0.77–1.28), p = 0.945 | |
| TT | 17 (3.2) | 16 (2.9) | 1.13 (0.76–1.66), p = 0.548 | |
| p for trend = 0.972 | ||||
| T allele vs. CC (ref) | 1.00 (0.78–1.31), p = 0.953 | |||
| TT vs. C allele (ref) | 0.91 (0.45–1.82), p = 0.789 | |||
| rs1799983 eNOS c.894G>T | GG | 290 (53.8) | 282 (51.0) | 1 (Ref) |
| GT | 208 (38.6) | 228 (41.2) | 1.13 (0.88–1.45), p = 0.346 | |
| TT | 41 (7.6) | 43 (7.8) | 1.08 (0.68–1.71), p = 0.746 | |
| p for trend = 0.437 | ||||
| GG (ref) vs. T allele | 1.12 (0.88–1.42), p = 0.353 | |||
| G allele (ref) vs. TT | 1.02 (0.66–1.60), p = 0.917 | |||
| rs2070744 eNOS c.−786C>T | TT | 223 (42.7) | 214 (40.1) | 1 (Ref) |
| CT | 231 (44.3) | 236 (44.3) | 1.06 (0.82–1.38), p = 0.638 | |
| CC | 68 (13.0) | 83 (15.6) | 1.27 (0.88–1.84), p = 0.204 | |
| p for trend = 0.232 | ||||
| CC vs. T allele (ref) | 1.23 (0.87–1.74), p = 0.238 | |||
| C allele vs. TT (ref) | 1.11 (0.87–1.42), p = 0.397 | |||
| rs4673 CYBA c.214T>C | CC | 230 (43.3) | 234 (42.9) | 1 (Ref) |
| CT | 239 (45.0) | 241 (44.1) | 0.99 (0.77–1.28), p = 0.945 | |
| TT | 62 (11.7) | 71 (13.0) | 1.13 (0.76–1.66), p = 0.548 | |
| p for trend = 0.666 | ||||
| T allele vs. CC (ref) | 1.02 (0.80–1.30), p = 0.880 | |||
| TT vs. C allele (ref) | 1.13 (0.79–1.63), p = 0.508 | |||
| rs9932581 CYBA c.−930G>A | AA | 84 (16.0) | 93 (17.0) | 1 (Ref) |
| AG | 262 (50.0) | 248 (45.4) | 0.85 (0.61–1.2), p = 0.369 | |
| GG | 178 (34.0) | 205 (37.6) | 1.04 (0.73–1.49), p = 0.828 | |
| p for trend = 0.546 | ||||
| A allele (ref) vs. GG | 1.17 (0.91–1.50), p = 0.222 | |||
| AA (ref) vs. G allele | 0.93 (0.67–1.28), p = 0.659 | |||
| Genetic Risk Score (GRS) | 1~2 | 40 (7.9) | 26 (5.0) | 1 (Ref) |
| 3~4 | 147 (29.1) | 147 (28.2) | 1.53 (0.89–2.67), p = 0.119 | |
| 5~6 | 205 (40.5) | 194 (37.2) | 1.45 (0.86–2.50), p = 0.164 | |
| 7~8 | 93 (18.4) | 120 (23.1) | 1.98 (1.13–3.51), p = 0.016 | |
| 9~12 | 21 (4.1) | 34 (6.5) | 2.47 (1.19–5.23), p = 0.014 | |
| p value for trend 0.008 |
| PARAMETERS | GENSINI SCORE | |||||
|---|---|---|---|---|---|---|
| Univariate Analysis | Multivariate Regression Analysis | |||||
| Spearman’s Rank Correlation Rho ρ | p Value | PON1 Model | GRS Model | |||
| β | p Value for F test | β | p Value for F test | |||
| Age (years) | 0.038 | 0.212 | 0.031 | 0.004 | 0.031 | 0.006 |
| Male sex (%) | 0.151 | <0.0001 | 0.965 | <0.0001 | 0.907 | <0.0001 |
| BMI (kg/m2) | −0.008 | 0.787 | - | - | - | - |
| Hypertension (%) | −0.019 | 0.535 | - | - | - | - |
| Diabetes (%) | −0.020 | 0.523 | - | - | - | - |
| Total cholesterol (mg%) | 0.013 | 0.673 | 0.007 | 0.001 | 0.007 | 0.003 |
| LDL cholesterol (mg%) | 0.039 | 0.236 | - | - | - | - |
| HDL cholesterol (mg%) | −0.116 | 0.0003 | −0.036 | <0.0001 | −0.037 | <0.0001 |
| Triglycerides (mg%) | 0.074 | 0.021 | - | - | - | - |
| History of smoking (%) | 0.093 | 0.003 | - | - | - | - |
| PON1 c.575A>G | 0.069 | 0.022 | 0.43 | 0.007 | NA | NA |
| GRS | 0.068 | 0.030 | NA | NA | 0.128 | 0.014 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Racis, M.; Stanisławska-Sachadyn, A.; Sobiczewski, W.; Wirtwein, M.; Krzemiński, M.; Krawczyńska, N.; Limon, J.; Rynkiewicz, A.; Gruchała, M. Association of Genes Related to Oxidative Stress with the Extent of Coronary Atherosclerosis. Life 2020, 10, 210. https://doi.org/10.3390/life10090210
Racis M, Stanisławska-Sachadyn A, Sobiczewski W, Wirtwein M, Krzemiński M, Krawczyńska N, Limon J, Rynkiewicz A, Gruchała M. Association of Genes Related to Oxidative Stress with the Extent of Coronary Atherosclerosis. Life. 2020; 10(9):210. https://doi.org/10.3390/life10090210
Chicago/Turabian StyleRacis, Milena, Anna Stanisławska-Sachadyn, Wojciech Sobiczewski, Marcin Wirtwein, Michał Krzemiński, Natalia Krawczyńska, Janusz Limon, Andrzej Rynkiewicz, and Marcin Gruchała. 2020. "Association of Genes Related to Oxidative Stress with the Extent of Coronary Atherosclerosis" Life 10, no. 9: 210. https://doi.org/10.3390/life10090210
APA StyleRacis, M., Stanisławska-Sachadyn, A., Sobiczewski, W., Wirtwein, M., Krzemiński, M., Krawczyńska, N., Limon, J., Rynkiewicz, A., & Gruchała, M. (2020). Association of Genes Related to Oxidative Stress with the Extent of Coronary Atherosclerosis. Life, 10(9), 210. https://doi.org/10.3390/life10090210