Abstract
Tissue fibrosis is characterized by excessive deposition of extracellular matrix (ECM) components that result from the disruption of regulatory processes responsible for ECM synthesis, deposition, and remodeling. Fibrosis develops in response to a trigger or injury and can occur in nearly all organs of the body. Thus, fibrosis leads to severe pathological conditions that disrupt organ architecture and cause loss of function. It has been estimated that severe fibrotic disorders are responsible for up to one-third of deaths worldwide. Although intensive research on the development of new strategies for fibrosis treatment has been carried out, therapeutic approaches remain limited. Since stem cells, especially mesenchymal stem cells (MSCs), show remarkable self-renewal, differentiation, and immunomodulatory capacity, they have been intensively tested in preclinical studies and clinical trials as a potential tool to slow down the progression of fibrosis and improve the quality of life of patients with fibrotic disorders. In this review, we summarize in vitro studies, preclinical studies performed on animal models of human fibrotic diseases, and recent clinical trials on the efficacy of allogeneic and autologous stem cell applications in severe types of fibrosis that develop in lungs, liver, heart, kidney, uterus, and skin. Although the results of the studies seem to be encouraging, there are many aspects of cell-based therapy, including the cell source, dose, administration route and frequency, timing of delivery, and long-term safety, that remain open areas for future investigation. We also discuss the contemporary status, challenges, and future perspectives of stem cell transplantation for therapeutic options in fibrotic diseases as well as we present recent patents for stem cell-based therapies in organ fibrosis.
1. Introduction
Fibrosis is the excessive accumulation of extracellular matrix (ECM), which leads to impairment of organ function and is consequently associated with high morbidity and mortality. The fibrotic process affects nearly all solid tissues and organs, including the heart, kidney, lung, liver, and skin, and in the majority of cases, this condition results from an ongoing disease (e.g., asthma, hypertension, diabetes, myocardial infarction (MI)) that commonly triggers these tissues [1,2]. Most fibrotic diseases have a relatively well-described etiology, including genetic predisposition, lifestyle, or systemic disorders (Figure 1). Despite showing different clinical features, the majority of fibrotic diseases share common pathological processes characterized by persistent inflammation leading to the production of growth factors, cytokines, and proteolytic enzymes. Altogether, these factors affect myofibroblast differentiation and stimulate excessive deposition of connective tissue components [3].
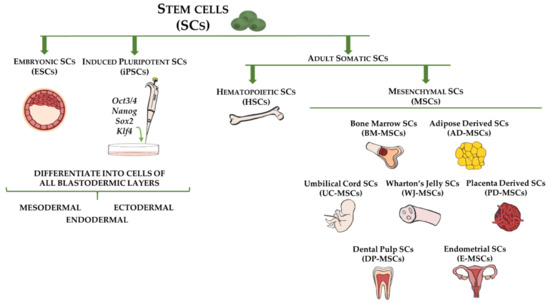
Figure 1.
Stem cell types and sources used for cell-based therapy for fibrotic disorders. Embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs) are capable of differentiating into cells of all three germ layers (ectoderm, mesoderm, and endoderm). Adult somatic stem cells include hematopoietic stem cells (HSCs), which are multipotent and reside in bone marrow, and mesenchymal stem cells (MSCs), which represent mesodermal progenitors existing in multiple tissues, including bone marrow, adipose tissue, umbilical cord blood, Wharton’s jelly, placenta, dental pulp, and endometrium.
It has been estimated that severe fibrotic disorders are responsible for up to one-third of deaths worldwide [1]. Researchers and clinicians all over the world have worked on developing efficient therapeutic strategies to treat these life-threatening diseases. However, despite constant efforts, there are still no effective therapies for the treatment of fibrosis in any organ (Figure 1). Stem cell-based therapy shows great promise for restoring injured tissues and treating/reversing fibrotic changes. The vast therapeutic potential of this type of therapy in the treatment of neurodegenerative, autoimmune, and genetic disorders has been reported [4,5]. Moreover, clinically relevant studies have included the application of mesenchymal stem cells (MSCs) in a broad range of degenerative, post-injury, and fibrosis-related diseases [6,7,8].
Mesenchymal stem cells are present in various tissues and organs, including bone marrow, adipose tissue, umbilical cord, and endometrium [9,10,11,12]. According to the standard definition, MSCs are fibroblast-like, adherent, clonogenic cells that express a number of surface markers, such as CD29, CD73, CD90, and CD105, and exhibit the capacity to differentiate into adipogenic, chondrogenic, and osteogenic lineages in vitro [13]. The mechanisms through which transplanted MSCs show their therapeutic effects rely on cell differentiation into tissue-specific cell types that replace defective cells or on the secretion of growth factors that lead to an enhanced renewal of target tissue [14,15]. Notably, in vivo studies using rodent models of degenerative neurological diseases have shown that MSCs might contribute to tissue repair by acting as immunomodulatory, neuroprotective, and anti-inflammatory agents [16,17].
In this review, we summarize the results of recent studies regarding the use of stem cells in experimentally induced and clinical cases of fibrosis in multiple organs as well as we present recent patents for stem cell-based therapies in organ fibrosis. We also discuss challenges, limitations, and future perspectives of cell-based therapies that should be considered before conducting large-scale trials.
2. Cellular and Molecular Basis of Tissue Fibrosis
It has been long established that myofibroblasts are the cells responsible for tissue fibrosis [18,19]. Myofibroblasts are heterogeneous cell populations that are defined by the expression of the contractile protein α-smooth muscle actin (αSMA or ACTA2) and the ability to synthesize and release ECM proteins such as collagen (COL) and fibronectin (FN) [18]. A variety of cell types, including resident fibroblasts, vascular pericytes, and bone marrow-derived cells, can be precursors of myofibroblasts [18]. Moreover, in the skin and the lung, epithelial cells can also contribute to the myofibroblast pool as they undergo the epithelial-to-mesenchymal transition (EMT) process [20,21]. Furthermore, as demonstrated in a study by Marangoni et al. [22], subcutaneous adipocytes can transdifferentiate into myofibroblast-like cells upon transforming growth factor beta (TGF)-β stimulation.
The development of fibrotic diseases is associated with abnormal accumulation of myofibroblasts. This aberration leads to excessive deposition of ECM components, which impairs organ structure and function. In healthy tissues, there is a balance between the synthesis and degradation of collagen and other ECM components. These processes are disrupted in fibrotic organs and directed toward ECM accumulation, which is partially evoked by an increased expression of tissue inhibitors of matrix metalloproteinases (TIMPs) versus a decrease in matrix metalloproteinases (MMPs) [23,24]. Indeed, numerous fibrotic-related human liver diseases, including biliary atresia, primary biliary cirrhosis, and primary sclerosing cholangitis, are associated with TIMP-1 and TIMP-2 overexpression [25]. Moreover, membrane-bound matrix metalloproteinase (MT1-MMP)-deficient mice that show a deficiency of membrane-type MMP-14, the metalloproteinase responsible for pericellular proteolysis of ECM, develop progressive fibrosis of the dermis and hair follicles [26].
Although multiple growth factors and pathways contribute to myofibroblast differentiation and EMT process, TGFβ signaling is the major inducer of fibrotic lesions, as has been evident in several in vitro and in vivo studies [27,28]. The activation of TGFβ signaling by overexpression of constitutively active TGFβ receptor type 1 (TGFβRI) is sufficient to induce a fibrotic phenotype characteristic of systemic sclerosis (SS) in mice [29]. In contrast, inhibition of the TGFβ pathway by phosphodiesterase (PDE) inhibitors exert potent antifibrotic effects in human lung fibroblasts [30]. TGFβ signaling is transduced through Smad and non-Smad pathways, such as mitogen-activated protein kinase pathways mediated by extracellular-signal-regulated kinase (ERK), RHO-associated kinase (ROCK), p38 and JUN N-terminal kinase (JNK), and RAC-α serine/threonine-protein kinase pathways [31]. Moreover, TGFβ1 stimulates the secretion of secondary signals, such as connective tissue growth factor (CTGF), plasminogen activator inhibitor 1 (PAI-1), endothelin-1, and NADPH oxidase 4 [32]. These agents show the capacity to mediate and/or mimic the effects of TGFβ1 and ultimately participate in signaling crosstalk, which consequently results in abnormal tissue remodeling and fibrosis in several organs [32].
At present, there is a large amount of data indicating that Wnt and hedgehog signaling, well-described developmental (morphogen) pathways, are potent modulators of fibrotic tissue remodeling across organs [33,34]. Overexpression of Wnt1 and Wnt10b proteins has been observed in human samples from SS, idiopathic pulmonary fibrosis (IPF), and liver cirrhosis [33]. Moreover, it has been shown that β-catenin, the central component of the canonical Wnt signaling, accumulates in the nuclei of fibroblasts from patients with SS and is overexpressed in the skin of mice with bleomycin (BLM)-induced SS. Interestingly, in parallel with the upregulation of Wnt ligands, a significant decrease in the endogenous Wnt antagonist Dickkopf-1 (Dkk-1) was reported [33]. Similarly, other studies demonstrated increased expression of sonic hedgehog (SHH) and the hedgehog transcription factor GLI2 in the skin of patients with SS [35]. At the same time, in the serum of those patients, high SHH concentrations were observed, correlating with the fibrotic burden [36]. Besides the aforementioned key regulators of fibrosis, there is a wide range of well-established factors that stimulate fibrogenesis, such as inflammatory mediators and cells, hypoxia, and DNA methylation of antifibrotic genes [37,38,39]. Importantly, it must be highlighted that tissue fibrosis is a complex process that involves the action of many factors and the activation of multiple signaling cascades that cross-react at multiple levels, and consequently, this condition presents a challenge in the development of novel antifibrotic approaches.
3. Stem Cell Types for Fibrotic Disorders Therapy
Although stem cells exist in relatively low quantities in adult tissues, they comprise a unique cell population that greatly contributes to tissue homeostasis and drives tissue regeneration [40]. By definition, stem cells are characterized by their ability to self-renew and differentiate into multiple cell lineages, which could provide therapeutic solutions for numerous diseases [6,41]. Although multiple criteria are used to classify stem cells (e.g., origin, differentiation potency), generally they can be categorized as embryonic stem cells (ESCs), collected from pre-implantation blastocysts, induced pluripotent stem cells (iPSCs), representing genetically reprogrammed adult somatic stem cells to an ESC-like state, and postnatal adult somatic stem cells, obtained from adult tissues (Figure 1) [42].
Embryonic stem cells are pluripotent cells that possess unlimited developmental potential, reflected by their capacity to form all types of tissues in the body. This ability to differentiate into multiple mature somatic cell types is maintained in cell cultures. The pluripotency fate of ESCs is driven by transcription factors Oct4, Sox2, and Nanog, which are termed pluripotency factors. However, upon appropriate stimulation, ESCs initially form 3D spherical structures termed embryoid bodies (EBs), then differentiate into precursor cells, and further give rise to various somatic cell lineages [43]. Indeed, the in vitro capacity of mouse ESCs to produce functional cardiomyocytes, chondrocytes, osteoblasts, endothelial cells, alveolar epithelium, and neuroectodermal cells, among others, has been demonstrated [44,45,46,47,48]. At present, ESC lines are available from the embryos of numerous mammalian species, including humans [49]. Although the generation of human ESC lines and their use in scientific research and clinical practice has raised an ethical controversy, it is important to acknowledge that differentiated cell lineages from ESCs have had an enormous impact in many fields of research, and they can serve as a promising tool for regenerative medicine in the treatment of a wide range of degenerative disorders.
The development of methods for reprogramming somatic cells into a pluripotent-like state and generating iPSCs via genomic integration and high expression of four Yamanaka factors (Oct4, Sox2, Klf4, and c-Myc) allowed researchers to overcome the obstacles related to the use of ESCs [50]. Even more important, since iPSCs share the same regenerative properties as ESCs, they hold great promise as custom-made pluripotent cells that could be produced for individual patients and further used in autologous transplantation. One of the advantages of iPSCs in clinical application is the fact that the cells are unlikely to cause host immune rejection [51]. Recent in vitro studies demonstrated the ability of iPSCs to differentiate into beating cardiomyocyte-like cells, insulin-producing islet-like clusters (ILCs), and neuronal and glial cell types [52,53,54]. Of particular interest, several iPSC-based clinical trials, mostly targeting eye diseases, are under way, highlighting the enormous progress that has been made in stem cell biology and regenerative medicine [55].
The most common adult somatic stem cells, which have been used for the longest time, are MSCs. They represent mesodermal progenitors existing in multiple tissues, including bone marrow, adipose tissue, umbilical cord blood, dental pulp, and endometrium (Figure 1) [11,12,56,57,58]. The use of MSCs allows researchers to overcome the ethical and legal issues associated with the application of ESCs and the uncontrolled mutational effects related to iPSCs. Although bone marrow was the first organ studied as a source of MSCs (BM-MSCs), cells isolated from other tissues, including adipose tissue, periodontal ligament, and trabecular bone, demonstrated comparable characteristics in terms of morphology, surface markers, and differentiation potential in vitro [59]. Moreover, limitations related to BM-MSC procurement, such as the high risk of morbidity associated with the bone marrow aspiration procedure and the relatively low yield of isolated MSCs (0.001–0.01% of harvested bone marrow cells), have led to a search for alternative sources of MSCs [59,60]. In this regard, adipose-derived stem cells (ASCs) overcome the obstacles of BM-MSCs, as these cells can be isolated from multiple fat depots in large quantities using a minimally invasive procedure, such as liposuction or other surgical interventions that in humans are used for the removal of excess fat [61]. Several studies have demonstrated the plasticity of human and animal ASCs [62,63,64]. Consistently, there is extensive literature regarding the safety and efficacy of ASC-based therapies for the treatment of numerous diseases and injuries created in animal models of human diseases, including cardiovascular disease and musculoskeletal and cutaneous injuries [65]. The therapeutic potential of ASCs or Stromal Vascular Fraction cells (SVFs) has been confirmed not only in experimental animal models but also in an increasing number of human clinical trials [66,67,68].
3.1. Pulmonary Fibrosis
Pulmonary fibrosis (PF) is associated with over 200 chronic lung diseases, which differ in the severity of their course, the degree of inflammation, and the advancement of fibrotic foci. The most common and lethal form of idiopathic interstitial pneumonia is idiopathic pulmonary fibrosis, which affects 3–9 per 100,000 people annually, with poor median survival rates at 2–3 years and 5-year survival ranging between 30% and 50% [69]. Many PF disorders emerge from underlying causes such as autoimmune diseases, including SS and rheumatoid arthritis. The important factors that contribute to the development of PF include smoking, hazardous chemicals, air pollution, exposure to cancer radiation therapy or chemotherapy, and genetic and epigenetic factors (Figure 2).
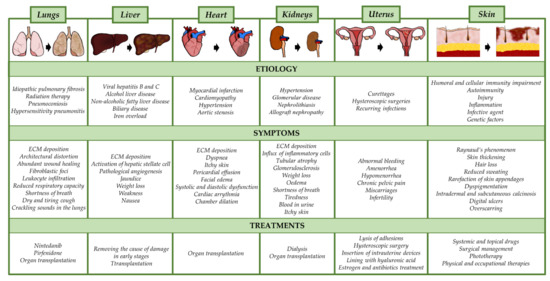
Figure 2.
Etiology, symptoms, and current treatment options for fibrotic diseases in individual body organs. ECM; extracellular matrix.
So far, only pirfenidone and nintedanib have been approved as pharmacological therapies for the treatment of IPF (Figure 2). These drugs used clinically are effective in prolonging the time of disease progression, slowing down the decline in lung function, and improving the quality of life of patients with IPF [70,71]. However, damaged lung tissue is not restored with these drugs, and both drugs are associated with gastrointestinal adverse events. Hence, there is an urgent need to establish novel treatment strategies for lung fibrosis, such as stem cell therapy.
Several in vivo studies using animal models of human PF demonstrated promising effects of using stem cells [72,73]. Indeed, a study by Ortiz et al. [72] showed that systemic administration of 5 × 105 BM-MSCs in mice with BLM-induced lung fibrosis protects the lungs from injury by reducing inflammation, collagen deposition, and MMP (MMP-2, -9, -13) activation in lung tissue. More interestingly, MSCs have been shown to engraft in lung tissue, where they acquire epithelial-like morphology, indicating that they might exert their effect not only via regulation of the local environment but differentiation and structural support of injured tissue [72]. These findings were further supported in a study by Rojas et al. [73], which demonstrated the presence of green fluorescent protein (GFP)-positive BM-MSCs in lungs 14 days after BLM treatment and subsequent cell transplantation. Furthermore, following BM-MSC delivery, the authors observed an increase in circulating levels of granulocyte colony stimulating factor (G-CSF) and granulocyte-macrophage colony-stimulating factor (GM-CSF), which may be responsible for mobilizing stem cells from bone marrow pools, suggesting that the therapeutic effect of BM-MSCs might rely, at least in part, on the mobilization of endogenous stem cells. The action of BM-MSCs was also manifested by the suppression of prolonged BLM-caused inflammatory response in the lungs, as this effect was shown by a decrease in mRNA expression of interferon (IFN)-γ, and interleukin (IL)-2, IL-1β, and IL-4 in lung tissue at 14 days after exposure to BLM and stem cell delivery [73].
Many investigators have used allogeneic mouse or human ASCs (hASCs) as possible treatments for BLM-induced lung fibrosis in mice [74,75]. In one such study, hASCs pooled from five donors were administrated intravenously at a dose of 4 × 107 cells/kg body weight on days 3, 6, and 9 post-BLM administration [75]. hASC administration resulted in significant inhibition of BLM-induced lung fibrosis, reflected by a reduction in collagen deposition and downregulation of the mRNA of proinflammatory cytokines (IL-2, IL-1β, tumor necrosis factor (TNF), TGFβ1), leading to a drop in mRNA expression of factors responsible for ECM deposition and remodeling, such as basic fibroblast growth factor (bFGF); Timp-1, -2, and -3; Mmp-2 and -3; CTGF; and Col1a1 and Col1a3 in lung tissue. Notably, histological analysis of lungs confirmed by high-resolution computed tomography exhibited attenuation of lung fibrosis accompanied by thickening of alveolar septa and maintenance of alveolar architecture [75]. Similarly, Kotani et al. [74] demonstrated inhibition of pulmonary inflammation and fibrosis upon intravenous infusion of mouse ASCs (mASCs) at a dose of 2.5 × 104 or 2.5 × 105 in mice with BLM-induced lung fibrosis. The anti-inflammatory effect of mASCs was exhibited by reduced numbers of infiltrated macrophages, neutrophils, and T lymphocytes in lung tissue as compared to control (BLM injected but not mASC-treated animals). Moreover, an in vitro study carried out on macrophages co-cultured with mASCs revealed significant downregulation of the mRNA of proinflammatory cytokines, TNF-α and IL-12 in activated macrophages and increased macrophage apoptosis. Additionally, the results of this study indicated that mASCs inhibited the differentiation and proliferation of Th2 cells, suggesting their role in promoting the differentiation and proliferation of T-regs, which might provide the mechanism based on which mASCs reduce pulmonary inflammation [74].
Important results indicating that the therapeutic efficacy of ASCs may vary with age were obtained by Tashiro et al. [76], who used 5 × 105 mASCs from young (4 months) and old (22 months) male mice as a treatment for BLM-induced lung fibrosis in an aged mouse model (>22 months). Decreased fibrosis, evidenced by reduced interstitial and perivascular collagen deposition estimated histologically and based on hydroxyproline content, was found in aged mice 21 days after transplantation of mASCs obtained from young animals. Moreover, delivery of mASCs from young donor mice led to multiple changes in fibrotic lungs, as manifested by a drop in mRNA expression of TGFβ, inhibition of Mmp-2, and protein kinase B (Akt activity, maintenance of redox balance, and reduced cell apoptosis. In contrast, delivery of old-donor ASCs into aged BLM-treated mice did not exert such remarkable effects, as they did not reduce fibrosis and its related markers. Undoubtedly, this study indicates that the antifibrotic properties of ASCs are age-dependent, and this should be considered in clinical trials in patients with PF [76].
There have also been studies investigating the use of umbilical cord MSCs (UC-MSCs) derived from Wharton’s jelly for the treatment of lung fibrosis that support the antifibrotic and immunomodulatory properties of stem cells [77,78]. As reported by Moodley et al. [78], systemically administered human UC-MSCs (hUC-MSCs; 1 × 106) in mice with BLM-induced lung fibrosis reduced levels of inflammatory and profibrotic markers at 14 days. Indeed, a decrease in mRNA expression of IL-10, TNF-α, TGFβ, and IFN-γ in lung tissue was shown following UC-MSC treatment. Furthermore, the cell delivery attenuated collagen deposition and improved the balance between MMP-2 and TIMP 1–4 in lung tissue that was abrogated upon BLM treatment [78]. Interestingly, others showed that intraperitoneal or intratracheal application of fetal membrane-derived MSCs from both humans and mice led to decreased neutrophil infiltration and a significant reduction in the severity of BLM-induced lung fibrosis regardless of the cell source (allogeneic or xenogeneic) or delivery route [77].
Preclinical results showed that iPSCs might provide new therapeutic opportunities in damaged lungs [79]. In a mouse model of BLM-induced lung fibrosis, Zhou et al. [79] demonstrated a significant therapeutic effect mediated by alveolar epithelial cells (AECs) differentiated from iPSCs. After 12 days, it was shown that the cells integrated into the lung alveolar structure and expressed lung progenitor markers such as surfactant protein C and T1α. Importantly, differentiated iPSCs contributed to the reconstitution of BLM-damaged lung tissue by diminishing inflammation and fibrosis [79]. Similar results have been shown using iPSCs generated from mouse embryonic fibroblasts and delivered at a dose of 2 × 106 in a mouse model of BLM-induced lung fibrosis. In that study, inhibition of collagen accumulation in the lungs decreased infiltration of inflammatory cells, and downregulation of TNF-α, IL-1β, and IL-6 at 21 days was reported. Moreover, iPSC transplantation blocked TGFβ1/Smad2/3 signaling, which is a key profibrotic pathway and potential therapeutic target in lung fibrosis [80]. In another study that focused on iPSC secretome, the culture medium containing hepatocyte growth factor (HGF) secreted by iPSCs contributed to AECs repair in vitro and attenuated BLM-induced lung fibrosis in vivo [81].
Many successful preclinical studies in rodents have encouraged the translation of stem cell-based therapy into clinical settings [82,83,84,85]. Currently, multiple clinical trials are being conducted to ensure the safety of stem cell treatment for IPF (https://ClinicalTrials.gov; accessed 16 September 2021) (Table 1). A non-randomized, non-placebo-controlled phase I trial named idiopathic pulmonary fibrosis via intravenous delivery (AETHER) showed the safety and efficacy of intravenous delivery of allogeneic BM-MSCs. At 36 weeks follow-up, improvement in lung function was found based on the results of 6-min walk test (6MWT), and at 60 weeks post transplantation, a decrease in the predicted value of forced vital capacity (FVC) by 3.0% on average and a 5.4% mean decline in carbon monoxide diffusing capacity (DLCO) were observed [84]. Another study carried out on IPF patients who received 1 × 107 or 1 × 108 of allogeneic BM-MSCs demonstrated slower progression of lung fibrosis, especially with the higher cell dose [83]. Averyanov et al. [82] conducted a phase I study in which allogeneic BM-MSCs were administrated to IPF patients in a high cumulative dose of 2 × 108 cells every 3 months (1.6 × 109 cells total). Patients were followed for 52 weeks, and no significant adverse effects were reported. In the group of MSC recipients, lung function increased compared to the placebo group. Indeed, improvements in 6MWT at 13 weeks, DLCO at 26 weeks, and FVC at 39 weeks were observed compared with placebo. Significantly, in the MSC therapy group, at 12 months, FVC increased by 7.8% from baseline, whereas it declined by 5.9% in the placebo group [82]. Tzouvelekis et al. [85] conducted a non-randomized, no placebo-controlled, phase Ib clinical trial in which IPF patients received autologous SVFs in three endobronchial infusions at a dose of 0.5 × 106 cells/kg body weight. They found at 12 months follow-up that 86% of patients had a stable function and exercise capacity. Safety monitoring indicated that cell-administered patients did not deteriorate in either functional parameters or indicators of quality of life [85].
Although two new drugs, nintedanib, and pirfenidone, have been found to be clinically effective in reducing the progression of PF and improving patients’ quality of life, the damaged lung tissue does not recover with these drugs. Therefore, taking into consideration the advantages of using stem cell therapy, their use in the treatment of PF seems to be highly desired. Notably, the potential effects of MSCs in PF mostly rely on their ability to secrete biologically active agents that show immunosuppressive, anti-inflammatory, and pro-angiogenic properties.
3.2. Liver Fibrosis
Liver fibrosis, a condition that frequently progresses to cirrhosis, is one of the major challenges of global health. It is estimated that nearly 7% of the population suffers from liver fibrosis, and every year more than 2 million people die worldwide due to fibrosis-associated liver failure [86]. The main causes of hepatic fibrosis include non-alcoholic steatohepatitis (NASH), non-alcoholic fatty liver disease (NAFLD), alcohol abuse, hepatitis B (HBV) and hepatitis C (HCV) virus infections, autoimmune hepatitis, iron overload, and biliary obstruction (Figure 2) [86]. While the early stages of hepatic damage are reversible, the severe stages of fibrotic diseases, especially advanced cirrhosis, are not, and they lead to subsequent morbidity and mortality [87].
Healthy liver tissue consists of around 3% ECM in the area of interest in a normal cross-section, mainly located around Glisson’s capsule, portal tracts, sinusoid walls, and central veins [88]. As fibrosis develops, the ECM content increases up to five-fold. Interestingly, the liver has a unique ability to slow down or even reverse fibrosis, and after removal of profibrotic factors, activated hepatic stellate cells undergo apoptosis, while MMPs break down excessive ECM. However, if the exposure to damaging factors is permanent, hepatic stellate cells migrate and proliferate drastically, altering their phenotype and secreting large amounts of ECM [89].
A study on a mouse model of liver fibrosis induced by carbon tetrachloride (CCl4) infusion demonstrated that administration of 1 × 106 BM-MSCs decreased liver fibrosis at 4 weeks follow-up [90]. Histological analysis of the liver demonstrated collagen reduction and recovery of tissue architecture as a consequence of BM-MSC administration. The mRNA expression of αSMA and Timp-1 was downregulated following BM-MSC treatment. Moreover, there were some engrafted cells in the recipient liver that were able to differentiate into albumin-positive cells [90]. These results are in agreement with studies performed on rats with CCl4-induced liver fibrosis that received BM-MSCs at a dose of 3 × 106 [91]. In the cell-treated group, a significant decrease in collagen deposition and a decrease in hydroxyproline content were evident in the liver. An elevation in serum albumin (ALB) and a decrease in alanine aminotransferase (ALT) levels were also detected following cell transplantation [91]. Likewise, Zhao et al. [92], using a rat model of CCl4 or dimethylnitrosamine-induced liver injury, demonstrated a reduced mortality rate mediated by BM-MSCs delivered at a dose of 3 × 106 cells. Consistent with other studies, decreased deposited collagen content and weaker αSMA staining in the liver of MSC-treated rats was found [92].
An important study underlining the source-specific potential of stem cells to exert a therapeutic effect was conducted using allogeneic MSCs, hematopoietic stem cells (HSCs), or combined MSCs + HSCs (1 × 106 cells) as a treatment for induced liver injury [93]. A comparable analysis revealed that MSCs demonstrated more efficient reparative activity than HSCs with no synergistic effects between the two types of cells. After transplantation of MSCs, the injured livers exhibited maximal restoration with thinner fibrotic areas and decreased collagen levels relative to the HSC- or MSC + HSC-treated groups. Mice receiving MSCs showed superior improvement of liver function, as demonstrated by decreased serum albumin (ALB), ALT, and aspartate aminotransferase (AST) levels in the peripheral blood. Moreover, in animals transplanted with MSCs, elevated serum concentration of anti-inflammatory IL-10 was observed, while the levels of IL-6 and TNF-α, which are considered as promoters of liver fibrosis, were downregulated when compared to the other cell-administered groups and non-cell-treated but CCL4 challenged control [93].
Several preclinical rodent studies have clearly demonstrated the beneficial effects of human BM-MSCs (hBM-MSCs) in chemically induced liver fibrosis [94,95]. In one such study, Chang et al. [94] transplanted 1 × 106 culture-expanded hBM-MSCs into CCl4-treated rats. At 4 weeks post-delivery, the levels of serum albumin and fibrinogen in the cell-treated group returned to normal levels, indicating restoration of specific liver functions. In addition, the incorporated hBM-MSCs expressed human α-fetoprotein, albumin, and cytokeratin-18, suggesting that transplanted cells can differentiate into albumin-secreting hepatocyte-like cells; hence, they contribute to the healing of the damaged liver [94]. Similarly, another study on a mouse model of CCl4-induced liver fibrosis showed that intravenous administration of hBM-MSCs at a dose of 5 × 105 significantly reduced fibrosis levels 4 weeks after the completion of surgery [95]. Moreover, in the group of animals receiving hBM-MSCs, increased MMP-9 expression and decreased αSMA, TNFα, and TGFβ expression in liver tissue was observed. Although the mechanisms by which BM-MSCs reduced hepatic fibrosis have not been well established, the authors postulated that it might occur, in part, through enhanced expression of MMP-9, which is important for ECM remodeling [95].
Several studies have shown that ASCs or human UC-MSCs (hUC-MSCs) administered in rodent models of hepatic damage also led to reduced tissue fibrosis and provided restoration of liver function [94,95,96,97,98]. A study in which ASCs were first pretreated in the hepatogenic medium and then transplanted into rats with liver injury showed a decrease in circulating liver enzymes such as ALT and AST along with improved serum albumin level as compared with PBS controls [97]. In thioacetamide-induced chronic liver fibrosis in rats, transplantation of hASCs at a dose of 1 × 106 by direct liver injection resulted in the recovery of organ function, as evidenced at 3 weeks by biochemical analysis of total bilirubin (TBIL), prothrombin time (PT), and albumin levels [96]. The authors found a significant decrease in liver fibrosis and inflammatory activity measured by metavir-based activity score in the ASC group. In addition, reduced expression of αSMA after 14 days and increased expression of MMP-9 in liver tissue at 7 and 14 days post transplantation were detected. Moreover, immunohistochemistry showed that hASCs injected directly into the liver differentiated into albumin- and α-fetoprotein-secreting liver-like cells as early as 1 week after transplantation [96]. Significantly, another group showed that ASCs were able to survive up to 4 months after engraftment into rat liver, and some of them acquired hepatocyte (asialoglycoprotein receptor ½ (ASGPR1/2)-positive) phenotype [98].
In the case of hUC-MSCs, it has been reported that administering them in a cirrhotic rat model at a dose of 1 × 10 6 mitigated the profibrotic action of CCl4 [99]. As demonstrated in this work, histopathological fibrosis score at 4 weeks follow-up showed improvement in the structure of the cirrhotic liver. Additionally, a significant decrease in mRNA and protein levels of TGFβ, collagen type I, and αSMA were observed in liver tissue after 2 and 4 weeks [99]. Similar results were obtained by Tsai et al. [100], who transplanted 5 × 105 hUC-MSCs into rats and, after 4 weeks, found a significant reduction in liver fibrosis, lower levels of serum glutamic oxaloacetic transaminase and glutamic pyruvate transaminase, and decreased protein expression of αSMA and TGFβ1 in the liver. However, engrafted hUC-MSCs were localized mostly in the hepatic connective tissue and did not differentiate into hepatocytes. Instead, these undifferentiated cells were able to secrete bioactive cytokines, including cutaneous T cell-attracting chemokine and leukemia inhibitory factor [100]. Overall, these data suggest that the effect of hUC-MSCs on reducing fibrosis might rely on bioactive factors released from the grafted cells rather than on their differentiation into hepatocytes. This concept was further supported by a study in which exosomes derived from hUC-MSCs were transplanted into a CCl4-induced mouse model of liver fibrosis [101]. The authors showed that 3 weeks after exosome delivery, liver fibrosis was significantly reduced. Similarly, the expression of collagen I and III mRNA and TGFβ1 protein decreased in liver tissue after 3 weeks. Notably, the results obtained from immunohistochemistry concerning E-cadherin, N-cadherin, and vimentin expression indicated that the exosomes contributed to the decline of the EMT process [101].
It should also be noted that there are studies reporting that MSCs might not be effective at providing improvements in hepatic fibrosis [102,103]. Carvalho et al. [102] investigated the effect of BM-MSCs administered at a dose of 1 × 107 in rats with liver fibrosis induced via CCl4 injection and associated with an alcoholic liquid diet. They showed no improvement in serum biochemical markers of liver disease, including ALT and AST, or in collagen deposition in the liver at 1 and 2 months post transplantation [102]. Likewise, Mannheimer et al. [103] used a similar model of rats with liver damage that were injected through the portal vein with 1.6 × 107 BM-MSCs isolated from cirrhotic rats. They found no significant differences in ALT and albumin blood levels between the cell-treated and placebo groups. Furthermore, no improvements were observed in portal vein diameter (PVD), liver parenchyma echogenicity, or collagen deposition. One explanation for the lack of efficacy of the therapy might be the use of BM-MSCs from cirrhotic rats, which may impact the functionality of MSCs [103].
Concerning clinical research, there have been several published stem cell-based trials that investigated the therapeutic effect and safety of MSCs in patients with liver fibrosis, mostly those with advanced cirrhosis [104,105,106,107,108,109,110,111,112]. The majority of trials indicated a clinical benefit of MSC transplantation in cirrhotic disease (Table 1). Indeed, a non-controlled phase I–II clinical trial with eight patients with end-stage liver disease (four with hepatitis B, one with hepatitis C, one with alcoholic, two cryptogenic) who received autologous BM-MSCs at approximately 3–5 × 107 exhibited improved liver function verified by the model for end-stage liver disease (MELD) score, which decreased from 17.9 ± 5.6 to 10.7 ± 6.3 [108]. Moreover, decreased prothrombin complex, serum creatinine, and bilirubin was detected at 24 weeks follow-up. Importantly, no side effects of this cell therapy were demonstrated, and all patients reported subjective improvements beginning 2 months after transplantation [108].
In another controlled trial, Peng et al. [110] enrolled 53 patients with liver failure due to chronic HBV infection, who received autologous BM-MSCs through the hepatic artery (105 patients served as the control group). The results showed that the levels of ALT and TBIL, PT, and MELD score improved significantly after 2–3 weeks compared to patients in the control group. A follow-up study at almost 4 years showed no major differences in the mortality of patients and the incidence of hepatocellular carcinomas between patients with and without cirrhosis in the BM-MSC group [110]. Similarly, benefits were reported in a randomized controlled trial carried out on 20 patients with end-stage liver failure due to chronic HCV infection who underwent transplantation of autologous BM-MSCs stimulated to hepatic lineage (approximately 2 × 107 hepatic lineage-committed cells). Compared to the control group, in patients who received cell therapy, parameters such as Child score, MELD score, fatigue scale, and performance status significantly improved at 6 weeks follow-up. Moreover, lower limb edema and serum albumin decreased in the cell-treated group relative to the control group [104].
Many similar beneficial effects were observed in two other clinical trials in which autologous BM-MSCs were transplanted into patients with liver cirrhosis induced by HCV [105,106]. In a separate phase II trial, 11 patients with alcoholic liver cirrhosis received two injections in the hepatic artery with 5 × 107 autologous BM-MSCs, at weeks 4 and 8 [107]. Histological examination of liver biopsy specimens collected 12 weeks post transplantation showed improvement in 6 out of 11 patients (54.5%). The Child–Pugh score, which is a direct marker of hepatic fibrosis and the development of side effects, improved in 10 patients (90.9%). Furthermore, mRNA levels of Tgfβ1, Col1, and αSMA significantly decreased in liver tissue following BM-MSC transplantation. Importantly, no significant complications or side effects were reported [107]. Another controlled clinical trial was related to patients with decompensated liver cirrhosis who received hUC-MSCs at a dose of 5 × 105 kg/body weight. Decreased hypogastric ascites volume and serum levels of laminin, procollagen III, COLIV, and hyaluronic acid (HA) were found, while the serum level of HGF was elevated [112]. On the other hand, there are also studies demonstrating a lack of efficacy of MSC therapy for the treatment of liver cirrhosis. Indeed, Mohamadnejad et al. [109] conducted a randomized placebo-controlled trial in which autologous BM-MSCs were peripherally infused into patients with decompensated cirrhosis. The results at 12 months post transplantation revealed no beneficial effect on the biochemical parameters of patients. Moreover, the absolute changes in MELD and Child scores, serum albumin, international normalized ratio (INR), serum transaminases, and liver volume did not differ significantly between the BM-MSC recipients and the placebo group [109].
Mesenchymal stem cells have emerged as a promising agent for the treatment of liver fibrosis and cirrhosis, mostly due to their immunomodulatory capacity and ability to differentiate into tissue-specific cells. Cell-based therapy seems to be extremely important because advanced fibrosis causes cirrhosis, for which liver transplantation is the only effective treatment. However, due to the limitations of liver transplantation, alternative therapeutic options are needed. However, there are some concerns about the efficacy and safety of using MSCs in patients with liver fibrosis, as these cells might show undesirable effects or even worsen the disease due to their profibrogenic potential [113,114].
3.3. Cardiac Fibrosis
Cardiac fibrosis is a common pathological condition related to heart injury and nearly all types of heart disease, including myocardial infarction (MI), coronary heart disease, hypertension, and genetic disorders associated with cardiomyopathies [115]. The mechanism underlying cardiac fibrosis is comparable to that in other organs. In brief, after heart injury, the death of cardiomyocytes triggers an inflammatory and fibrogenic response, leading to the formation of scar tissue, which preserves the structural and functional integrity of the myocardium (Figure 2) [116]. Regardless of the cause, fibrosis leads to cardiac tissue stiffness, contractile dysfunction, impaired myocardial function, and arrhythmogenicity, and subsequently, the condition progresses to heart failure [117]. Depending on the location and etiology, cardiac fibrosis can be divided into four types: reactive interstitial, replacement, infiltrative interstitial, and endomyocardial fibrosis (Figure 2) [118]. According to the World Health Organization’s list of the top 10 causes of death, heart diseases are the leading cause of death in the world. Since cardiac fibrosis is commonly associated with cardiovascular disease, considerable efforts are being devoted to the search for antifibrotic treatments. Researchers and clinicians are focused mainly on the inhibition of molecules that activate cardiac fibroblasts and affect the development of cardiac fibrosis.
The results of numerous studies with CTGF, galectin 3 (Gal-3), TGFβ, endothelin, MMPs, mineralocorticoid receptors, and the renin-angiotensin-aldosterone system as targets for antifibrotic therapy demonstrated attenuation of cardiac fibrosis and reduced ECM protein synthesis in different animal models [119,120,121,122]. However, these pharmacological therapies failed due to the occurrence of harmful side effects, such as adverse cardiac remodeling, gastrointestinal and liver dysfunction, and even death. Thus, effective pharmacotherapy for preventing or reversing cardiac fibrosis is presently unavailable. Therefore, novel approaches involving stem cell-based therapy have been introduced into the field of cardiovascular research.
Mesenchymal stem cells provide an attractive therapeutic approach for cardioprotection, particularly due to their ability to differentiate into cardiovascular cells in vivo and in vitro and by exerting immunomodulatory and angiogenic properties [123,124,125]. Moreover, the absence of cell surface histocompatibility complex (HLA) class II and T cell co-stimulatory molecules reduces the risk of transplant rejection, which makes MSCs useful in both autologous and allogeneic cell therapy [123,126,127]. The great majority of studies examined the therapeutic effect of BM-MSCs in the treatment of cardiac fibrosis. One such study showed that transplantation of BM-MSCs in mouse hearts after MI contributed to reduced fibrosis by modulating abundantly deposited ECM [128]. Collectively, the results of all of these studies demonstrate that transplantation of BM-MSCs or cardiac stem cells in a rat heart failure model resulted in reduced total collagen volume, decreased mRNA levels of Col1 and -3, TIMP-1, and TGFβ, and expression of gelatinases in the myocardium [129,130,131,132]. There is also an increasing body of work supporting the idea that the effect of MSCs on fibrosis may be mediated through reduced inflammation. The results of a study conducted by Du et al. [133] showed that delivery of BM-MSCs into peri-infarct rat myocardium immediately after induction of MI led to inhibition of NF-kappa B activity, attenuated protein levels of TNF-α and IL-6, and increased expression of anti-inflammatory IL-10 in the myocardium [133]. Another study showed that the use of 3 × 106 BM-MSCs in a rat model of MI reduced the levels of CD68-positive inflammatory cells and monocyte chemotactic protein-1 (MCP-1) in the myocardium at 3 weeks; thus, the cells contributed to improved cardiac function [134].
There is considerable evidence that MSCs have a therapeutic function in cardiovascular diseases primarily through paracrine action [135,136,137,138,139]. Kishore et al. [137] showed that transplanted bone marrow progenitor cells (BMPCs) had an antifibrotic effect by paracrine regulation of cardiac miRNAs in mice. Intramyocardially transplanted BMPCs at a dose of 1 × 106 in db/db (diabetic) mice subjected to MI released HGF, which inhibited the miR-155-mediated pro-fibrosis response, leading to decreased mRNA expression of Col1A1, Col3A1, and αSMA, and concomitantly improved cardiac function [137]. Another study showed that conditioned medium from BM-MSCs markedly increased the mRNA expression of negative regulators for cell proliferation, such as elastin (Eln), myocardin, and DNA-damage inducible transcript 3 (DDIT3) [138]. Moreover, there are data demonstrating that exosomes secreted by cardiac MSCs improved cardiac function by enhancing capillary density and cardiomyocyte proliferation in ischemic mouse myocardium [136].
Another study showed that exosomes from BM-MSCs are enriched with miR-22, and injecting them into ischemic hearts of MI mice significantly downregulated Mecp2, consequently diminishing apoptosis in ischemic myocardium and leading to a reduced fibrotic area [135]. Similarly, it has been shown that exosomes derived from rat BM-MSCs overexpressing GATA4 displayed antifibrotic properties in vivo, partially related to the high expression of miR-19a [139]. In addition, the administration of exosomes secreted from hypoxic cardiac stem cells resulted in reduced heart fibrosis in rats, while the administration of exosomes in TGFβ1-stimulated rat cardiac fibroblasts decreased mRNA transcription of profibrotic factors [140]. Accumulative data demonstrate that genetically engineered BM-MSCs that overexpress Akt, HGF, insulin growth factor 1 (IGF-1), miR-133, or stromal-cell-derived factor 1 alpha (SDF-1α) reduced myocardial fibrosis and restored cardiac function more efficiently than their non-engineered counterparts [131,141,142,143,144]. Of note, pretreating MSCs with melatonin, sildenafil, or anti-ischemic drugs (trimetazidine) or exposing the cells to hyperbaric oxygen (HBO) or even anoxic preconditioning enhanced their survival and led to amplified antifibrotic activity following myocardial implantation in vivo [124,145,146,147,148].
Because MSCs injected into infarcted hearts show low efficacy due to their low retention in the cardiac tissue, a variety of improvements were undertaken to enhance their survival rate. Indeed, encapsulating cells in injectable materials or loading them into porous scaffolds or hydrogels have been found to be innovative solutions to augments the therapeutic activity of transplanted cells [149,150,151,152]. The results of cumulative studies with the use of different scaffolds for MSC transplantation, including platelet-rich fibrin, microporous alginate-chitosan, co-polymers, and hyaluronan-based scaffolds, demonstrated that these biomaterials enhanced the antifibrotic effect of MSCs by increasing myocardial vascularization or reducing the degree of fibrosis in the scar area, thereby improving heart function after MI [149,150,151,152].
Embryonic stem cells, due to their ability to differentiate into functional cardiomyocytes represented by all types of specialized heart cells, including atrial-, ventricular-, sinus nodal-, and Purkinje-like cells, have been shown to exert a therapeutic effect in rodent models of cardiac fibrosis [153]. In a series of papers, Singla et al. [154,155,156] showed that mouse ESCs or their conditioned media improved cardiac function by multiple mechanisms. Upon transplantation into the post-MI myocardium, they both led to inhibition of cardiac myocyte cell death and enhanced activation of the Akt cell survival pathway in the myocardium. Their therapeutic effect was achieved through decreased MMP-9 signaling and increased levels of HGF and insulin growth factor 1 (IGF-1) in the heart [154,155,156]. Other studies have shown that transplantation of mouse ESCs overexpressing TIMP-1 significantly enhanced cardiac myocyte differentiation, leading to reduced cell apoptosis, increased Akt activity, and decreased MMP-9 in infracted myocardium compared with animals that received ESCs or control [157]. At the functional level, echocardiography showed that fractional shortening and ejection fraction (EF) were significantly improved in the group administered ESCs overexpressing TIMP-1 [157]. Furthermore, mouse and human ESCs lethally inactivated with irradiation injected into ischemic myocardial tissue of mice and rhesus macaque monkeys, respectively, were reported to result in improved myocardial function and decreased myocardial infarct size [158]. Interestingly, injection of mouse ESC-derived exosomes into infarcted mouse hearts augmented the survival of cardiac progenitor cells and stimulated the formation of cardiomyocytes in ischemic hearts. These improvements were probably mediated by the miR290–295 cluster, specifically miR-294, which highly enriched exosomes from mouse ESCs [159].
Based on the experimental success of using MSCs as a treatment for cardiovascular diseases in animal models, many clinical trials have been conducted to investigate the efficacy of different types of MSCs (Table 1) [160,161,162,163,164]. In detail, in a phase I/II randomized pilot trial (the POSEIDON trial), Hare et al. [127] tested the safety and effectiveness of autologous and allogeneic BM-MSCs in patients with ischemic cardiomyopathy. The cells were delivered by a transendocardial injection into 10 left ventricular sites in doses of 2 × 107, 1 × 108, and 2 × 108 of each cell type. The administration of both cell types was safe and associated with low rates of serious adverse events in the 13-month follow-up. Following the cell transplantation, reduced scar size accompanied by decreased end-diastolic and end-systolic volume and sphericity index was observed. However, improvements in 6MWT and quality of life were associated only with autologous, not allogeneic, MSC therapy [127]. The same group conducted a randomized controlled trial in which they compared the safety and efficacy of autologous versus allogeneic BM-MSCs in patients with nonischemic dilated cardiomyopathy (NIDCM) [126]. The superior effect of allogeneic over autologous BM-MSC therapy was demonstrated at 12 months. Indeed ejection fraction EF and 6MWT increased significantly in the allogeneic group compared to the autologous group. In addition, endothelial function was improved only in the group treated with allogeneic BM-MSCs, and TNFα suppression was also greater in this group. Consistently, the occurrence of serious adverse events was 28.2% in allogeneic BM-MSC recipients and 63.5% in the autologous group [126]. Collectively, these findings provide evidence of a clinically relevant effect of allogeneic BM-MSCs of greater magnitude than that observed using autologous cells.
Florea et al. [162] conducted a study (the TRIDENT study) using allogeneic human BM-MSCs in two doses (2 × 107 and 1 × 108) administered via transendocardial injection to patients with ischemic cardiomyopathy. Twelve months after MSC administration, no serious treatment-related adverse events were observed. The allogeneic MSC therapy improved cardiac function, with scar size reduction in both groups and an increase in ejection fraction only in the group that received the higher dose [162]. These findings underline the importance of using the proper cell dose in response to cell therapy.
Besides BM-MSCs, the safety and efficacy of other cell types, including ASCs, UC-MSCs, and Wharton’s jelly-derived MSCs, were evaluated in clinical trials [165,166,167]. Direct intramyocardial injection of ASCs and intravenous infusion of UC-MSCs or Wharton’s jelly-derived MSCs did not cause any complications or serious adverse events in patients with heart failure. Moreover, increased myocardial viability, enhanced function of the left ventricle, and improved quality of life were reported in patients treated with MSCs [165,166,167]. Although these clinical trials reported some beneficial effects of the treatment of cardiac failure, none of them indicated that stem cell transplantation entirely reduced heart fibrosis. There are a few ongoing registered clinical trials (https://clinicaltrials.gov/; 16 September 2021) concerning the use of MSCs in the treatment of cardiac fibrosis, which so far have not provided any results.
Despite many pieces of evidence indicating the advantages of MSCs in heart failure treatment, cell-based therapy still faces some challenges, such as the poorly targeted migration, the low rate of MSC differentiation into cardiomyocytes, and the low survival rate. On the other hand, the use of ESCs, despite their documented effectiveness in the treatment of cardiac fibrosis in animals, is limited by an ethical controversy related to their source of origin in humans, immunogenic potential, risk of cancer formation (teratomas formed in approximately 50% of rats injected with ESCs; [168], and low rate of differentiation into cardiomyocytes (cardiomyocytes usually represent less than 1% of the total cells in cell cultures [161]). Therefore, elaborate protocols may be needed for immunosuppressive therapy in order to reduce the risk of cell rejection. In vitro differentiation of ESCs before transplantation might be an option to increase the production of cardiac cells and avoid tumor development post-delivery.
3.4. Renal Fibrosis
Renal fibrosis is considered as a common final consequence of a variety of chronic renal diseases, including chronic glomerulonephritis, diabetic nephropathy, hypertensive nephropathy, and chronic renal allograft injury [169]. All of the functional compartments of the kidney can be affected by fibrosis, which is specifically termed interstitial fibrosis in the tubulointerstitium, glomerulosclerosis in the glomeruli, and arteriosclerosis or perivascular fibrosis in the vasculature [170,171].
The aging population and the rising prevalence of diabetes, obesity, atherosclerosis, and hypertension increase the incidence of kidney diseases, and consequently, renal fibrosis. In turn, renal fibrosis leads to eventual end-stage renal disease, organ failure, and the need for renal replacement therapy (i.e., dialysis or transplantation) (Figure 2) [172]. Inhibiting renal fibrosis from progressing is crucial in order to prevent progressive deterioration of kidney function, and it could avoid the final solution in the form of renal replacement therapy. In recent years, many efforts have been made to identify mediators and targets for renal fibrosis therapy. The results of many animal studies have revealed efficiency in reducing renal fibrosis and improved kidney function after the inhibition of molecules directly involved in the development of fibrosis [171,172,173,174]. However, such therapies failed in clinical trials due to an inappropriate balance between antifibrotic efficacy and adverse effects [173,174]. In the face of the lack of an effective method of treating renal fibrosis, the employment of stem cell therapy represents a promising treatment strategy.
A large body of data has demonstrated the therapeutic effects of BM-MSCs in rodent models of renal fibrosis [175,176,177,178,179,180,181,182,183,184,185,186,187,188,189,190,191]. Administration of BM-MSCs into renal parenchyma or intravenous delivery led to reduced interstitial fibrosis reflected by decreased expression of ECM components and vimentin, reduced MMP-2 activity, and inhibited TGFβ/SMAD signaling in the damaged kidney [175,176,177,178]. Several studies showed that the renal expression of E-cadherin was upregulated, while the expression of αSMA and desmin were downregulated after BM-MSC injection [175,177,178,179]. Additionally, BM-MSCs were found to attenuate renal inflammation by downregulating inflammatory mediators such as c-c motif chemokine ligands (CCL)-4, -7, -19, IFN-α/β, TNFα, IL-1β, and IL-6 [175,177,180]. There is also evidence that one of the mechanisms by which MSCs might exert their antifibrotic activity in the kidney involves regulation of the cell cycle of tubular epithelial cells (TECs). Generally, injury to TECs results in the arrest of cells in the G2/M phase of the cell cycle. This cell cycle arrest mediates fibrosis by excessive production of profibrotic factors, including TGFβ, epidermal growth factor (EGF), TNFα, nuclear factor kappa B (NF-κB), lipocalin 2 (NGAL), and hepatitis A virus cellular receptor 1 (KIM-1) [171,181,182]. Zhu et al. [183] demonstrated that transplanting human ASCs into mice with induced acute kidney injury (AKI) caused a significant decrease in the number of TECs arrested in G2/M, correlated with reduced pathological renal damage [183].
A growing body of evidence indicates that BM-MSCs mediate their effects mainly through paracrine mechanisms [184,185]. Extracellular vesicles (Evs) derived from MSCs have been shown to contribute to kidney repair [184,185,186,187]. Several studies on rodents demonstrated attenuation of renal fibrosis after injection of BM-MSC-conditioned medium or BM-MSC-derived Evs, through their involvement in inhibiting apoptosis, stimulating tubular epithelial cell proliferation, and decreasing the expression of COLI, αSMA, TGFβ1, TNFα, TIMP-1, MMP-3, and snail family transcriptional repressor 1 (SNAI1) in the kidney [184,185,186,187]. Similarly, the administration of ASCs or ASC-derived Evs to damaged pig or rodent kidneys caused reduced renal fibrosis and decreased inflammation [188,189,190]. Additionally, such therapy has been shown to decrease the occurrence of EMT, inhibit TGFβ/SMAD signaling and the renin-angiotensin system, and induce a shift in the macrophage phenotype from inflammatory to reparative in renal tissue [183,188,189,190]. Likewise, delivery of 2 × 107 microparticles produced by kidney-derived MSCs into mice with unilateral ureteral obstruction, a model of tubulointerstitial scarring, inhibited the infiltration of inflammatory cells and suppressed tubulointerstitial fibrosis, as demonstrated by a decrease in F4/80- and αSMA-positive cells [191].
A significant reduction in renal fibrosis in mice was also demonstrated after administration of ASCs overexpressing glial cell line-derived neurotrophic factor (GDNF) or exosomes derived from GDNF-transfected ASCs [192]. This effect was linked to suppression of inflammation and activation of the sirtuin 1/ nitric oxide synthase (SIRT1/eNOS) signaling pathway in renal tissue [192]. Several studies also reported that administration of BM-MSCs overexpressing HGF, ACE2, vascular endothelial growth factor (VEGF), or miRNA-let7c or in combination with antifibrotic serelaxin effectively reduced renal fibrosis in a rat model [193,194,195]. Indeed, such an approach was effective at inhibiting fibrosis, manifested by decreased expression of αSMA, COLI, COLIV, MMP-9, and FN, inhibition of TGFβ signaling, and upregulation of MMP-2 expression in renal tissue [193,194,195,196,197]. Additionally, recent studies demonstrated that MSC culture in vitro and preparation prior to transplantation might affect the therapeutic properties. Indeed, two independent studies showed that the use of BM-MSCs pretreated with melatonin or IFNγ ameliorated interstitial fibrosis compared with control groups [198,199]. Similarly, amniotic fluid-derived stem cells (AFSCs), which exhibit characteristics of both ESCs and MSCs, preconditioned with GDNF, were shown to abrogate the degree of renal interstitial fibrosis in mice [200]. Intravenous delivery of 3.5 × 105 GDNF-AFSCs suppressed oxidative stress and inflammation, repaired renal microvessels, and relieved tissue hypoxia and mitochondrial damage in the kidney [200].
The results of both in vitro studies and preclinical in vivo investigations conducted on rodent models of renal fibrosis demonstrated that administration of MSCs from the umbilical cord and placenta or their conditioned media was effective at inhibiting renal fibrosis [201,202,203,204,205,206]. This effect has been linked to decreased expression of COLI, FN, αSMA, CTGF, and proinflammatory cytokines in renal tissue. Moreover, inhibition of the EMT process or TGFβ/Smad and TLR4/NF-κB signaling or reinforcement of the Akt signaling pathway has been demonstrated in kidneys [201,202,203,204,205,206].
Although human ESCs have the ability to differentiate into aquaporin (AQP) 1- or AQP 2-positive cells showing morphological and functional features typical of specific renal cells, there is a limited number of studies concerning the impact of ESCs on renal fibrosis [207,208]. In work performed by Geng et al. [209], murine ESCs were loaded into gelatin microcryogels, and the structures were packed into pedicled greater omentum flaps in a rat model of chronic kidney disease. At 12 weeks after transplantation, reduced glomerulosclerosis and tubular injury were found. Moreover, the levels of plasma creatinine and urea nitrogen were decreased in ESC-treated animals compared to the control group [209]. In a separate study, De Chiara et al. [210] examined the impact of tubular-like cells (GTCs) that arise from germline cell-derived pluripotent stem cells (GPSCs) on mice with ischemic renal injury. They found that the cell treatment reduced cortical damage, tubular apoptosis, and renal oxidative stress while it upregulated tubular expression of the antioxidant enzyme hemeoxygenase-1. Moreover, at 6 weeks, kidneys of the mice that received GTCs showed less fibrosis, and reduced inflammatory infiltrate compared to kidneys of vehicle-treated counterparts [210]. Besides ESCs, iPSs were also shown to have the capacity to differentiate into renal lineages or cells that exhibit a renal-like phenotype and gene signature [211,212]. A recent study showed that human iPSCs (hiPSCs) generated from peripheral blood, along with renal progenitor cells (RPCs), differentiated from them, reduced interstitial fibrosis, tubular atrophy, and glomerulosclerosis in a rat model of chronic kidney disease [213]. Caldas et al. [214] used a similar experimental rat model of kidney disease and injected into 0.5 × 106 iPSCs derived from rat skin fibroblasts into renal parenchyma. They found elevated serum creatinine, reduced glomerulosclerosis, and decreased macrophage infiltration. However, histopathological analysis of liver specimens revealed tumors with characteristics of nephroblastoma, suggesting that although iPSCs seem to be encouraging as cell therapy, they carry a risk of Wilms’ tumor development [214].
Despite the experimental success of MSCs in the treatment of renal diseases in animal models and the results from a limited number of human clinical trials demonstrating the safety and feasibility of MSC-based kidney therapy, the efficacy of the studies remains controversial (Table 1). Swaminathan et al. [215] conducted a phase II randomized placebo-controlled trial to determine the safety and efficacy of allogeneic MSCs in reducing the recovery time from AKI after cardiac surgery. Intra-aortic administration of MSCs at a dose of 2 × 106 cells/kg body weight was safe and well tolerated but did not markedly improve renal function or patient mortality. Further, MSC transplantation did not decrease the time to recovery of kidney function [215]. Similarly, the results of another clinical study in which autologous BM-MSCs at a dose of 2 × 106 cells/kg body weight were given to patients with autosomal-dominant polycystic kidney disease (ADPKD) revealed no cell-related adverse effects at 12 months [216]. Concomitantly, MSC infusion did not induce any significant changes in estimated glomerular filtration rate (eGFR) or reductions in serum creatinine compared to baseline in all patients [216].
Saad et al. [68] investigated the role of intravenously infused autologous ASCs at doses of 1 × 105 and 2.5 × 105 for the treatment of atherosclerotic renovascular disease (RVD). They found that ASC infusions were well tolerated by the patients. Three months after cell transplantation, increased cortical perfusion and renal blood flow were reported. Increased kidney perfusion was accompanied by decreased fractional tissue hypoxia and stabilization of the glomerular filtration rate [68]. In a separate non-randomized and placebo-free phase I clinical trial, Alatab et al. [66] treated patients receiving peritoneal dialysis with autologous ASCs at a dose of 1.2 ± 0.1 × 106 cells/kg. While hematological and systemic biochemical parameters were stable over 24 weeks, a significant change in the ASC group was decreased body mass index (BMI), which probably resulted from the decrease in the degree of edema due to the increased ultrafiltration rate [66]. The results of other studies verifying the validity of autologous MSCs (https://clinicaltrials.gov/, accessed 16 September 2021) in patients suffering from chronic kidney disease, renovascular hypertension, or occlusive kidney disease have been not published yet. Currently, among all registered clinical trials concerning the use of MSCs for the treatment of kidney diseases (https://clinicaltrials.gov/, accessed 16 September 2021), only a few focus on the treatment of renal fibrosis. To date, none of these studies have provided results.
The standard methods of treating renal fibrosis are limited; however, the use of stem cells seems to have potential as a therapeutic approach for renal fibrosis-related diseases. In particular, MSC-based therapy, which has been demonstrated as being safe and feasible, has vital potential to improve renal function and patient mortality. Further studies should be conducted to improve the long-term therapeutic activity of transplanted cells.
3.5. Uterine Fibrosis (Asherman Syndrome)
Asherman syndrome (AS) is a complex gynecological disorder characterized by intrauterine adhesions (IUAs) that lead to defective function of the endometrium. Surgical procedures that disrupt the endometrial basalis layer, such as excessive postpartum or post-abortion curettage and frequent hysteroscopic surgery, as well as recurring infections, are the main factors responsible for the development of IUAs (Figure 2) [217,218]. Endometrial trauma causes molecular changes that may lead to fibrosis. As a result, the endometrium stops responding to hormonal stimulation, and the uterine cavity becomes obstructed, leading to the creation of IUAs [217]. Severe cases of AS lead to abnormal uterine bleeding, amenorrhea, hypomenorrhea, chronic pelvic pain, abnormal placentation, recurrent miscarriages, or even infertility. So far, the main therapy for AS relies on surgical intervention, with hysteroscopic surgery performed to re-establish a normal uterine cavity and restore uterine function [219]. However, traumatized endometrium tends to develop post-surgical intrauterine adhesions, so the actual challenge in handling AS is to prevent or minimize the development of new adhesions [220]. Therefore, post-surgical methods, including inserting a balloon or other type of intrauterine device or creating a protective lining with HA, have been applied to provide a physical barrier inside the uterus that prevents the occurrence of new adhesions. Moreover, to promote the healing process, estrogen and antibiotic treatment are also recommended [221,222,223].
The development of IUAs involves the destruction of the basalis layer of the endometrium, which is rich in somatic stem cells responsible for its cyclic regeneration. Therefore, stem cell transplantation might be a promising approach for the reconstruction of the stem cell pool, which in turn might contribute to endometrial renewal and AS resistance. Indeed, several lines of evidence indicate that transplanting MSCs to the endometrium contributes to the renewal of this tissue [224,225]. Carvelló et al. [226] showed that transplanted human CD133+ BM-MSCs in the uterine horn in an AS mouse model successfully engrafted around blood vessels and induced proliferation of surrounding cells via paracrine factors such as thrombospondin 1 (Thbs1) and IGF-1 [226]. A study by Alawadhi et al. [224] also conducted on a mouse model of AS demonstrated that transplantation of 1 × 107 BM-MSCs improved fertility. In the BM-MSC transplant group, 9 of 10 mice conceived, whereas, in the non-transplanted control group, only 3 of 10 females were pregnant [224].
A recent report by Çil et al. [67] showed that a single dose of ASCs in female rats with experimentally induced AS maintained epithelial integrity in the endometrium, reduced leukocyte infiltration and fibrosis, and increased vascular proliferation. Interestingly, similar improvements were observed in mice that received combined therapy consisting of ASCs and oral estrogen. Of note, the authors assumed that the therapeutic effect might be mediated through IGF-1 and VEGF [67]. Interestingly, current data show that menstrual blood is a noninvasive and easily available source of endometrial MSCs (eMSCs) [227]. Domnina et al. [227] performed functional studies in which adult female Wistar rats received a suspension of human eMSCs, human eMSCs spheroids, or rat BM-MSCs via the intravenous or intrauterine route (eMSC spheroids were transplanted into the uterus). Three estrous cycles after AS induction and the delivery of cells, the females were mated with males, and finally, the number of pregnant animals and their litter size were recorded. It was found that the number of pregnant females significantly increased after stem cell administration compared to the PBS-treated group. A higher pregnancy rate was achieved with intrauterine than intravenous administration of cells. Notably, transplantation of eMSC spheroids produced a superior effect, manifested by the highest conception rate and the highest number of pups born [227].
Functional restoration of endometrium in a mouse AS model was shown to be mediated by human amniotic mesenchymal stromal cells (hAMSCs) [228]. Besides increases in the pregnancy rate and the number of fetuses upon hAMSC administration, the authors found several post-transplanted changes, including increased microvessel density and elevated expression of VEGF, proliferating cell nuclear antigen (PCNA), estrogen, and progesterone receptors, indicating endometrial repair responsible for the restoration of endometrial function [228].
The efficacy of MSCs for AS treatment has also been demonstrated in clinical research. (Table 1). Santamaria et al. [229] showed that autologous CD133+ BM-MSC administration in conjunction with hormonal replacement therapy affected patients with IUA. At the third month after treatment, increased epidermal thickness and neoangiogenesis and prolonged duration of menses were shown. Notably, 3 out of 16 patients became pregnant spontaneously, resulting in birth, while 7 positive pregnancies were obtained after 14 embryo transfers [229]. These findings were further supported by Singh et al. [225], who showed that transplantation of human BM-MSCs into women with AS led to a significant increase in endometrial thickness and resumption of menses in a majority of amenorrheic patients and may have allowed 3 out of 25 patients to spontaneously conceive, with positive pregnancy outcomes [225].
In a recent paper, Lee et al. [230] showed the results of a trial in which autologous SVFs at a dose of 4.6 ± 0.7 × 106 were transplanted into the uteruses of six infertile women with severe AS. They found that SVF delivery followed by estrogen hormone therapy increased endometrial thickness from 3 mm to around 6.9 mm. After the combined therapy, five of the women had an embryo transfer; among them, one woman conceived but aborted spontaneously at 9 weeks gestation [230]. Another group demonstrated a therapeutic effect mediated by allogeneic UC-MSCs supported by a collagen scaffold transplanted into the uterine cavities of patients with AS-related infertility [231]. In agreement with other studies, the authors revealed that the average endometrial thickness improved at 3 months, and, importantly, there was a decrease in intrauterine adhesion scores compared to those before the treatment. In endometrial biopsies, the expression of estrogen receptor alpha (ERα), vimentin, Ki67, and von Willebrand factor increased, suggesting post-transplantation improvement in endometrial proliferation, differentiation, and neovascularization. Even more important, by the end of the 30-month follow-up period, 10 out of 26 patients had become pregnant, and 8 of them had positive pregnancy outcomes [231]. Similarly, autologous menstrual blood-derived stromal cells (menSCs) were demonstrated to improve endometrial thickness in women with severe AS, and notably, two of the four patients who underwent frozen embryo transfer conceived successfully, while one patient had a spontaneous pregnancy after a second menSCs transplantation [232].
Based on the above studies, MSCs exhibit a positive treatment effect in women with AS. The effects were reflected by morphological improvement of the endometrium, and functional enhancement was manifested by the resumption of menstruation and increased fertility outcomes of infertile AS women. Certainly, AS prevents implantation of the blastocyst and impairs the blood supply to the uterus and early fetus, and finally, the disease results in recurrent miscarriage or infertility. Although AS is not fatal, its effects on the psyche and quality of life of women make further research on stem cell-based therapy extremely important. Clinical trials have demonstrated the initial safety and effectiveness profiles of different types of stem cells that might provide potential options for treating women with severe AS.
3.6. Skin Fibrosis
Fibrotic skin disorders, although of unknown etiology, share a similar set of abnormal processes manifested by the presence of activated fibroblasts and excessive deposition of ECM [233]. The development of fibrotic skin lesions, such as in SS mainly occurs in association with metabolic and immunological disorders or can arise in response to dermal injury that might lead to the formation of hypertrophic scars or keloids (Figure 2) [233]. Experimental data on rodents showed that dermal fibrosis can be induced by local administration of drugs and chemicals such as bleomycin, vinyl chloride, or hypochlorous acid (HOCl) [234,235,236]. Moreover, there are several established genetic mouse models with a fibrotic skin phenotype, including fibrillin-1 mutant tight skin mice (Tsk), transgenic animals that include those that express a kinase-deficient type II TGFβ receptor, overexpress Wnt 10b, or are deficient in caveolin [236,237].
The prototype of fibrotic skin diseases is scleroderma, which can be classified into localized (LSc, morphea) and systemic (SS) types. Systemic sclerosis can be further divided into two clinical subtypes: limited cutaneous (lcSS), with fibrotic skin changes present in fingers, hands, and face, and diffuse cutaneous (dcSS), which initiates in fingers and hands and gradually spreads to limbs and trunk [233]. SS is a chronic multisystem disorder manifested by autoimmunity, microvascular dysfunction, and fibrosis of the skin and internal organs, including lungs and kidneys. The disease more frequently affects women than men and causes high mortality, with a survival rate between 34% and 73% [238]. The clinical features of SS primarily represent consequences of vascular impairment and ischemia, such as Raynaud’s phenomenon and digital ulcers [233]. Despite collagen overproduction, the pathophysiology of SS is characterized by damage to small vessels and dysregulation of the humoral and cellular immunity, reflected, at least in part, by the production of autoantibodies to nuclear, nucleolar, and cytoplasmic antigens as well as endothelial cells. Accordingly, emerging data demonstrate that the sera of individuals with scleroderma have antibodies against centromere, Scl-70/topoisomerase I, anti-fibrillarin/anti-U3 nucleolar antigens RNA polymerase I–III, and those that block MMP-1 and MMP-3 [233,239].
SS is characterized by fibrotic lesions that not only develop in the skin but also involve internal organs, and the disease is life-threatening, intractable, and resistant to standard immunosuppressive therapy. In addition to functional challenges of affected organs, cutaneous symptoms are often associated with pain and with psychological and esthetic distress and cause body image dissatisfaction. Importantly, many other scleroderma-like conditions, including scleredema, eosinophilic fasciitis (Shulman’s disease), porphyria cutanea tarda, diabetic stiff-hand syndrome, lichen sclerosis, and graft-versus-host disease (GVHD), show distinct cutaneous manifestations, pathological skin histology, and systemic implications (Figure 2). It should also be emphasized that the most common manifestation of cutaneous fibrosis is scarring that occurs after injury, which is a natural consequence of the wound healing process [240]. In contrast to mammalian fetuses, which heal cutaneous wounds without scars via regeneration, regular wound healing in adult mammals results in scar formation [241,242]. Overscarring is a frequent wound healing-associated fibrotic disorder that is clinically recognized as hypertrophic scars or keloids [243]. Although both generate excessive scar tissue, keloids are characterized by their extensive growth beyond the original wound site, while hypertrophic scars are raised but remain within the confines of the initial wound border [243].
Given that fibrotic skin diseases are complex, chronic, and heterogeneous and the evaluation of therapy is difficult, effective treatments have not been established so far. Nevertheless, among potential systemic antifibrotic therapies under investigation, those that employ MSCs have emerged as among the most promising [244,245,246,247,248].
Recent studies on HOCl-induced mouse models of SS revealed that systemic infusion of allogeneic BM-MSCs at a dose of 2.5 × 105, 5 × 105, or 106 cells led to a reduction in inflammatory, oxidative, and fibrotic processes in skin and lungs [246]. Delivery of the cell concomitantly with HOCI resulted in 25%–30% lower skin thickness in the BM-MSC-treated group at day 21 post treatment. In addition, at 42 days, decreases in both collagen content and transcript levels of Col1, Col3, Tgfβ1, and αSma in the skin were observed, and the effect was inversely proportional to the cell dose. Notably, the second infusion of cells at day 21 led to further inhibition of skin thickening progression from days 28 to 42. Moreover, a significant reduction in the expression of mRNA of Col1, Col3, Tgfβ1, and αSma in the skin and Col3 and Tgfβ1 in the lung was detected [246]. This study indicated that not only the cell dose but also the time of cell injection may be an important factor controlling the efficacy of cell-based therapy. In a separate study, Chen et al. [245] indicated that systemic administration of allogeneic mouse BM-MSCs or BM-MSC-derived exosomes ameliorated autoimmune and dermal phenotypes of fibrosis in Tsk/+ mice through miR151-5p transfer to recipient cells. Specifically, in terms of skin, reduced hypodermal thickness was found. This effect was accompanied by a significant improvement in the bone formation rate, rescuing osteopenia in Tsk/+ mice. It was indicated that the therapeutic effect was achieved via IL-4 receptor alpha (IL4Rα) and decreased mTOR signaling gene expression in Tsk/+ BM-MSCs [245]. In a similar study, Akiyama et al. [244] demonstrated that BM-MSCs reduced hypodermal thickness and exerted an immunomodulatory effect via FAS/FASL-mediated apoptosis of T cells.
To evaluate the therapeutic effect of ASCs in a systemic model of scleroderma, Rubio et al. [248] employed a murine model of BLM-induced SS. As demonstrated in this work, intravenous ASC treatment at a dose of 5 × 105 cells prevented the development of fibrosis in the lung and skin. Regarding skin, the therapeutic effect was evidenced by well-arranged collagen fibers parallel to the epidermis that resembled the collagen network observed in healthy, saline-treated control mice [248]. ASC administration attenuated the impairment in wound healing in BLM-treated mice, reflected by a significant reduction in total wound size by 58.8% as compared to the control group. The ASC-primed improvement in wound healing resolution was associated with decreased expression of miR-199–3p and upregulation of its corresponding target caveolin 1 in both lung and skin wounds [248]. These findings support results reported by other authors and indicate that one mechanism by which MSCs may regulate fibrotic pathways is by modulating miRNA expression in target tissues [245,248]. Other studies demonstrated that ASC administration was sufficient to attenuate BLM-induced scleroderma by suppressing the infiltration of CD4+ and CD8+ T cells and macrophages into the dermis, but it also reduced the frequency of CD4+ T cells and effector B cells in the spleens of SS mice [247]. Moreover, there was a decrease in mRNA expression of Col1a2 and profibrotic cytokines such as IL-6 and IL-13, suggesting an immunomodulatory and anti-inflammatory effect of ASCs in fibrotic skin [247].
To test the therapeutic potential of MSCs in wound healing-associated fibrosis, Domergue et al. [249] created a humanized animal hypertrophic scar model by delivering healthy human split-thickness skin grafts into the backs of immunocompetent nude mice. Next, at week 7 after graft implantation, human SVF suspension containing 1 × 106 of hASCs, 1 × 106 of cultured hASCs, or PBS used as a control was subcutaneously injected into the scar. The follow-up study revealed a reduction in hypertrophic scarring in the SVF- and hASC-treated group [249]. The reduction in post-wound skin thickness was more significant in the hASC-treated group than in the cohort that received SVFs. This effect corresponded with decreased collagen content in skin specimens. Moreover, cell-treated mice had higher mRNA levels of antifibrotic markers TGFβ3 and HGF, elevated expression of MMP-2, and a higher MMP-2/TIMP-2 ratio in hypertrophic scars, reflecting the remodeling activity contributing to fibrosis resorption [249]. A similar model, in which fragments of human keloid tissue were subcutaneously implanted into skin pockets created on the dorsal skin of athymic mice, was used to investigate whether the secretome of hASCs could provide a strategy against keloid formation [250]. Given that one mechanism by which MSCs exert their effect on a target tissue is paracrine secretions, in the aforementioned study, conditioned medium from ASCs (ASC-CM) was injected within keloid xenografts. At 4 weeks post treatment, increased keloid shrinkage along with better-organized collagen fibers was found in the ASC-CM-treated group. In addition, mice exposed to ASC-CM showed a reduced presence of CD31- and CD68-positive cells in keloid tissue [250]. Together, these results demonstrate the therapeutic potential of hASCs for clinical application in the treatment of hypertrophic scars and keloids.
Several clinical studies demonstrated the possible benefits of HSC and MSC (mostly SVF/ASC) therapy in SS patients (Table 1). In a recent paper, Henrique-Neto et al. [251] reported the results of a longitudinal study that included 70 adult SS patients with a severe SS profile who received autologous HSCs at a mean dose of 5.63 (3.21) × 106 CD34+ cells/kg body weight. The most clinically detectable outcome of HSC administration at 5 years post transplantation was improved skin thickness, assessed by modified Rodnan’s skin score (mRSS). Of note, enhanced pulmonary function (forced vital capacity and diffusing capacity of carbon monoxide) was also observed [251]. These data are consistent with previous results from three randomized controlled trials that demonstrated the superiority of autologous HSC transplantation over the standard cyclophosphamide treatment: American Scleroderma Stem Cell versus Immune Suppression Trial (ASSIST, phase II) [252], Autologous Stem Cell Transplantation International Scleroderma Trial (ASTIS, phase III) [253], and Scleroderma: Cyclophosphamide or Transplantation (SCOT, phase II) [254].
Preclinical and clinical investigations have demonstrated promising therapeutic effects of fat grafting or autologous SVF/ASC-based therapy for patients with cutaneous manifestations of SS (Table 1). In the initial case report, patients with cutaneous manifestations of SS received a local injection of ASCs at a dose of 4 × 106 to 8 × 106 suspended cells in HA solution. Over 1 year of follow-up, all individuals exhibited arrest of local disease [255]. Specifically, four out of six patients showed dyschromia regression (67%), five patients showed improved skin softening (83%), four patients showed better sensitivity (67%), and one patient showed reduced erythema (17%). The study was one of the first to demonstrate a beneficial effect of locally implanted ASCs in patients with severe fibrosis. The first open phase I clinical trial was conducted among 12 SS patients with impaired hand function who received an injection of 3.76 ± 1.85 × 106 autologous SVFs into each finger [256]. Preliminary assessment at 6 months post transplantation revealed reduced pain, increased grip strength, reduced finger circumference, and an average reduction in Raynaud’s severity by 67.5%. Moreover, significant decreases in dystrophic capillaries and vascular suppression scores were observed [256].
There are currently data demonstrating that besides enzymatically isolated SVFs/ASCs, an autologous lipoaspirate rich in multipotent stem cells can also be used as a cell-based therapy for SS [257,258]. As presented by Almadori et al. [257], the lipoaspirate injected directly into fibrotic oro-facial tissues of SS patients significantly improved mouth function and facial volumetric appearance. Similarly, another report showed that fat grafting into patients with advanced SS-related perioral thickening and mouth opening limitation resulted in a significant increase in interincisal distance and oral perimeter along with improved neovascularization [258].
The studies described above provide encouraging and supportive data for the use of MSCs, in particular SVFs or ASCs, as a vital therapeutic strategy against SS. Nonetheless, the results of larger, randomized double-blind and placebo-controlled trials are needed to obtain a meticulous clinical evaluation of MSCs.

Table 1.
Clinical trials of stem cell therapies in fibrotic diseases in lungs, liver, heart, kidney, uterus, and skin.
Table 1.
Clinical trials of stem cell therapies in fibrotic diseases in lungs, liver, heart, kidney, uterus, and skin.
| Organ | Disease | Stem Cell Type | Number of Cells | Delivery Route | Effects | References |
|---|---|---|---|---|---|---|
| Lung | Idiopathic pulmonary fibrosis | Allogeneic BM-MSCs | 2 × 108 cells/infusion (4 infusions in 3 month intervals, total 1.6 × 109) | Intravenous | Improved lung function based on FVC, DLCO, and 6MWT | [82] |
| Idiopathic pulmonary fibrosis | Allogeneic BM-MSCs | 2 × 107 or 1 × 108 | Intravenous | Higher cell dose alleviated fibrosis progression | [83] | |
| Idiopathic pulmonary fibrosis | Allogeneic BM-MSCs | 2 × 107, 1 × 108, or 2 × 108 | Intravenous | Improved lung function assessed by 6MWT at 36 weeks | [84] | |
| Liver | Alcoholic cirrhosis | Autologous BM-MSCs | 5 × 107/injection (2 injections at study weeks 4 and 8) | Intra-arterial (right hepatic) | Histological improvement of liver biopsy based on Laennec fibrosis scoring system; decreased collagen deposition, mRNA expression of TGFβ1, Col1, and αSMA in liver biopsy, and MELD score; improved Child–Pugh score | [107] |
| Hepatitis C-induced liver cirrhosis | Autologous BM-MSCs | 1 × 107 | Intrasplenic injection | Improved liver function assessed by decreased TBIL, AST, ALT, PT, and INR levels and increased albumin and PC levels | [105] | |
| Hepatitis C-induced liver cirrhosis | Autologous BM-MSCs | 1 × 106/kg | Intravenous | Decreased jaundice, lower limb edema, MELD score, and serum creatinine level; improved encephalopathic manifestation, ascites, serum bilirubin, and albumin levels | [106] | |
| End-stage liver failure due to chronic hepatitis C | Autologous BM-MSCs | 2 × 108 | Intrasplenic or intrahepatic injection | Improved liver function based on MELD and Child scores, fatigue impact scale, and performance status | [104] | |
| Liver failure caused by hepatitis B | Autologous BM-MSCs | N/A | Intra-arterial (hepatic) | Improved liver function assessed by ALB, TBIL, and PT levels and MELD score | [110] | |
| Liver cirrhosis | Autologous BM-MSCs | 3–5 × 107 | Intravenous (peripheral or portal vein) | Improved liver function assessed by MELD score and decreased prothrombin complex, serum creatinine, and bilirubin at 24 weeks | [108] | |
| Liver cirrhosis | Autologous ASCs | 3.3 × 105 or 6.6 × 105 cells/kg | Intra-arterial (hepatic) | Increased concentrations of serum HGF, IL-6, IL-18, M-CSF, and MIF at 1 day post treatment | [111] | |
| Decompensated liver cirrhosis | Allogeneic UC-MSC | 5 × 105 cells/k | Intravenous | Decreased hypogastric ascites volume and serum levels of plasma laminin, procollagen III, COLIV, and HA | [112] | |
| Heart | Ischemic cardiomyopathy | Allogeneic BM-MSCs | 2 × 107, 1 × 108 | Transendocardial injection | Reduced scar size and improved NYHA classification for both groups; increased ejection fraction (1 × 108 group) and proBNP (2 × 107 group) | [162] |
| Ischemic cardiomyopathy | Autologous or allogeneic BM-MSCs | 2 × 107, 1 × 108, or 2 × 108 | Transendocardial injection in 10 left ventricular sites | Reduced scar size accompanied by decreased end-diastolic and end-systolic volume, increased ejection fraction; improved sphericity index; functional improvement in autologous group assessed by 6MWT | [127] | |
| Nonischemic dilated cardiomyopathy | Autologous or allogeneic BM-MSCs | 1 × 108 | Transendocardial injection in 10 left ventricular sites | Increased ejection fraction (EF) and 6MWT in allogeneic BM-MCSc group compared to autologous group; allogeneic BM-MCSc group showed improved endothelial function, suppression of TNFα, functional capacity, and quality of life compared to allogeneic group; serious adverse events occurred in 28.2% of allogeneic group and 63.5% of autologous group | [126] | |
| Ischemic heart disease and ischemic heart failure (IHF) | Allogeneic ASCs | 1.1 × 108 | Intramyocardial injection | Decrease left ventricular end-systolic volume, increased LVEF and exercise capacity | [167] | |
| Heart failure (HF) with reduced ejection fraction (HFrEF) | Allogeneic UC-MSCs | 1 × 106/kg | Intravenous | Increased LVEF, improved NYHA classification and MLHFQ | [165] | |
| Acute myocardial infarction (AMI) | Wharton’s jelly-derived MSCs | 6 × 106 | Intracoronary infusion into the infarct artery | Improved cardiac function reflected by absolute increase in myocardial viability, perfusion within infarcted territory (SPECT); absolute increase in LVEF; absolute decreases in LV end-systolic and end-diastolic volume | [166] | |
| Kidney | Renal fibrosis on peritoneal dialysis (PD) | Autologous ASCs | 1.2 ± 0.1 × 106/kg | Intravenous (cubital vein) | Decline in rate of solute transport across peritoneum determined by PET and D/Pcr | [66] |
| Atherosclerotic renovascular disease (RVD) | Autologous ASCs | 1 × 105, 2.5 × 105/kg | Intra-arterial infusion | Increased renal tissue oxygenation and cortical blood flow | [68] | |
| Uterus | Asherman’s syndrome and endometrial atrophy | Autologous BM-MSCs | Mean 6.53 × 107 (range: 1.9 × 107 to 2 × 108) | Transmyometrial implant in subendometrial zone | Improved uterine cavity, increased endometrial thickness; improved menstrual duration and intensity and pregnancy rates | [225] |
| Asherman’s syndrome and endometrial atrophy | Autologous CD133+ BM-SCs | Mean 1.23 × 108 (range: 4.2 × 107 to 2 × 108) | Intra-arterial catherization | Improved uterine cavity and increased endometrial thickness; increased mature vessel density, duration and intensity of menses, and pregnancy rates | [229] | |
| Asherman’s syndrome | Autologous SVFs | 4.6 ± 0.7 × 106 | Intrauterine | Increased endometrial thickness and menstrual bleeding | [230] | |
| Recurrent intrauterine adhesions (IUAs) | Allogeneic UC-MSCs in collagen scaffold | 1 × 107 | Infusion in uterine wall via catheter placed in uterine cavity | Increased endometrial thickness and expression levels of ERα, Ki67, and vWF; resumed menses and increased menstrual bleeding; improved uterine cavity and pregnancy rates | [231] | |
| Asherman’s syndrome | Autologous menstrual blood-derived stromal cells (menSCs) | 1.0 × 106 | Intrauterine | Increased endometrial thickness and pregnancy rates | [232] | |
| Skin | Systemic sclerosis | Autologous CD34+ HSCs | Mean dose 5.63 × 106 /kg | Intravenous | Improved skin thickness assessed by mRSS | [251] |
| Systemic sclerosis | Autologous CD34+ HSCs | Median dose 5.6 × 106/kg | N/A (infusion) | Improved skin thickness based on mRSS | [254] | |
| Systemic sclerosis | Autologous CD34+ HSCs | ≥2 × 106/kg | N/A (infusion) | Improved skin thickness based on mRSS | [253] | |
| Systemic sclerosis | Autologous HSCs | N/A | N/A (infusion) | Improved skin thickness based on mRSS | [252] | |
| Systemic sclerosis | Autologous SVFs | Mean 3.76 ± 1.85 × 106 | Injection into subcutaneous tissue in contact with neurovascular pedicles | Reduced pain, finger circumference, and Raynaud’s severity; increased grip strength; decreased dystrophic capillaries and vascular suppression score | [256] | |
| Systemic sclerosis | Autologous ASCs | 4 × 106 to 8 × 106 | Injection into patient-specific area (face, arm, foot, limb) | Regression of dyschromia and reduced erythema; improved skin softening and sensitivity | [255] | |
| Systemic sclerosis | Autologous fat-enriched multipotent stem cells | N/A | Injection into oro-facial tissues | Improved mouth function and facial volumetric appearance | [257] | |
| Systemic sclerosis | Autologous fat-enriched multipotent stem cells | N/A | Local injection into oro-facial tissues | Increased interincisal distance and oral perimeter; improved neovascularization | [258] |
Abbreviations: αSMA, α-smooth muscle actin; ASCs, adipose-derived stem cells; ALB, serum albumin; ALT, alanine aminotransferase; AST, aspartate transaminase; BM-MSCs, bone marrow mesenchymal stem cells; COL, collagen; D/P cr, dialysate-to-creatinine ratio; HGF, hepatocyte growth factor; HA, hyaluronic acid; HSCs, hematopoietic stem cells; IL, interleukin; INR, international normalized ratio; DLCO, diffusing capacity of the lung for carbon monoxide; EF, ejection fraction; ERα, estrogen receptor alpha; FVC, forced ventilation capacity; LVEF, left ventricular ejection fraction; N/A, not available; MELD, model for end-stage liver disease; MIF, macrophage migration inhibitory factor; MLHFQ, Minnesota Living with Heart Failure Questionnaire; NYHA, New York Heart Association class; mRSS, modified Rodnan’s skin score; PET, peritoneal equilibration test; proBNP, pro-brain natriuretic peptide; PT, prothrombin time; SPECT, single-photon emission computed tomography; SVFs, stromal vascular fraction cells; TBIL, total bilirubin; TGFβ1, transforming growth factor-1; TNFα, tumor necrosis factor alpha; 6MWT, six minute walk test; UC-MSCs, umbilical cord mesenchymal stem cells; vWF, von Willebrand factor.
4. Limitations
The clinical application of stem cells, especially human ESCs (hESCs), iPSCs, and iPSC-derived cells, raises several ethical and safety concerns. Ethical issues concerning stem cells are mainly related to the source of hESCs and iPSCs, while safety issues are related to the potential of all stem cells to transform into undesirable cell types. The main ethical dilemma concerning hESCs is their origin and the destruction of human embryos to obtain them. Other controversies are related to their immunogenic potential and risk of cancer formation. The plasticity of hESCs allows them to create several cell types; however, this ability makes them challenging to control after in vivo transplantation [259]. Once undifferentiated hESCs are transplanted, there is a huge risk that teratomas and tumors containing all three germ layers will develop [260]. It is estimated that teratomas develop in 33–100% of hESC-transplanted immunodeficient mice [261,262]. To be sure that teratomas will not develop, hESCs should be differentiated into desired and mature cell types before transplantation and then monitored for the presence of undifferentiated cells [263].
Despite the similarities between iPSs and hESCs in terms of karyotype, phenotype, telomerase activity, and differentiation capacity, they are ethically superior to hESCs, as their generation does not involve the destruction of embryos [264]. The main safety issue regarding the transplantation of iPSCs and iPSC-derived cells is their undesired differentiation and malignant transformation. Moreover, the genomic instability of iPSCs, along with uncontrolled proliferation and differentiation, may lead to the formation of tumors and/or undesired differentiation into a variety of somatic cells [54]. Thus, effective methods for the generation of purified populations of autologous iPSC-derived differentiated cells still remain a challenge for personalized and regenerative medicine [265].
Along with the promising results of MSC-based therapy, safety issues regarding treatment with MSCs are still a matter of debate. Similar to the aforementioned ESCs, iPSC, and iPSC-derived cells, MSCs also have the capacity to differentiate into undesired tissue, such as bone and cartilage [266,267]. Probably the local microenvironment in the tissues of recipients contains factors that promote unwanted differentiation of transplanted MSCs. In addition, MSCs promote metastasis by inducing angiogenesis, as they have the potential to differentiate into endothelial cells and create capillary tubular networks [268,269].
A very important issue in the context of the treatment of fibrosis is myofibroblast differentiation from stem cells. Indeed, using genetic lineage tracing technology, it has been demonstrated that diverse organ-resident perivascular MSC-like cells and BM-MSCs are implicated in myofibroblast generation during fibrosis [270,271]. Moreover, there is experimental evidence demonstrating that BM-MSCc transplanted into mice with chemically induced cirrhosis differentiate into myofibroblasts and contribute to hepatic fibrosis rather than providing an antifibrotic therapeutic element [114]. Thus, follow-up studies should pay special attention to this aspect of stem cell properties [18]. Since the results of studies indicate that MSCs can differentiate into undesired cell types and can also transform into tumors [272], studies that use MSCs in fibrotic disorders should be more focused on monitoring and long-term follow-up of MSC recipients. Concerning clinical trials conducted using stem cells as a treatment for organ fibrosis, the results have generally been optimistic, as several early-phase clinical studies have demonstrated encouraging effects on organ function and an acceptable safety profile in relatively short-term follow-up. Nevertheless, for the best clinical outcome, there is still a need to optimize therapeutic regimens, including the cell source, dose, administration route and frequency, the timing of delivery, and long-term safety.
5. Conclusions
In recent years, the understanding of the pathophysiology of organ fibrosis has significantly progressed. However, efficient therapy to stop the progression of fibrotic diseases or reverse them does not exist. At present, antifibrotic therapies are aimed at stabilizing the fibrotic process, halting inflammation, relieving symptoms, and improving the quality of life of patients, but the long-term goal is to reverse these destructive diseases and develop novel strategies to cure them. Results obtained from completed and ongoing clinical studies demonstrate the promising therapeutic potential of stem cell-based therapy in the treatment of fibrotic diseases in the lungs, liver, kidney, heart, and even the uterus and skin. Moreover, a number of patent applications for stem cell-based therapies in organ fibrosis have been filed worldwide (see examples in Table 2). As we described in this review, stem cells obtained from different sources such as bone marrow, adipose tissue, the pluripotent inner cell mass of the pre-implantation embryo, placenta, umbilical cord, Wharton’s jelly, and even menstrual blood have been used by researchers and clinicians to develop antifibrotic therapy. As we show here, several preclinical and clinical studies on stem cell-based therapy have been carried out to date. Nevertheless, there is still no specific stem cell-based therapy or extra-clinical treatment available to the general public.
Table 2.
Examples of patent applications for stem cell-based therapies in organ fibrosis (5 October 2021; Espacenet Patent search; https://worldwide.espacenet.com).
The results of a large body of studies indicate a strong impact of the application of stem cells on fibrotic markers in in vitro research and clinical trials. However, the exact mechanisms of action of stem cells remain poorly understood and demand further investigation. Ascertaining these mechanisms would help to improve the efficiency of stem cell-based therapies and the survival rate of people suffering from organ fibrosis. Furthermore, many issues still have to be addressed before stem cells can be used clinically for fibrotic diseases, including sufficient cell numbers, optimal time, and optimal delivery route for cell transplantation. The lack of standardized and optimized protocols for stem cell isolation and culture expansion must also be resolved to assure reproducibility in clinical settings. Additionally, taking into consideration the safety issues, the development of stem cell-based therapy for fibrotic disorders should be more focused on constant observation and long-term follow-up of treated animal models to define possible detrimental effects and increase the clinical safety and efficacy.
Author Contributions
Conceptualization, A.S.-M. and J.W.; writing—original draft preparation, J.W., A.S., A.W., M.S., A.S.-M.; writing—review and editing A.S.-M., J.W.; visualization, M.S.; supervision, A.S.-M., J.W.; funding acquisition, A.S.-M. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by National Science Centre, Poland, Grant SONATA 15 No 2019/35/D/NZ9/02989.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
The authors thank Barbara Gawrońska-Kozak, Institute of Animal Reproduction and Food Research, Polish Academy of Sciences, Olsztyn, Poland for a revision of manuscript and constructive comments.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| 6MWT | 6-min walk test |
| αSMA | α-smooth muscle actin |
| ACTA2 | α-smooth muscle actin |
| ADPKD | autosomal-dominant polycystic kidney disease |
| AECs | alveolar epithelial cells |
| AFSCs | amniotic fluid-derived stem cells |
| AKI | acute kidney injury |
| Akt | protein kinase B |
| ALB | serum albumin |
| ALT | alanine aminotransferase |
| AS | Asherman syndrome |
| AST | aspartate aminotransferase |
| ASCs | adipose-derived stem cells |
| ASCs-CM | conditioned medium from ASCs |
| AQP | aquaporin |
| BLM | bleomycin |
| bFGF | basic fibroblast growth factor |
| BMI | body mass index |
| BM-MSCs | bone marrow mesenchymal stem cells |
| BMPCs | bone marrow progenitor cells |
| CCl4 | carbon tetrachloride |
| COL | collagen |
| CTGF | connective tissue growth factor |
| dcSS | diffuse cutaneous systemic sclerosis |
| DDIT3 | DNA-damage inducible transcript 3 |
| Dkk-1 | Dickkopf-1 |
| DLCO | carbon monoxide diffusing capacity |
| D/P cr | dialysate-to-creatinine ratio |
| EBs | embryoid bodies |
| ECM | extracellular matrix |
| EF | ejection fraction |
| eGFR | estimated glomerular filtration rate |
| Eln | elastin |
| eMSC | endometrial MSCs |
| EMT | epithelial-to-mesenchymal transition process |
| ERα | estrogen receptor alpha |
| ERK | extracellular-signal-regulated kinase |
| ESCs | embryonic stem cells |
| Ev | extracellular vesicles |
| FN | fibronectin |
| FVC | forced vital capacity |
| G-CSF | granulocyte colony-stimulating factor |
| GDNF | glial cell line-derived neurotrophic factor |
| GFP | green fluorescent protein |
| GM-CSF | granulocyte-macrophage colony-stimulating factor |
| GPSCs | germline cell-derived pluripotent stem cells |
| GTCs | tubular-like cells |
| GVHD | graft-versus-host disease |
| HA | hyaluronic acid |
| hAMSCs | human amniotic mesenchymal stromal cells |
| HBO | hyperbaric oxygen |
| HBV | hepatitis B |
| HCV | hepatitis C |
| HGF | hepatocyte growth factor |
| HLA | histocompatibility complex |
| HOCl | hypochlorous acid |
| HSCs | hematopoietic stem cells |
| IGF-1 | insulin growth factor 1 |
| IL4Rα | IL-4 receptor alpha |
| IL | interleukin |
| ILCs | insulin-producing islet-like clusters |
| INR | international normalized ratio |
| IPF | idiopathic pulmonary fibrosis |
| iPSCs | induced pluripotent stem cells |
| IUAs | intrauterine adhesions |
| JNK | JUN N-terminal kinase |
| lcSS | limited cutaneous systemic sclerosis |
| LVEF | left ventricular ejection fraction |
| MCP-1 | monocyte chemotactic protein-1 |
| MELD | model for end-stage liver disease |
| MI | myocardial infarction |
| MIF | macrophage migration inhibitory factor |
| MLHFQ | Minnesota living with heart failure questionnaire |
| MMPs | matrix metalloproteinases |
| mRSS | modified Rodnan’s skin score |
| MSCs | mesenchymal stem cells |
| MT1-MMP | membrane-bound matrix metalloproteinases |
| NAFLD | non-alcoholic fatty liver disease |
| NASH | non-alcoholic steatohepatitis |
| NYHA | New York Heart Association class |
| PAI-1 | plasminogen activator inhibitor 1 |
| PCNA | proliferating cell nuclear antigen |
| PDE | phosphodiesterase |
| PET | peritoneal equilibration test |
| PF | pulmonary fibrosis |
| proBNP | pro-brain natriuretic peptide |
| PT | prothrombin time |
| PVD | portal vein diameter |
| ROCK | RHO-associated kinase |
| RPCs | renal progenitor cells |
| RVD | renovascular disease |
| SDF-1α | stromal cell-derived factor 1 alpha |
| SHH | sonic hedgehog |
| SPC | surfactant protein C |
| SPECT | single-photon emission computed tomography |
| SS | systemic sclerosis |
| SVF | stromal vascular fraction |
| TBIL | total bilirubin |
| TECs | tubular epithelial cells |
| TGF- β | transforming growth factor β |
| TGFβRI | TGFβ receptor type 1 |
| Thbs1 | thrombospondin 1 |
| TIMPs | tissue inhibitors of matrix metalloproteinases |
| TNF | tumor necrosis factor |
| TTA | thioacetamide |
| UC-MSC | umbilical cord MSCs |
| vWF | von Willebrand factor |
References
- Jun, J.I.; Lau, L.F. Resolution of organ fibrosis. J. Clin. Investig. 2018, 128, 97–107. [Google Scholar] [CrossRef]
- Zeisberg, M.; Kalluri, R. Cellular mechanisms of tissue fibrosis. 1. Common and organ-specific mechanisms associated with tissue fibrosis. Am. J. Physiol. Cell Physiol. 2013, 304, C216–C225. [Google Scholar] [CrossRef]
- Wynn, T.A. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat. Rev. Immunol. 2004, 4, 583–594. [Google Scholar] [CrossRef]
- Muzes, G.; Sipos, F. Issues and opportunities of stem cell therapy in autoimmune diseases. World J. Stem Cells 2019, 11, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Sivandzade, F.; Cucullo, L. Regenerative stem cell therapy for neurodegenerative diseases: An overview. Int. J. Mol. Sci. 2021, 22, 2153. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.; McKee, C.; Bakshi, S.; Walker, K.; Hakman, E.; Halassy, S.; Svinarich, D.; Dodds, R.; Govind, C.K.; Chaudhry, G.R. Mesenchymal stem cells: Cell therapy and regeneration potential. J. Tissue Eng. Regen. Med. 2019, 13, 1738–1755. [Google Scholar] [CrossRef]
- Han, Y.; Li, X.; Zhang, Y.; Han, Y.; Chang, F.; Ding, J. Mesenchymal Stem Cells for Regenerative Medicine. Cells 2019, 8, 886. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.-S.; Jeong, E.-J.; Kim, J.-Y.; Park, S.J.; Ju, W.S.; Kim, C.-H.; Kim, J.-S.; Choo, Y.-K. Application of Mesenchymal Stem Cells in Inflammatory and Fibrotic Diseases. Int. J. Mol. Sci. 2020, 21, 8366. [Google Scholar] [CrossRef] [PubMed]
- Friedenstein, A.J.; Piatetzky, S., II; Petrakova, K.V. Osteogenesis in transplants of bone marrow cells. J. Embryol. Exp. Morphol. 1966, 16, 381–390. [Google Scholar]
- Romanov, Y.A.; Svintsitskaya, V.A.; Smirnov, V.N. Searching for alternative sources of postnatal human mesenchymal stem cells: Candidate MSC-like cells from umbilical cord. Stem Cells 2003, 21, 105–110. [Google Scholar] [CrossRef]
- Schwab, K.E.; Gargett, C.E. Co-expression of two perivascular cell markers isolates mesenchymal stem-like cells from human endometrium. Hum. Reprod. 2007, 22, 2903–2911. [Google Scholar] [CrossRef]
- Zuk, P.A.; Zhu, M.; Mizuno, H.; Huang, J.; Futrell, J.W.; Katz, A.J.; Benhaim, P.; Lorenz, H.P.; Hedrick, M.H. Multilineage cells from human adipose tissue: Implications for cell-based therapies. Tissue Eng. 2001, 7, 211–228. [Google Scholar] [CrossRef]
- Dominici, M.; le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef]
- Nie, C.; Yang, D.; Xu, J.; Si, Z.; Jin, X.; Zhang, J. Locally Administered Adipose-Derived Stem Cells Accelerate Wound Healing through Differentiation and Vasculogenesis. Cell Transplant. 2011, 20, 205–216. [Google Scholar] [CrossRef]
- Park, S.-R.; Kim, J.-W.; Jun, H.-S.; Roh, J.Y.; Lee, H.-Y.; Hong, I.-S. Stem Cell Secretome and Its Effect on Cellular Mechanisms Relevant to Wound Healing. Mol. Ther. 2018, 26, 606–617. [Google Scholar] [CrossRef]
- Kim, H.W.; Lee, H.S.; Kang, J.M.; Bae, S.H.; Kim, C.; Lee, S.H.; Schwarz, J.; Kim, G.J.; Kim, J.S.; Cha, D.H.; et al. Dual Effects of Human Placenta-Derived Neural Cells on Neuroprotection and the Inhibition of Neuroinflammation in a Rodent Model of Parkinson’s Disease. Cell Transpl. 2018, 27, 814–830. [Google Scholar] [CrossRef]
- Zhang, R.; Liu, Y.; Yan, K.; Chen, L.; Chen, X.R.; Li, P.; Chen, F.F.; Jiang, X.D. Anti-inflammatory and immunomodulatory mechanisms of mesenchymal stem cell transplantation in experimental traumatic brain injury. J. Neuroinflammation 2013, 10, 106. [Google Scholar] [CrossRef]
- El Agha, E.; Kramann, R.; Schneider, R.K.; Li, X.; Seeger, W. BD Humphreys and S Bellusci. Mesenchymal Stem Cells in Fibrotic Disease. Cell Stem Cell 2017, 21, 166–177. [Google Scholar] [CrossRef] [PubMed]
- Hinz, B.; Phan, S.; Thannickal, V.J.; Prunotto, M.; Desmoulière, A.; Varga, J.; De Wever, O.; Mareel, M.M.; Gabbiani, G. Recent Developments in Myofibroblast Biology: Paradigms for Connective Tissue Remodeling. Am. J. Pathol. 2012, 180, 1340–1355. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Neilson, E.G. Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Investig. 2003, 112, 1776–1784. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.K.; Kugler, M.C.; Wolters, P.J.; Robillard, L.; Galvez, M.G.; Brumwell, A.N.; Sheppard, D.; Chapman, H.A. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc. Natl. Acad. Sci. USA 2006, 103, 13180–13185. [Google Scholar] [CrossRef]
- Marangoni, R.G.; Korman, B.; Wei, J.; Wood, T.A.; Graham, L.V.; Whitfield, M.L.; Scherer, P.E.; Tourtellotte, W.G.; Varga, J. Myofibroblasts in Murine Cutaneous Fibrosis Originate From Adiponectin-Positive Intradermal Progenitors. Arthritis Rheumatol. 2015, 67, 1062–1073. [Google Scholar] [CrossRef] [PubMed]
- Hemmann, S.; Graf, J.; Roderfeld, M.; Roeb, E. Expression of MMPs and TIMPs in liver fibrosis—A systematic review with special emphasis on anti-fibrotic strategies. J. Hepatol. 2007, 46, 955–975. [Google Scholar] [CrossRef]
- Hynes, R.O. The extracellular matrix: Not just pretty fibrils. Science 2009, 326, 1216–1219. [Google Scholar] [CrossRef] [PubMed]
- Benyon, R.C.; Iredale, J.P.; Goddard, S.; Winwood, P.J.; Arthur, M.J. Expression of tissue inhibitor of metalloproteinases 1 and 2 is increased in fibrotic human liver. Gastroenterology 1996, 110, 821–831. [Google Scholar] [CrossRef]
- Holmbeck, K.; Bianco, P.; Caterina, J.; Yamada, S.; Kromer, M.; Kuznetsov, S.A.; Mankani, M.; Robey, P.; Poole, A.; Pidoux, I.; et al. MT1-MMP-deficient mice develop dwarfism, osteopenia, arthritis, and connective tissue disease due to inadequate collagen turnover. Cell 1999, 99, 81–92. [Google Scholar] [CrossRef]
- Grupp, C.; Troche, I.; Klass, C.; Köhler, M.; Müller, G.A. A novel model to study renal myofibroblast formation in vitro. Kidney Int. 2001, 59, 543–553. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.; Foreman, D.; Ferguson, M. Neutralisation of TGF-beta 1 and TGF-beta 2 or exogenous addition of TGF-beta 3 to cutaneous rat wounds reduces scarring. J. Cell Sci. 1995, 108, 985–1002. [Google Scholar] [CrossRef]
- Sonnylal, S.; Denton, C.P.; Zheng, B.; Keene, D.R.; He, R.; Adams, H.P.; Vanpelt, C.S.; Geng, Y.J.; Deng, J.M.; Behringer, R.R.; et al. Postnatal induction of transforming growth factor beta signaling in fibroblasts of mice recapitulates clinical, histologic, and biochemical features of scleroderma. Arthritis Rheum. 2007, 56, 334–344. [Google Scholar] [CrossRef]
- Wojcik-Pszczola, K.; Chlon-Rzepa, G.; Jankowska, A.; Slusarczyk, M.; Ferdek, P.E.; Kusiak, A.A.; Swierczek, A.; Pociecha, K.; Koczurkiewicz-Adamczyk, P.; Wyska, E.; et al. A Novel, Pan-PDE Inhibitor Exerts Anti-Fibrotic Effects in Human Lung Fibroblasts via Inhibition of TGF-beta Signaling and Activation of cAMP/PKA Signaling. Int. J. Mol. Sci. 2020, 21, 4008. [Google Scholar] [CrossRef]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Lafyatis, R. Transforming growth factor beta—At the centre of systemic sclerosis. Nat. Rev. Rheumatol. 2014, 10, 706–719. [Google Scholar] [CrossRef] [PubMed]
- Akhmetshina, A.; Palumbo, K.; Dees, C.; Bergmann, C.; Venalis, P.; Zerr, P.; Horn, A.; Kireva, T.; Beyer, C.; Zwerina, J.; et al. Activation of canonical Wnt signalling is required for TGF-beta-mediated fibrosis. Nat. Commun. 2012, 3, 735. [Google Scholar] [CrossRef]
- He, W.; Dai, C.; Li, Y.; Zeng, G.; Monga, S.P.; Liu, Y. Wnt/beta-catenin signaling promotes renal interstitial fibrosis. J. Am. Soc. Nephrol. 2009, 20, 765–776. [Google Scholar] [CrossRef]
- Horn, A.; Palumbo, K.; Cordazzo, C.; Dees, C.; Akhmetshina, A.; Tomcik, M.; Zerr, P.; Avouac, J.; Gusinde, J.; Zwerina, J.; et al. Hedgehog signaling controls fibroblast activation and tissue fibrosis in systemic sclerosis. Arthritis Rheum. 2012, 64, 2724–2733. [Google Scholar] [CrossRef]
- Beyer, C.; Huscher, D.; Ramming, A.; Bergmann, C.; Avouac, J.; Guiducci, S.; Meier, F.; Vettori, S.; Siegert, E.; Jaeger, V.K.; et al. Elevated serum levels of sonic hedgehog are associated with fibrotic and vascular manifestations in systemic sclerosis. Ann. Rheum. Dis. 2018, 77, 626–628. [Google Scholar] [CrossRef]
- Chen, X.; Li, W.X.; Chen, Y.; Li, X.F.; Li, H.D.; Huang, H.M.; Bu, F.T.; Pan, X.Y.; Yang, Y.; Huang, C.; et al. Suppression of SUN2 by DNA methylation is associated with HSCs activation and hepatic fibrosis. Cell Death Dis. 2018, 9, 1021. [Google Scholar] [CrossRef] [PubMed]
- Corpechot, C.; Barbu, V.; Wendum, D.; Kinnman, N.; Rey, C.; Poupon, R.; Housset, C.; Rosmorduc, O. Hypoxia-induced VEGF and collagen I expressions are associated with angiogenesis and fibrogenesis in experimental cirrhosis. Hepatology 2002, 35, 1010–1021. [Google Scholar] [CrossRef]
- Tang, P.M.K.; Nikolic-Paterson, D.J.; Lan, H.-Y. Macrophages: Versatile players in renal inflammation and fibrosis. Nat. Rev. Nephrol. 2019, 15, 144–158. [Google Scholar] [CrossRef]
- Blanpain, C.; Fuchs, E. Plasticity of epithelial stem cells in tissue regeneration. Science 2014, 344, 6189. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Fuentes, D.E.; Fernandez-Garza, L.E.; Samia-Meza, J.A.; Barrera-Barrera, S.A.; Caplan, A.I.; Barrera-Saldana, H.A. Mesenchymal Stem Cells Current Clinical Applications: A Systematic Review. Arch. Med. Res. 2021, 52, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Mahla, R.S. Stem Cells Applications in Regenerative Medicine and Disease Therapeutics. Int. J. Cell Biol. 2016, 2016, 1–24. [Google Scholar] [CrossRef]
- Rippon, H.J.; Bishop, A.E. Embryonic stem cells. Cell Prolif. 2004, 37, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Ali, N.N.; Edgar, A.J.; Samadikuchaksaraei, A.; Timson, C.M.; Romanska, H.M.; Polak, J.M.; Bishop, A.E. Derivation of Type II Alveolar Epithelial Cells from Murine Embryonic Stem Cells. Tissue Eng. 2002, 8, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Buttery, L.D.K.; Bourne, S.; Xynos, J.D.; Wood, H.; Hughes, F.J.; Hughes, S.P.F.; Episkopou, V.; Polak, J.M. Differentiation of Osteoblasts andin VitroBone Formation from Murine Embryonic Stem Cells. Tissue Eng. 2001, 7, 89–99. [Google Scholar] [CrossRef]
- Kramer, J.; Hegert, C.; Guan, K.; Wobus, A.M.; Müller, P.K.; Rohwedel, J. Embryonic stem cell-derived chondrogenic differentiation in vitro: Activation by BMP-2 and BMP-4. Mech. Dev. 2000, 92, 193–205. [Google Scholar] [CrossRef]
- Stavridis, M.P.; Smith, A.G. Neural differentiation of mouse embryonic stem cells. Biochem. Soc. Trans. 2003, 31, 45–49. [Google Scholar] [CrossRef]
- Yurugi-Kobayashi, T.; Itoh, H.; Yamashita, J.; Yamahara, K.; Hirai, H.; Kobayashi, T.; Ogawa, M.; Nishikawa, S.; Nishikawa, S.-I.; Nakao, K. Effective contribution of transplanted vascular progenitor cells derived from embryonic stem cells to adult neovascularization in proper differentiation stage. Blood 2003, 101, 2675–2678. [Google Scholar] [CrossRef]
- Lerou, P. Embryonic Stem Cell Derivation from Human Embryos. Methods Mol. Biol. 2011, 767, 31–35. [Google Scholar]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Bellin, M.; Marchetto, M.C.; Gage, F.H.; Mummery, C.L. Induced pluripotent stem cells: The new patient? Nat. Rev. Mol. Cell Biol. 2012, 13, 713–726. [Google Scholar] [CrossRef]
- Ahmed, R.P.; Haider, H.K.; Buccini, S.; Li, L.; Jiang, S.; Ashraf, M. Reprogramming of skeletal myoblasts for induction of pluripotency for tumor-free cardiomyogenesis in the infarcted heart. Circ. Res. 2011, 109, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Tateishi, K.; He, J.; Taranova, O.; Liang, G.; D’Alessio, A.C.; Zhang, Y. Generation of Insulin-secreting Islet-like Clusters from Human Skin Fibroblasts. J. Biol. Chem. 2008, 283, 31601–31607. [Google Scholar] [CrossRef]
- Wernig, M.; Zhao, J.P.; Pruszak, J.; Hedlund, E.; Fu, D.; Soldner, F.; Broccoli, V.; Constantine-Paton, M.; Isacson, O.; Jaenisch, R. Neurons derived from reprogrammed fibroblasts functionally integrate into the fetal brain and improve symptoms of rats with Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2008, 105, 5856–5861. [Google Scholar] [CrossRef] [PubMed]
- Sayed, N.; Liu, C.; Wu, J.C. Translation of Human-Induced Pluripotent Stem Cells: From Clinical Trial in a Dish to Precision Medicine. J. Am. Coll. Cardiol. 2016, 67, 2161–2176. [Google Scholar] [CrossRef] [PubMed]
- Friedenstein, A.J.; Chailakhjan, R.K.; Lalykina, K.S. The development of fibroblast colonies in monolayer cultures of guinea-pig bone marrow and spleen cells. Cell. Tissue Kinet. 1970, 3, 393–403. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, R.; Gopal, S.; Masood, H.; Vivek, P.; Deb, K. Regenerative potential of dental pulp mesenchymal stem cells harvested from high caries patient’s teeth. J. Stem. Cells 2013, 8, 25–41. [Google Scholar]
- Weiss, M.L.; Troyer, D.L. Stem cells in the umbilical cord. Stem Cell Rev. 2006, 2, 155–162. [Google Scholar] [CrossRef]
- Kern, S.; Eichler, H.; Stoeve, J.; Kluter, H.; Bieback, K. Comparative analysis of mesenchymal stem cells from bone marrow, umbilical cord blood, or adipose tissue. Stem Cells 2006, 24, 1294–1301. [Google Scholar] [CrossRef]
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage potential of adult human mesenchymal stem cells. Science 1999, 284, 143–147. [Google Scholar] [CrossRef]
- Zuk, P.A.; Zhu, M.; Ashjian, P.; de Ugarte, D.A.; Huang, J.I.; Mizuno, H.; Alfonso, Z.C.; Fraser, J.K.; Benhaim, P.; Hedrick, M.H. Human adipose tissue is a source of multipotent stem cells. Mol. Biol. Cell 2002, 13, 4279–4295. [Google Scholar] [CrossRef]
- Gimble, J.M.; Guilak, F.; Nuttall, M.E.; Sathishkumar, S.; Vidal, M.; Bunnell, B.A. In vitro Differentiation Potential of Mesenchymal Stem Cells. Transfus. Med. Hemother. 2008, 35, 228–238. [Google Scholar] [CrossRef]
- Gimble, J.M.; Katz, A.J.; Bunnell, B.A. Adipose-derived stem cells for regenerative medicine. Circ Res. 2007, 100, 1249–1260. [Google Scholar] [CrossRef]
- Xie, L.; Zhang, N.; Marsano, A.; Vunjak-Novakovic, G.; Zhang, Y.; Lopez, M.J. In vitro mesenchymal trilineage differentiation and extracellular matrix production by adipose and bone marrow derived adult equine multipotent stromal cells on a collagen scaffold. Stem Cell Rev. Rep. 2013, 9, 858–872. [Google Scholar] [CrossRef] [PubMed]
- Bukowska, J.; Szostek-Mioduchowska, A.Z.; Kopcewicz, M.; Walendzik, K.; Machcinska, S.; Gawronska-Kozak, B. Adipose-Derived Stromal/Stem Cells from Large Animal Models: From Basic to Applied Science. Stem Cell Rev. Rep. 2021, 17, 719–738. [Google Scholar] [CrossRef] [PubMed]
- Alatab, S.; Shekarchian, S.; Najafi, I.; Moghadasali, R.; Ahmadbeigi, N.; Pourmand, M.R.; Bolurieh, T.; Jaroughi, N.; Pourmand, G.; Aghdami, N. Systemic Infusion of Autologous Adipose Tissue-Derived Mesenchymal Stem Cells in Peritoneal Dialysis Patients: Feasibility and Safety. Cell J. 2019, 20, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Cil, N.; Yaka, M.; Unal, M.S.; Dodurga, Y.; Tan, S.; Secme, M.; Karagur, E.R.; Mete, G.A. Adipose derived mesenchymal stem cell treatment in experimental asherman syndrome induced rats. Mol. Biol. Rep. 2020, 47, 4541–4552. [Google Scholar] [CrossRef] [PubMed]
- Saad, A.; Dietz, A.B.; Herrmann, S.M.S.; Hickson, L.J.; Glockner, J.F.; McKusick, M.A.; Misra, S.; Bjarnason, H.; Armstrong, A.S.; Gastineau, D.A.; et al. Autologous Mesenchymal Stem Cells Increase Cortical Perfusion in Renovascular Disease. J. Am. Soc. Nephrol. 2017, 28, 2777–2785. [Google Scholar] [CrossRef] [PubMed]
- Lim, R.; Ricardo, S.D.; Sievert, W. Cell-Based Therapies for Tissue Fibrosis. Front. Pharmacol. 2017, 8, 633. [Google Scholar] [CrossRef] [PubMed]
- Cameli, P.; Refini, R.M.; Bergantini, L.; d’Alessandro, M.; Alonzi, V.; Magnoni, C.; Rottoli, P.; Sestini, P.; Bargagli, E. Long-Term Follow-Up of Patients With Idiopathic Pulmonary Fibrosis Treated With Pirfenidone or Nintedanib: A Real-Life Comparison Study. Front. Mol. Biosci. 2020, 7, 581828. [Google Scholar] [CrossRef]
- Flaherty, K.R.; Fell, C.D.; Huggins, J.T.; Nunes, H.; Sussman, R.; Valenzuela, C.; Petzinger, U.; Stauffer, J.L.; Gilberg, F.; Bengus, M.; et al. Safety of nintedanib added to pirfenidone treatment for idiopathic pulmonary fibrosis. Eur. Respir. J. 2018, 52, 1800230. [Google Scholar] [CrossRef]
- Ortiz, L.A.; Gambelli, F.; McBride, C.; Gaupp, D.; Baddoo, M.; Kaminski, N.; Phinney, D.G. Mesenchymal stem cell engraftment in lung is enhanced in response to bleomycin exposure and ameliorates its fibrotic effects. Proc. Natl. Acad. Sci. USA 2003, 100, 8407–8411. [Google Scholar] [CrossRef]
- Rojas, M.; Xu, J.; Woods, C.R.; Mora, A.L.; Spears, W.; Roman, J.; Brigham, K.L. Bone marrow-derived mesenchymal stem cells in repair of the injured lung. Am. J. Respir. Cell Mol. Biol. 2005, 33, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Kotani, T.; Masutani, R.; Suzuka, T.; Oda, K.; Makino, S.; Ii, M. Anti-inflammatory and anti-fibrotic effects of intravenous adipose-derived stem cell transplantation in a mouse model of bleomycin-induced interstitial pneumonia. Sci. Rep. 2017, 7, 14608. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.; Fonseca, L.; Gowda, S.; Chougule, B.; Hari, A.; Totey, S. Human Adipose-derived Mesenchymal Stem Cells Attenuate Early Stage of Bleomycin Induced Pulmonary Fibrosis: Comparison with Pirfenidone. Int. J. Stem. Cells 2016, 9, 192–206. [Google Scholar] [CrossRef] [PubMed]
- Tashiro, J.; Elliot, S.J.; Gerth, D.J.; Xia, X.; Pereira-Simon, S.; Choi, R.; Catanuto, P.; Shahzeidi, S.; Toonkel, R.L.; Shah, R.H.; et al. Therapeutic benefits of young, but not old, adipose-derived mesenchymal stem cells in a chronic mouse model of bleomycin-induced pulmonary fibrosis. Transl. Res. 2015, 166, 554–567. [Google Scholar] [CrossRef]
- Cargnoni, A.; Gibelli, L.; Tosini, A.; Signoroni, P.B.; Nassuato, C.; Arienti, D.; Lombardi, G.; Albertini, A.; Wengler, G.S.; Parolini, O. Transplantation of allogeneic and xenogeneic placenta-derived cells reduces bleomycin-induced lung fibrosis. Cell Transpl. 2009, 18, 405–422. [Google Scholar] [CrossRef]
- Moodley, Y.; Atienza, D.; Manuelpillai, U.; Samuel, C.S.; Tchongue, J.; Ilancheran, S.; Boyd, R.; Trounson, A. Human umbilical cord mesenchymal stem cells reduce fibrosis of bleomycin-induced lung injury. Am. J. Pathol. 2009, 175, 303–313. [Google Scholar] [CrossRef]
- Zhou, Q.; Ye, X.; Sun, R.; Matsumoto, Y.; Moriyama, M.; Asano, Y.; Ajioka, Y.; Saijo, Y. Differentiation of mouse induced pluripotent stem cells into alveolar epithelial cells in vitro for use in vivo. Stem Cells Transl. Med. 2014, 3, 675–685. [Google Scholar] [CrossRef]
- Zhou, Y.; He, Z.; Gao, Y.; Zheng, R.; Zhang, X.; Zhao, L.; Tan, M. Induced Pluripotent Stem Cells Inhibit Bleomycin-Induced Pulmonary Fibrosis in Mice through Suppressing TGF-beta1/Smad-Mediated Epithelial to Mesenchymal Transition. Front. Pharmacol. 2016, 7, 430. [Google Scholar] [CrossRef]
- Gazdhar, A.; Grad, I.; Tamo, L.; Gugger, M.; Feki, A.; Geiser, T. The secretome of induced pluripotent stem cells reduces lung fibrosis in part by hepatocyte growth factor. Stem Cell Res. Ther. 2014, 5, 123. [Google Scholar] [CrossRef] [PubMed]
- Averyanov, A.; Koroleva, I.; Konoplyannikov, M.; Revkova, V.; Lesnyak, V.; Kalsin, V.; Danilevskaya, O.; Nikitin, A.; Sotnikova, A.; Kotova, S.; et al. First-in-human high-cumulative-dose stem cell therapy in idiopathic pulmonary fibrosis with rapid lung function decline. Stem Cells Transl. Med. 2020, 9, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Fishman, J.E.; Kim, G.J.; Kyeong, N.Y.; Goldin, J.G.; Glassberg, M.K. Intravenous stem cell dose and changes in quantitative lung fibrosis and DLCO in the AETHER trial: A pilot study. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 7568–7572. [Google Scholar] [PubMed]
- Glassberg, M.K.; Minkiewicz, J.; Toonkel, R.L.; Simonet, E.S.; Rubio, G.A.; DiFede, D.; Shafazand, S.; Khan, A.; Pujol, M.V.; LaRussa, V.F.; et al. Allogeneic Human Mesenchymal Stem Cells in Patients With Idiopathic Pulmonary Fibrosis via Intravenous Delivery (AETHER): A Phase I Safety Clinical Trial. Chest 2017, 151, 971–981. [Google Scholar] [CrossRef]
- Tzouvelekis, A.; Paspaliaris, V.; Koliakos, G.; Ntolios, P.; Bouros, E.; Oikonomou, A.; Zissimopoulos, A.; Boussios, N.; Dardzinski, B.; Gritzalis, D.; et al. A prospective, non-randomized, no placebo-controlled, phase Ib clinical trial to study the safety of the adipose derived stromal cells-stromal vascular fraction in idiopathic pulmonary fibrosis. J. Transl. Med. 2013, 11, 171. [Google Scholar] [CrossRef] [PubMed]
- Gines, P.; Graupera, I.; Lammert, F.; Angeli, P.; Caballeria, L.; Krag, A.; Guha, I.N.; Murad, S.D.; Castera, L. Screening for liver fibrosis in the general population: A call for action. Lancet Gastroenterol. Hepatol. 2016, 1, 256–260. [Google Scholar] [CrossRef]
- Friedman, S.L. Mechanisms of hepatic fibrogenesis. Gastroenterology 2008, 134, 1655–1669. [Google Scholar] [CrossRef]
- Nielsen, M.J.; Leeming, D.J.; Karsdal, M.A.; Krag, A. Biomarkers of Extracellular Matrix Remodeling in Liver Diseases. In Biomarkers in Liver Disease. Biomarkers in Disease: Methods, Discoveries and Applications; Patel, V., Preedy, V., Eds.; Springer: Berlin/Heidelberg, Germany, 2017. [Google Scholar]
- Marra, F. Hepatic stellate cells and the regulation of liver inflammation. J. Hepatol. 1999, 31, 1120–1130. [Google Scholar] [CrossRef]
- Rabani, V.; Shahsavani, M.; Gharavi, M.; Piryaei, A.; Azhdari, Z.; Baharvand, H. Mesenchymal stem cell infusion therapy in a carbon tetrachloride-induced liver fibrosis model affects matrix metalloproteinase expression. Cell Biol. Int. 2010, 34, 601–605. [Google Scholar] [CrossRef]
- Abdel Aziz, M.T.; Atta, H.M.; Mahfouz, S.; Fouad, H.H.; Roshdy, N.K.; Ahmed, H.H.; Rashed, L.A.; Sabry, D.; Hassouna, A.A.; Hasan, N.M. Therapeutic potential of bone marrow-derived mesenchymal stem cells on experimental liver fibrosis. Clin. Biochem. 2007, 40, 893–899. [Google Scholar] [CrossRef]
- Zhao, D.C.; Lei, J.X.; Chen, R.; Yu, W.H.; Zhang, X.M.; Li, S.N.; Xiang, P. Bone marrow-derived mesenchymal stem cells protect against experimental liver fibrosis in rats. World J. Gastroenterol. 2005, 11, 3431–3440. [Google Scholar] [CrossRef]
- Li, Q.; Zhou, X.; Shi, Y.; Li, J.; Zheng, L.; Cui, L.; Zhang, J.; Wang, L.; Han, Z.; Han, Y.; et al. In vivo tracking and comparison of the therapeutic effects of MSCs and HSCs for liver injury. PLoS ONE 2013, 8, e62363. [Google Scholar] [CrossRef]
- Chang, Y.J.; Liu, J.W.; Lin, P.C.; Sun, L.Y.; Peng, C.W.; Luo, G.H.; Chen, T.M.; Lee, R.P.; Lin, S.Z.; Harn, H.J.; et al. Mesenchymal stem cells facilitate recovery from chemically induced liver damage and decrease liver fibrosis. Life Sci. 2009, 85, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Tanimoto, H.; Terai, S.; Taro, T.; Murata, Y.; Fujisawa, K.; Yamamoto, N.; Sakaida, I. Improvement of liver fibrosis by infusion of cultured cells derived from human bone marrow. Cell Tissue Res. 2013, 354, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Harn, H.J.; Lin, S.Z.; Hung, S.H.; Subeq, Y.M.; Li, Y.S.; Syu, W.S.; Ding, D.C.; Lee, R.P.; Hsieh, D.K.; Lin, P.C.; et al. Adipose-derived stem cells can abrogate chemical-induced liver fibrosis and facilitate recovery of liver function. Cell Transplant. 2012, 21, 2753–2764. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Ma, T.; Chen, W.; Hu, J.; Bai, X.; Li, J.; Liang, T. Therapeutic potential and related signal pathway of adipose-derived stem cell transplantation for rat liver injury. Hepatol. Res. 2009, 39, 822–832. [Google Scholar] [CrossRef]
- Munoz, M.F.; Arguelles, S.; Guzman-Chozas, M.; Guillen-Sanz, R.; Franco, J.M.; Pintor-Toro, J.A.; Cano, M.; Ayala, A. Cell tracking, survival, and differentiation capacity of adipose-derived stem cells after engraftment in rat tissue. J. Cell Physiol. 2018, 233, 6317–6328. [Google Scholar] [CrossRef] [PubMed]
- Jung, K.H.; Shin, H.P.; Lee, S.; Lim, Y.J.; Hwang, S.H.; Han, H.; Park, H.K.; Chung, J.H.; Yim, S.V. Effect of human umbilical cord blood-derived mesenchymal stem cells in a cirrhotic rat model. Liver Int. 2009, 29, 898–909. [Google Scholar] [CrossRef]
- Tsai, P.C.; Fu, T.W.; Chen, Y.M.; Ko, T.L.; Chen, T.H.; Shih, Y.H.; Hung, S.C.; Fu, Y.S. The therapeutic potential of human umbilical mesenchymal stem cells from Wharton’s jelly in the treatment of rat liver fibrosis. Liver Transpl. 2009, 15, 484–495. [Google Scholar] [CrossRef]
- Li, T.; Yan, Y.; Wang, B.; Qian, H.; Zhang, X.; Shen, L.; Wang, M.; Zhou, Y.; Zhu, W.; Li, W.; et al. Exosomes derived from human umbilical cord mesenchymal stem cells alleviate liver fibrosis. Stem Cells Dev. 2013, 22, 845–854. [Google Scholar] [CrossRef]
- Carvalho, A.B.; Quintanilha, L.F.; Dias, J.V.; Paredes, B.D.; Mannheimer, E.G.; Carvalho, F.G.; Asensi, K.D.; Gutfilen, B.; Fonseca, L.M.; Resende, C.M.; et al. Bone marrow multipotent mesenchymal stromal cells do not reduce fibrosis or improve function in a rat model of severe chronic liver injury. Stem Cells 2008, 26, 1307–1314. [Google Scholar] [CrossRef]
- Mannheimer, E.G.; Quintanilha, L.F.; Carvalho, A.B.; Paredes, B.D.; de Carvalho, F.G.; Takyia, C.M.; Resende, C.M.; Rezende, G.F.d.; de Carvalho, A.C.C.; Schanaider, A.; et al. Bone marrow cells obtained from cirrhotic rats do not improve function or reduce fibrosis in a chronic liver disease model. Clin. Transpl. 2011, 25, 54–60. [Google Scholar] [CrossRef]
- Amer, M.E.; El-Sayed, S.Z.; El-Kheir, W.A.; Gabr, H.; Gomaa, A.A.; El-Noomani, N.; Hegazy, M. Clinical and laboratory evaluation of patients with end-stage liver cell failure injected with bone marrow-derived hepatocyte-like cells. Eur. J. Gastroenterol. Hepatol. 2011, 23, 936–941. [Google Scholar] [CrossRef]
- Amin, M.A.; Sabry, D.; Rashed, L.A.; Aref, W.M.; el-Ghobary, M.A.; Farhan, M.S.; Fouad, H.A.; Youssef, Y.A. Short-term evaluation of autologous transplantation of bone marrow-derived mesenchymal stem cells in patients with cirrhosis: Egyptian study. Clin. Transplant. 2013, 27, 607–612. [Google Scholar] [CrossRef]
- El-Ansary, M.; Abdel-Aziz, I.; Mogawer, S.; Abdel-Hamid, S.; Hammam, O.; Teaema, S.; Wahdan, M. Phase II trial: Undifferentiated versus differentiated autologous mesenchymal stem cells transplantation in Egyptian patients with HCV induced liver cirrhosis. Stem Cell Rev. Rep. 2012, 8, 972–981. [Google Scholar] [CrossRef]
- Jang, Y.O.; Kim, Y.J.; Baik, S.K.; Kim, M.Y.; Eom, Y.W.; Cho, M.Y.; Park, H.J.; Park, S.Y.; Kim, B.R.; Kim, J.W.; et al. Histological improvement following administration of autologous bone marrow-derived mesenchymal stem cells for alcoholic cirrhosis: A pilot study. Liver Int. 2014, 34, 33–41. [Google Scholar] [CrossRef]
- Kharaziha, P.; Hellstrom, P.M.; Noorinayer, B.; Farzaneh, F.; Aghajani, K.; Jafari, F.; Telkabadi, M.; Atashi, A.; Honardoost, M.; Zali, M.R.; et al. Improvement of liver function in liver cirrhosis patients after autologous mesenchymal stem cell injection: A phase I-II clinical trial. Eur. J. Gastroenterol. Hepatol. 2009, 21, 1199–1205. [Google Scholar] [CrossRef] [PubMed]
- Mohamadnejad, M.; Alimoghaddam, K.; Bagheri, M.; Ashrafi, M.; Abdollahzadeh, L.; Akhlaghpoor, S.; Bashtar, M.; Ghavamzadeh, A.; Malekzadeh, R. Randomized placebo-controlled trial of mesenchymal stem cell transplantation in decompensated cirrhosis. Liver Int. 2013, 33, 1490–1496. [Google Scholar] [CrossRef]
- Peng, L.; Xie, D.Y.; Lin, B.L.; Liu, J.; Zhu, H.P.; Xie, C.; Zheng, Y.B.; Gao, Z.L. Autologous bone marrow mesenchymal stem cell transplantation in liver failure patients caused by hepatitis B: Short-term and long-term outcomes. Hepatology 2011, 54, 820–828. [Google Scholar] [CrossRef] [PubMed]
- Sakai, Y.; Takamura, M.; Seki, A.; Sunagozaka, H.; Terashima, T.; Komura, T.; Yamato, M.; Miyazawa, M.; Kawaguchi, K.; Nasti, A.; et al. Phase I clinical study of liver regenerative therapy for cirrhosis by intrahepatic arterial infusion of freshly isolated autologous adipose tissue-derived stromal/stem (regenerative) cell. Regen. Ther. 2017, 6, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Lin, H.; Shi, M.; Xu, R.; Fu, J.; Lv, J.; Chen, L.; Lv, S.; Li, Y.; Yu, S.; et al. Human umbilical cord mesenchymal stem cells improve liver function and ascites in decompensated liver cirrhosis patients. J. Gastroenterol. Hepatol. 2012, 27, 112–120. [Google Scholar] [CrossRef]
- Li, C.; Kong, Y.; Wang, H.; Wang, S.; Yu, H.; Liu, X.; Yang, L.; Jiang, X.; Li, L. Homing of bone marrow mesenchymal stem cells mediated by sphingosine 1-phosphate contributes to liver fibrosis. J. Hepatol. 2009, 50, 1174–1183. [Google Scholar] [CrossRef]
- Russo, F.P.; Alison, M.R.; Bigger, B.W.; Amofah, E.; Florou, A.; Amin, F.; Bou-Gharios, G.; Jeffery, R.; Iredale, J.P.; Forbes, S.J. The bone marrow functionally contributes to liver fibrosis. Gastroenterology 2006, 130, 1807–1821. [Google Scholar] [CrossRef]
- Hassan, S.; Barrett, C.J.; Crossman, D.J. Imaging tools for assessment of myocardial fibrosis in humans: The need for greater detail. Biophys. Rev. 2020, 12, 969–987. [Google Scholar] [CrossRef]
- Talman, V.; Ruskoaho, H. Cardiac fibrosis in myocardial infarction-from repair and remodeling to regeneration. Cell Tissue Res. 2016, 365, 563–581. [Google Scholar] [CrossRef]
- Okyere, A.D.; Tilley, D.G. Leukocyte-Dependent Regulation of Cardiac Fibrosis. Front. Physiol. 2020, 11, 301. [Google Scholar] [CrossRef]
- Ranjan, P.; Kumari, R.; Verma, S.K. Cardiac Fibroblasts and Cardiac Fibrosis: Precise Role of Exosomes. Front. Cell Dev. Biol. 2019, 7, 318. [Google Scholar] [CrossRef]
- Fan, Z.; Guan, J. Antifibrotic therapies to control cardiac fibrosis. Biomater. Res. 2016, 20, 13. [Google Scholar] [CrossRef]
- Sweeney, M.; Corden, B.; Cook, S.A. Targeting cardiac fibrosis in heart failure with preserved ejection fraction: Mirage or miracle? EMBO Mol. Med. 2020, 12, e10865. [Google Scholar] [CrossRef]
- Wang, B.; Wang, H.; Zhang, M.; Ji, R.; Wei, J.; Xin, Y.; Jiang, X. Radiation-induced myocardial fibrosis: Mechanisms underlying its pathogenesis and therapeutic strategies. J. Cell Mol. Med. 2020, 24, 7717–7729. [Google Scholar] [CrossRef]
- Webber, M.; Jackson, S.P.; Moon, J.C.; Captur, G. Myocardial Fibrosis in Heart Failure: Anti-Fibrotic Therapies and the Role of Cardiovascular Magnetic Resonance in Drug Trials. Cardiol. Ther. 2020, 9, 363–376. [Google Scholar] [CrossRef]
- Golpanian, S.; Wolf, A.; Hatzistergos, K.E.; Hare, J.M. Rebuilding the Damaged Heart: Mesenchymal Stem Cells, Cell-Based Therapy, and Engineered Heart Tissue. Physiol. Rev. 2016, 96, 1127–1168. [Google Scholar] [CrossRef]
- Mias, C.; Lairez, O.; Trouche, E.; Roncalli, J.; Calise, D.; Seguelas, M.H.; Ordener, C.; Piercecchi-Marti, M.D.; Auge, N.; Salvayre, A.N.; et al. Mesenchymal stem cells promote matrix metalloproteinase secretion by cardiac fibroblasts and reduce cardiac ventricular fibrosis after myocardial infarction. Stem Cells 2009, 27, 2734–2743. [Google Scholar] [CrossRef]
- Pei, Z.; Zeng, J.; Song, Y.; Gao, Y.; Wu, R.; Chen, Y.; Li, F.; Li, W.; Zhou, H.; Yang, Y. In vivo imaging to monitor differentiation and therapeutic effects of transplanted mesenchymal stem cells in myocardial infarction. Sci. Rep. 2017, 7, 6296. [Google Scholar] [CrossRef]
- Hare, J.M.; DiFede, D.L.; Rieger, A.C.; Florea, V.; Landin, A.M.; El-Khorazaty, J.; Khan, A.; Mushtaq, M.; Lowery, M.H.; Byrnes, J.J.; et al. Randomized Comparison of Allogeneic Versus Autologous Mesenchymal Stem Cells for Nonischemic Dilated Cardiomyopathy: POSEIDON-DCM Trial. J. Am. Coll. Cardiol. 2017, 69, 526–537. [Google Scholar] [CrossRef]
- Hare, J.M.; Fishman, J.E.; Gerstenblith, G.; Velazquez, D.L.D.; Zambrano, J.P.; Suncion, V.Y.; Tracy, M.; Ghersin, E.; Johnston, P.V.; Brinker, J.A.; et al. Comparison of allogeneic vs autologous bone marrow-derived mesenchymal stem cells delivered by transendocardial injection in patients with ischemic cardiomyopathy: The POSEIDON randomized trial. JAMA 2012, 308, 2369–2379. [Google Scholar] [CrossRef]
- Kudo, M.; Wang, Y.; Wani, M.A.; Xu, M.; Ayub, A.; Ashraf, M. Implantation of bone marrow stem cells reduces the infarction and fibrosis in ischemic mouse heart. J. Mol. Cell. Cardiol. 2003, 35, 1113–1119. [Google Scholar] [CrossRef]
- Li, L.; Zhang, Y.; Li, Y.; Yu, B.; Xu, Y.; Zhao, S.; Guan, Z. Mesenchymal stem cell transplantation attenuates cardiac fibrosis associated with isoproterenol-induced global heart failure. Transpl. Int. 2008, 21, 1181–1189. [Google Scholar] [CrossRef]
- Nagaya, N.; Kangawa, K.; Itoh, T.; Iwase, T.; Murakami, S.; Miyahara, Y.; Fujii, T.; Uematsu, M.; Ohgushi, H.; Yamagishi, M.; et al. Transplantation of mesenchymal stem cells improves cardiac function in a rat model of dilated cardiomyopathy. Circulation 2005, 112, 1128–1135. [Google Scholar] [CrossRef]
- Tang, X.L.; Rokosh, G.; Sanganalmath, S.K.; Yuan, F.; Sato, H.; Mu, J.; Dai, S.; Li, C.; Chen, N.; Peng, Y.; et al. Intracoronary administration of cardiac progenitor cells alleviates left ventricular dysfunction in rats with a 30-day-old infarction. Circulation 2010, 121, 293–305. [Google Scholar] [CrossRef]
- Xu, X.; Xu, Z.; Xu, Y.; Cui, G. Selective down-regulation of extracellular matrix gene expression by bone marrow derived stem cell transplantation into infarcted myocardium. Circ. J. 2005, 69, 1275–1283. [Google Scholar] [CrossRef][Green Version]
- Du, Y.Y.; Zhou, S.H.; Zhou, T.; Su, H.; Pan, H.W.; Du, W.H.; Liu, B.; Liu, Q.M. Immuno-inflammatory regulation effect of mesenchymal stem cell transplantation in a rat model of myocardial infarction. Cytotherapy 2008, 10, 469–478. [Google Scholar] [CrossRef]
- Ohnishi, S.; Yanagawa, B.; Tanaka, K.; Miyahara, Y.; Obata, H.; Kataoka, M.; Kodama, M.; Ishibashi-Ueda, H.; Kangawa, K.; Kitamura, S.; et al. Transplantation of mesenchymal stem cells attenuates myocardial injury and dysfunction in a rat model of acute myocarditis. J. Mol. Cell. Cardiol. 2007, 42, 88–97. [Google Scholar] [CrossRef]
- Feng, Y.; Huang, W.; Wani, M.; Yu, X.; Ashraf, M. Ischemic preconditioning potentiates the protective effect of stem cells through secretion of exosomes by targeting Mecp2 via miR-22. PLoS ONE 2014, 9, e88685. [Google Scholar] [CrossRef]
- Ju, C.; Shen, Y.; Ma, G.; Liu, Y.; Cai, J.; Kim, I.M.; Weintraub, N.L.; Liu, N.; Tang, Y. Transplantation of Cardiac Mesenchymal Stem Cell-Derived Exosomes Promotes Repair in Ischemic Myocardium. J. Cardiovasc. Transl. Res. 2018, 11, 420–428. [Google Scholar] [CrossRef]
- Kishore, R.; Verma, S.K.; Mackie, A.R.; Vaughan, E.E.; Abramova, T.V.; Aiko, I.; Krishnamurthy, P. Bone marrow progenitor cell therapy-mediated paracrine regulation of cardiac miRNA-155 modulates fibrotic response in diabetic hearts. PLoS ONE 2013, 8, e60161. [Google Scholar] [CrossRef]
- Ohnishi, S.; Sumiyoshi, H.; Kitamura, S.; Nagaya, N. Mesenchymal stem cells attenuate cardiac fibroblast proliferation and collagen synthesis through paracrine actions. FEBS Lett. 2007, 581, 3961–3966. [Google Scholar] [CrossRef]
- Yu, B.; Kim, H.W.; Gong, M.; Wang, J.; Millard, R.W.; Wang, Y.; Ashraf, M.; Xu, M. Exosomes secreted from GATA-4 overexpressing mesenchymal stem cells serve as a reservoir of anti-apoptotic microRNAs for cardioprotection. Int. J. Cardiol. 2015, 182, 349–360. [Google Scholar] [CrossRef]
- Gray, W.D.; French, K.M.; Ghosh-Choudhary, S.; Maxwell, J.T.; Brown, M.E.; Platt, M.O.; Searles, C.D.; Davis, M.E. Identification of therapeutic covariant microRNA clusters in hypoxia-treated cardiac progenitor cell exosomes using systems biology. Circ. Res. 2015, 116, 255–263. [Google Scholar] [CrossRef]
- Chen, H.; Xia, R.; Li, Z.; Zhang, L.; Xia, C.; Ai, H.; Yang, Z.; Guo, Y. Mesenchymal Stem Cells Combined with Hepatocyte Growth Factor Therapy for Attenuating Ischaemic Myocardial Fibrosis: Assessment using Multimodal Molecular Imaging. Sci. Rep. 2016, 6, 33700. [Google Scholar] [CrossRef]
- Chen, Y.; Zhao, Y.; Chen, W.; Xie, L.; Zhao, Z.A.; Yang, J.; Lei, W.; Shen, Z. MicroRNA-133 overexpression promotes the therapeutic efficacy of mesenchymal stem cells on acute myocardial infarction. Stem Cell Res. Ther. 2017, 8, 268. [Google Scholar] [CrossRef]
- Mangi, A.A.; Noiseux, N.; Kong, D.; He, H.; Rezvani, M.; Ingwall, J.S.; Dzau, V.J. Mesenchymal stem cells modified with Akt prevent remodeling and restore performance of infarcted hearts. Nat. Med. 2003, 9, 1195–1201. [Google Scholar] [CrossRef] [PubMed]
- Silva, D.N.; Souza, B.S.F.; Azevedo, C.M.; Vasconcelos, J.F.; de Jesus, P.G.; Feitoza, M.S.; Meira, C.S.; Carvalho, G.B.; Cavalcante, B.R.; Ribeiro-Dos-Santos, R.; et al. IGF-1-Overexpressing Mesenchymal Stem/Stromal Cells Promote Immunomodulatory and Proregenerative Effects in Chronic Experimental Chagas Disease. Stem Cells Int. 2018, 2018, 9108681. [Google Scholar] [CrossRef]
- Hoke, N.N.; Salloum, F.N.; Kass, D.A.; Das, A.; Kukreja, R.C. Preconditioning by phosphodiesterase-5 inhibition improves therapeutic efficacy of adipose-derived stem cells following myocardial infarction in mice. Stem Cells 2012, 30, 326–335. [Google Scholar] [CrossRef]
- Khan, M.; Meduru, S.; Mohan, I.K.; Kuppusamy, M.L.; Wisel, S.; Kulkarni, A.; Rivera, B.K.; Hamlin, R.L.; Kuppusamy, P. Hyperbaric oxygenation enhances transplanted cell graft and functional recovery in the infarct heart. J. Mol. Cell. Cardiol. 2009, 47, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Uemura, R.; Xu, M.; Ahmad, N.; Ashraf, M. Bone marrow stem cells prevent left ventricular remodeling of ischemic heart through paracrine signaling. Circ. Res. 2006, 98, 1414–1421. [Google Scholar] [CrossRef] [PubMed]
- Wisel, S.; Khan, M.; Kuppusamy, M.L.; Mohan, I.K.; Chacko, S.M.; Rivera, B.K.; Sun, B.C.; Hideg, K.; Kuppusamy, P. Pharmacological preconditioning of mesenchymal stem cells with trimetazidine (1-[2,3,4-trimethoxybenzyl]piperazine) protects hypoxic cells against oxidative stress and enhances recovery of myocardial function in infarcted heart through Bcl-2 expression. J. Pharmacol. Exp. Ther. 2009, 329, 543–550. [Google Scholar] [CrossRef]
- Ceccaldi, C.; Bushkalova, R.; Alfarano, C.; Lairez, O.; Calise, D.; Bourin, P.; Frugier, C.; Rouzaud-Laborde, C.; Cussac, D.; Parini, A.; et al. Evaluation of polyelectrolyte complex-based scaffolds for mesenchymal stem cell therapy in cardiac ischemia treatment. Acta Biomater. 2014, 10, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Fiumana, E.; Pasquinelli, G.; Foroni, L.; Carboni, M.; Bonafe, F.; Orrico, C.; Nardo, B.; Tsivian, M.; Neri, F.; Arpesella, G.; et al. Localization of mesenchymal stem cells grafted with a hyaluronan-based scaffold in the infarcted heart. J. Surg. Res. 2013, 179, e21–e29. [Google Scholar] [CrossRef]
- Sun, C.K.; Zhen, Y.Y.; Leu, S.; Tsai, T.H.; Chang, L.T.; Sheu, J.J.; Chen, Y.L.; Chua, S.; Chai, H.T.; Lu, H.I.; et al. Direct implantation versus platelet-rich fibrin-embedded adipose-derived mesenchymal stem cells in treating rat acute myocardial infarction. Int. J. Cardiol. 2014, 173, 410–423. [Google Scholar] [CrossRef]
- Xia, Y.; Zhu, K.; Lai, H.; Lang, M.; Xiao, Y.; Lian, S.; Guo, C.; Wang, C. Enhanced infarct myocardium repair mediated by thermosensiive copolymer hydrogel-based stem cell transplantation. Exp. Biol. Med. 2015, 240, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Boheler, K.R.; Czyz, J.; Tweedie, D.; Yang, H.T.; Anisimov, S.V.; Wobus, A.M. Differentiation of pluripotent embryonic stem cells into cardiomyocytes. Circ. Res. 2002, 91, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Singla, D.K. Akt-mTOR Pathway Inhibits Apoptosis and Fibrosis in Doxorubicin-Induced Cardiotoxicity Following Embryonic Stem Cell Transplantation. Cell Transpl. 2015, 24, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Singla, D.K.; Ahmed, A.; Singla, R.; Yan, B. Embryonic stem cells improve cardiac function in Doxorubicin-induced cardiomyopathy mediated through multiple mechanisms. Cell Transpl. 2012, 21, 1919–1930. [Google Scholar] [CrossRef] [PubMed]
- Singla, D.K.; Lyons, G.E.; Kamp, T.J. Transplanted embryonic stem cells following mouse myocardial infarction inhibit apoptosis and cardiac remodeling. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H1308–H1314. [Google Scholar] [CrossRef]
- Glass, C.; Singla, D.K. Overexpression of TIMP-1 in embryonic stem cells attenuates adverse cardiac remodeling following myocardial infarction. Cell Transpl. 2012, 21, 1931–1944. [Google Scholar] [CrossRef]
- Burt, R.K.; Chen, Y.H.; Verda, L.; Lucena, C.; Navale, S.; Johnson, J.; Han, X.; Lomasney, J.; Baker, J.M.; Ngai, K.L.; et al. Mitotically inactivated embryonic stem cells can be used as an in vivo feeder layer to nurse damaged myocardium after acute myocardial infarction: A preclinical study. Circ. Res. 2012, 111, 1286–1296. [Google Scholar] [CrossRef]
- Khan, M.; Nickoloff, E.; Abramova, T.; Johnson, J.; Verma, S.K.; Krishnamurthy, P.; Mackie, A.R.; Vaughan, E.; Garikipati, V.N.; Benedict, C.; et al. Embryonic stem cell-derived exosomes promote endogenous repair mechanisms and enhance cardiac function following myocardial infarction. Circ. Res. 2015, 117, 52–64. [Google Scholar] [CrossRef]
- Bagno, L.; Hatzistergos, K.E.; Balkan, W.; Hare, J.M. Mesenchymal Stem Cell-Based Therapy for Cardiovascular Disease: Progress and Challenges. Mol. Ther. 2018, 26, 1610–1623. [Google Scholar] [CrossRef]
- Boudoulas, K.D.; Hatzopoulos, A.K. Cardiac repair and regeneration: The Rubik’s cube of cell therapy for heart disease. Dis. Model. Mech. 2009, 2, 344–358. [Google Scholar] [CrossRef]
- Florea, V.; Rieger, A.C.; DiFede, D.L.; El-Khorazaty, J.; Natsumeda, M.; Banerjee, M.N.; Tompkins, B.A.; Khan, A.; Schulman, I.H.; Landin, A.M.; et al. Dose Comparison Study of Allogeneic Mesenchymal Stem Cells in Patients with Ischemic Cardiomyopathy (The TRIDENT Study). Circ. Res. 2017, 121, 1279–1290. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Yu, Y.; Hu, S.; Chen, Y.; Shen, Z. The therapeutic potential of mesenchymal stem cells for cardiovascular diseases. Cell Death Dis. 2020, 11, 349. [Google Scholar] [CrossRef] [PubMed]
- Michler, R.E. The current status of stem cell therapy in ischemic heart disease. J. Card. Surg. 2018, 33, 520–531. [Google Scholar] [CrossRef] [PubMed]
- Bartolucci, J.; Verdugo, F.J.; Gonzalez, P.L.; Larrea, R.E.; Abarzua, E.; Goset, C.; Rojo, P.; Palma, I.; Lamich, R.; Pedreros, P.A.; et al. Safety and Efficacy of the Intravenous Infusion of Umbilical Cord Mesenchymal Stem Cells in Patients With Heart Failure: A Phase 1/2 Randomized Controlled Trial (RIMECARD Trial [Randomized Clinical Trial of Intravenous Infusion Umbilical Cord Mesenchymal Stem Cells on Cardiopathy]). Circ. Res. 2017, 121, 1192–1204. [Google Scholar] [PubMed]
- Gao, L.R.; Chen, Y.; Zhang, N.K.; Yang, X.L.; Liu, H.L.; Wang, Z.G.; Yan, X.Y.; Wang, Y.; Zhu, Z.M.; Li, T.C.; et al. Intracoronary infusion of Wharton’s jelly-derived mesenchymal stem cells in acute myocardial infarction: Double-blind, randomized controlled trial. BMC Med. 2015, 13, 162. [Google Scholar] [CrossRef] [PubMed]
- Kastrup, J.; Haack-Sorensen, M.; Juhl, M.; Sondergaard, R.H.; Follin, B.; Lund, L.D.; Johansen, E.M.; Qayyum, A.A.; Mathiasen, A.B.; Jorgensen, E.; et al. Cryopreserved Off-the-Shelf Allogeneic Adipose-Derived Stromal Cells for Therapy in Patients with Ischemic Heart Disease and Heart Failure-A Safety Study. Stem Cells Transl. Med. 2017, 6, 1963–1971. [Google Scholar] [CrossRef]
- Caspi, O.; Huber, I.; Kehat, I.; Habib, M.; Arbel, G.; Gepstein, A.; Yankelson, L.; Aronson, D.; Beyar, R.; Gepstein, L. Transplantation of human embryonic stem cell-derived cardiomyocytes improves myocardial performance in infarcted rat hearts. J. Am. Coll. Cardiol. 2007, 50, 1884–1893. [Google Scholar] [CrossRef]
- Vernon, M.A.; Mylonas, K.J.; Hughes, J. Macrophages and renal fibrosis. Semin. Nephrol. 2010, 30, 302–317. [Google Scholar] [CrossRef] [PubMed]
- Bulow, R.D.; Boor, P. Extracellular Matrix in Kidney Fibrosis: More Than Just a Scaffold. J. Histochem. Cytochem. 2019, 67, 643–661. [Google Scholar] [CrossRef]
- Djudjaj, S.; Boor, P. Cellular and molecular mechanisms of kidney fibrosis. Mol. Asp. Med. 2019, 65, 16–36. [Google Scholar] [CrossRef]
- Brennan, E.P.; Cacace, A.; Godson, C. Specialized pro-resolving mediators in renal fibrosis. Mol. Asp. Med. 2017, 58, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Francois, H.; Chatziantoniou, C. Renal fibrosis: Recent translational aspects. Matrix Biol. 2018, 68–69, 318–332. [Google Scholar] [CrossRef] [PubMed]
- Klinkhammer, B.M.; Goldschmeding, R.; Floege, J.; Boor, P. Treatment of Renal Fibrosis-Turning Challenges into Opportunities. Adv. Chronic Kidney Dis. 2017, 24, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Lee, J.S.; Lee, H.K.; Park, E.J.; Jeon, H.W.; Kang, Y.J.; Lee, T.Y.; Kim, K.S.; Bae, S.C.; Park, J.H.; et al. Mesenchymal Stem Cells Ameliorate Renal Inflammation in Adriamycin-induced Nephropathy. Immune Netw. 2019, 19, e36. [Google Scholar] [CrossRef] [PubMed]
- Lira, R.; Oliveira, M.; Martins, M.; Silva, C.; Carvalho, S.; Stumbo, A.C.; Cortez, E.; Verdoorn, K.; Einicker-Lamas, M.; Thole, A.; et al. Transplantation of bone marrow-derived MSCs improves renal function and Na(+)+K(+)-ATPase activity in rats with renovascular hypertension. Cell Tissue Res. 2017, 369, 287–301. [Google Scholar] [CrossRef]
- Semedo, P.; Correa-Costa, M.; Cenedeze, M.A.; Malheiros, D.M.A.C.; Reis, M.A.d.; Shimizu, M.H.; Seguro, A.C.; Pacheco-Silva, A.; Camara, N.O.S. Mesenchymal stem cells attenuate renal fibrosis through immune modulation and remodeling properties in a rat remnant kidney model. Stem Cells 2009, 27, 3063–3073. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.J.; Yiu, W.H.; Li, R.X.; Wong, D.W.; Leung, J.C.; Chan, L.Y.; Zhang, Y.; Lian, Q.; Lin, M.; Tse, H.F.; et al. Mesenchymal stem cells modulate albumin-induced renal tubular inflammation and fibrosis. PLoS ONE 2014, 9, e90883. [Google Scholar] [CrossRef]
- Alfarano, C.; Roubeix, C.; Chaaya, R.; Ceccaldi, C.; Calise, D.; Mias, C.; Cussac, D.; Bascands, J.L.; Parini, A. Intraparenchymal injection of bone marrow mesenchymal stem cells reduces kidney fibrosis after ischemia-reperfusion in cyclosporine-immunosuppressed rats. Cell Transpl. 2012, 21, 2009–2019. [Google Scholar] [CrossRef]
- Zhang, T.; Liu, G.; Sun, M.; Guan, G.; Chen, B.; Li, X. Functional, histological and biochemical consequences of renal lymph disorder in mononephrectomized rats. J. Nephrol. 2009, 22, 109–116. [Google Scholar]
- Boor, P.; Floege, J. Renal allograft fibrosis: Biology and therapeutic targets. Am. J. Transpl. 2015, 15, 863–886. [Google Scholar] [CrossRef]
- Canaud, G.; Bonventre, J.V. Cell cycle arrest and the evolution of chronic kidney disease from acute kidney injury. Nephrol. Dial. Transpl. 2015, 30, 575–583. [Google Scholar] [CrossRef]
- Zhu, F.; Shin, O.L.S.C.L.; Pei, G.; Hu, Z.; Yang, J.; Zhu, H.; Wang, M.; Mou, J.; Sun, J.; Wang, Y.; et al. Adipose-derived mesenchymal stem cells employed exosomes to attenuate AKI-CKD transition through tubular epithelial cell dependent Sox9 activation. Oncotarget 2017, 8, 70707–70726. [Google Scholar] [CrossRef]
- Gatti, S.; Bruno, S.; Deregibus, M.C.; Sordi, A.; Cantaluppi, V.; Tetta, C.; Camussi, G. Microvesicles derived from human adult mesenchymal stem cells protect against ischaemia-reperfusion-induced acute and chronic kidney injury. Nephrol. Dial. Transpl. 2011, 26, 1474–1483. [Google Scholar] [CrossRef] [PubMed]
- Kholia, S.; Sanchez, M.B.H.; Cedrino, M.; Papadimitriou, E.; Tapparo, M.; Deregibus, M.C.; Bruno, S.; Antico, F.; Brizzi, M.F.; Quesenberry, P.J.; et al. Mesenchymal Stem Cell Derived Extracellular Vesicles Ameliorate Kidney Injury in Aristolochic Acid Nephropathy. Front. Cell Dev. Biol. 2020, 8, 188. [Google Scholar] [CrossRef] [PubMed]
- Grange, C.; Tritta, S.; Tapparo, M.; Cedrino, M.; Tetta, C.; Camussi, G.; Brizzi, M.F. Stem cell-derived extracellular vesicles inhibit and revert fibrosis progression in a mouse model of diabetic nephropathy. Sci. Rep. 2019, 9, 4468. [Google Scholar] [CrossRef]
- Wang, Y.; Guo, Y.F.; Fu, G.P.; Guan, C.; Zhang, X.; Yang, D.G.; Shi, Y.C. Protective effect of miRNA-containing extracellular vesicles derived from mesenchymal stromal cells of old rats on renal function in chronic kidney disease. Stem Cell Res. Ther. 2020, 11, 274. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.J.; Zhang, X.; Jiang, K.; Krier, J.D.; Zhu, X.; Conley, S.; Lerman, A.; Lerman, L.O. Adjunctive mesenchymal stem/stromal cells augment microvascular function in poststenotic kidneys treated with low-energy shockwave therapy. J. Cell Physiol. 2020, 235, 9806–9818. [Google Scholar] [CrossRef]
- Eirin, A.; Zhang, X.; Zhu, X.Y.; Tang, H.; Jordan, K.L.; Grande, J.P.; Dietz, A.B.; Lerman, A.; Textor, S.C.; Lerman, L.O. Renal vein cytokine release as an index of renal parenchymal inflammation in chronic experimental renal artery stenosis. Nephrol. Dial. Transpl. 2014, 29, 274–282. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhu, X.; Zhang, L.; Ferguson, C.M.; Song, T.; Jiang, K.; Conley, S.M.; Krier, J.D.; Tang, H.; Saadiq, I.; et al. Mesenchymal Stem/Stromal Cells and their Extracellular Vesicle Progeny Decrease Injury in Poststenotic Swine Kidney Through Different Mechanisms. Stem Cells Dev. 2020, 29, 1190–1200. [Google Scholar] [CrossRef]
- Choi, H.Y.; Lee, H.G.; Kim, B.S.; Ahn, S.H.; Jung, A.; Lee, M.; Lee, J.E.; Kim, H.J.; Ha, S.K.; Park, H.C. Mesenchymal stem cell-derived microparticles ameliorate peritubular capillary rarefaction via inhibition of endothelial-mesenchymal transition and decrease tubulointerstitial fibrosis in unilateral ureteral obstruction. Stem Cell Res. Ther. 2015, 6, 18. [Google Scholar] [CrossRef]
- Chen, L.; Wang, Y.; Li, S.; Zuo, B.; Zhang, X.; Wang, F.; Sun, D. Exosomes derived from GDNF-modified human adipose mesenchymal stem cells ameliorate peritubular capillary loss in tubulointerstitial fibrosis by activating the SIRT1/eNOS signaling pathway. Theranostics 2020, 10, 9425–9442. [Google Scholar] [CrossRef]
- Ozbek, E.; Adas, G.; Otunctemur, A.; Duruksu, G.; Koc, B.; Polat, E.C.; Sarvan, A.K.; Okcu, A.; Kamali, G.; Subasi, C.; et al. Role of Mesenchymal Stem Cells Transfected With Vascular Endothelial Growth Factor in Maintaining Renal Structure and Function in Rats with Unilateral Ureteral Obstruction. Exp. Clin. Transpl. 2015, 13, 262–272. [Google Scholar]
- Wang, B.; Yao, K.; Huuskes, B.M.; Shen, H.H.; Zhuang, J.; Godson, C.; Brennan, E.P.; Wilkinson-Berka, J.L.; Wise, A.F.; Ricardo, S.D. Mesenchymal Stem Cells Deliver Exogenous MicroRNA-let7c via Exosomes to Attenuate Renal Fibrosis. Mol. Ther. 2016, 24, 1290–1301. [Google Scholar] [CrossRef]
- Xie, M.; Wan, J.; Zhang, F.; Zhang, R.; Zhou, Z.; You, D. Influence of hepatocyte growth factor-transfected bone marrow-derived mesenchymal stem cells towards renal fibrosis in rats. Indian J. Med. Res. 2019, 149, 508–516. [Google Scholar] [PubMed]
- Huuskes, B.M.; Wise, A.F.; Cox, A.J.; Lim, E.X.; Payne, N.L.; Kelly, D.J.; Samuel, C.S.; Ricardo, S.D. Combination therapy of mesenchymal stem cells and serelaxin effectively attenuates renal fibrosis in obstructive nephropathy. FASEB J. 2015, 29, 540–553. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Lv, S.; Liu, J.; Liu, S.; Wang, Y.; Liu, G. Mesenchymal stem cells modified with angiotensin-converting enzyme 2 are superior for amelioration of glomerular fibrosis in diabetic nephropathy. Diabetes Res. Clin. Pract. 2020, 162, 108093. [Google Scholar] [CrossRef] [PubMed]
- Kanai, R.; Nakashima, A.; Doi, S.; Kimura, T.; Yoshida, K.; Maeda, S.; Ishiuchi, N.; Yamada, Y.; Ike, T.; Doi, T.; et al. Interferon-gamma enhances the therapeutic effect of mesenchymal stem cells on experimental renal fibrosis. Sci. Rep. 2021, 11, 850. [Google Scholar] [CrossRef] [PubMed]
- Saberi, K.; Pasbakhsh, P.; Omidi, A.; Borhani-Haghighi, M.; Nekoonam, S.; Omidi, N.; Ghasemi, S.; Kashani, I.R. Melatonin preconditioning of bone marrow-derived mesenchymal stem cells promotes their engraftment and improves renal regeneration in a rat model of chronic kidney disease. J. Mol. Histol. 2019, 50, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhao, Y.; Wang, Z.; Wang, J.; Liu, C.; Sun, D. Transplantation of Amniotic Fluid-Derived Stem Cells Preconditioned with Glial Cell Line-Derived Neurotrophic Factor Gene Alleviates Renal Fibrosis. Cell Transpl. 2019, 28, 65–78. [Google Scholar] [CrossRef]
- Li, H.; Rong, P.; Ma, X.; Nie, W.; Chen, Y.; Zhang, J.; Dong, Q.; Yang, M.; Wang, W. Mouse Umbilical Cord Mesenchymal Stem Cell Paracrine Alleviates Renal Fibrosis in Diabetic Nephropathy by Reducing Myofibroblast Transdifferentiation and Cell Proliferation and Upregulating MMPs in Mesangial Cells. J. Diabetes Res. 2020, 2020, 3847171. [Google Scholar] [CrossRef]
- Liu, B.; Ding, F.; Hu, D.; Zhou, Y.; Long, C.; Shen, L.; Zhang, Y.; Zhang, D.; Wei, G. Human umbilical cord mesenchymal stem cell conditioned medium attenuates renal fibrosis by reducing inflammation and epithelial-to-mesenchymal transition via the TLR4/NF-kappaB signaling pathway in vivo and in vitro. Stem Cell Res. Ther. 2018, 9, 7. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Wu, Y.; Xu, Y.; Sun, L.; Zhang, X. Human umbilical mesenchymal stem cells attenuate the progression of focal segmental glomerulosclerosis. Am. J. Med. Sci. 2013, 346, 486–493. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Park, J.; Hwang, S.H.; Han, H.; Ha, H. Delayed treatment with human umbilical cord blood-derived stem cells attenuates diabetic renal injury. Transplant. Proc. 2012, 44, 1123–1126. [Google Scholar] [CrossRef] [PubMed]
- Xiang, E.; Han, B.; Zhang, Q.; Rao, W.; Wang, Z.; Chang, C.; Zhang, Y.; Tu, C.; Li, C.; Wu, D. Human umbilical cord-derived mesenchymal stem cells prevent the progression of early diabetic nephropathy through inhibiting inflammation and fibrosis. Stem Cell Res. Ther. 2020, 11, 336. [Google Scholar] [CrossRef]
- Yu, Y.; Hu, D.; Zhou, Y.; Xiang, H.; Liu, B.; Shen, L.; Long, C.; Liu, X.; Lin, T.; He, D.; et al. Human umbilical cord mesenchymal stem cell attenuates renal fibrosis via TGF-beta/Smad signaling pathways in vivo and in vitro. Eur. J. Pharmacol. 2020, 883, 173343. [Google Scholar] [CrossRef]
- Kobayashi, T.; Tanaka, H.; Kuwana, H.; Inoshita, S.; Teraoka, H.; Sasaki, S.; Terada, Y. Wnt4-transformed mouse embryonic stem cells differentiate into renal tubular cells. Biochem. Biophys. Res. Commun. 2005, 336, 585–595. [Google Scholar] [CrossRef]
- Narayanan, K.; Schumacher, K.M.; Tasnim, F.; Kandasamy, K.; Schumacher, A.; Ni, M.; Gao, S.; Gopalan, B.; Zink, D.; Ying, J.Y. Human embryonic stem cells differentiate into functional renal proximal tubular-like cells. Kidney Int. 2013, 83, 593–603. [Google Scholar] [CrossRef]
- Geng, X.D.; Zheng, W.; Wu, C.M.; Wang, S.Q.; Hong, Q.; Cai, G.Y.; Chen, X.M.; Wu, D. Embryonic Stem Cells-loaded Gelatin Microcryogels Slow Progression of Chronic Kidney Disease. Chin. Med. J. 2016, 129, 392–398. [Google Scholar] [CrossRef]
- De Chiara, L.; Fagoonee, S.; Ranghino, A.; Bruno, S.; Camussi, G.; Tolosano, E.; Silengo, L.; Altruda, F. Renal cells from spermatogonial germline stem cells protect against kidney injury. J. Am. Soc. Nephrol. 2014, 25, 316–328. [Google Scholar] [CrossRef]
- Ciampi, O.; Iacone, R.; Longaretti, L.; Benedetti, V.; Graf, M.; Magnone, M.C.; Patsch, C.; Xinaris, C.; Remuzzi, G.; Benigni, A.; et al. Generation of functional podocytes from human induced pluripotent stem cells. Stem Cell Res. 2016, 17, 130–139. [Google Scholar] [CrossRef]
- Qian, T.; Hernday, S.E.; Bao, X.; Olson, W.R.; Panzer, S.E.; Shusta, E.V.; Palecek, S.P. Directed Differentiation of Human Pluripotent Stem Cells to Podocytes under Defined Conditions. Sci. Rep. 2019, 9, 2765. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, P.C.; Lojudice, F.H.; Fernandes-Charpiot, I.M.M.; Baptista, M.; Araujo, S.D.; Mendes, G.E.F.; Sogayar, M.C.; Abbud-Filho, M.; Caldas, H.C. Therapeutic potential of human induced pluripotent stem cells and renal progenitor cells in experimental chronic kidney disease. Stem Cell Res. Ther. 2020, 11, 530. [Google Scholar] [CrossRef] [PubMed]
- Caldas, H.C.; Lojudice, F.H.; Dias, C.; Fernandes-Charpiot, I.M.M.; Baptista, M.; Kawasaki-Oyama, R.S.; Sogayar, M.C.; Takiya, C.M.; Abbud-Filho, M. Induced Pluripotent Stem Cells Reduce Progression of Experimental Chronic Kidney Disease but Develop Wilms’ Tumors. Stem Cells Int. 2017, 2017, 7428316. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, M.; Stafford-Smith, M.; Chertow, G.M.; Warnock, D.G.; Paragamian, V.; Brenner, R.M.; Lellouche, F.; Fox-Robichaud, A.; Atta, M.G.; Melby, S.; et al. Allogeneic Mesenchymal Stem Cells for Treatment of AKI after Cardiac Surgery. J. Am. Soc. Nephrol. 2018, 29, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Makhlough, A.; Shekarchian, S.; Moghadasali, R.; Einollahi, B.; Hosseini, S.E.; Jaroughi, N.; Bolurieh, T.; Baharvand, H.; Aghdami, N. Safety and tolerability of autologous bone marrow mesenchymal stromal cells in ADPKD patients. Stem Cell Res. Ther. 2017, 8, 116. [Google Scholar] [CrossRef]
- Evans-Hoeker, E.A.; Young, S.L. Endometrial receptivity and intrauterine adhesive disease. Semin. Reprod. Med. 2014, 32, 392–401. [Google Scholar] [PubMed]
- Yu, D.; Wong, Y.M.; Cheong, Y.; Xia, E.; Li, T.C. Asherman syndrome—One century later. Fertil. Steril. 2008, 89, 759–779. [Google Scholar] [CrossRef]
- March, C.M. Asherman’s syndrome. Semin. Reprod. Med. 2011, 29, 83–94. [Google Scholar] [CrossRef]
- Chen, F.P.; Soong, Y.K.; Hui, Y.L. Successful treatment of severe uterine synechiae with transcervical resectoscopy combined with laminaria tent. Hum. Reprod. 1997, 12, 943–947. [Google Scholar] [CrossRef]
- Hooker, A.B.; de Leeuw, R.; van de Ven, P.M.; Bakkum, E.A.; Thurkow, A.L.; Vogel, N.E.A.; van Vliet, H.; Bongers, M.Y.; Emanuel, M.H.; Verdonkschot, A.E.M.; et al. Prevalence of intrauterine adhesions after the application of hyaluronic acid gel after dilatation and curettage in women with at least one previous curettage: Short-term outcomes of a multicenter, prospective randomized controlled trial. Fertil. Steril. 2017, 107, 1223.e3–1231.e3. [Google Scholar] [CrossRef]
- Lin, X.N.; Zhou, F.; Wei, M.L.; Yang, Y.; Li, Y.; Li, T.C.; Zhang, S.Y. Randomized, controlled trial comparing the efficacy of intrauterine balloon and intrauterine contraceptive device in the prevention of adhesion reformation after hysteroscopic adhesiolysis. Fertil. Steril. 2015, 104, 235–240. [Google Scholar] [CrossRef]
- Schenker, J.G. Etiology of and therapeutic approach to synechia uteri. Eur. J. Obstet. Gynecol. Reprod. Biol. 1996, 65, 109–113. [Google Scholar] [CrossRef]
- Alawadhi, F.; Du, H.; Cakmak, H.; Taylor, H.S. Bone Marrow-Derived Stem Cell (BMDSC) transplantation improves fertility in a murine model of Asherman’s syndrome. PLoS ONE 2014, 9, e96662. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Shekhar, B.; Mohanty, S.; Kumar, S.; Seth, T.; Girish, B. Autologous Bone Marrow-Derived Stem Cell Therapy for Asherman’s Syndrome and Endometrial Atrophy: A 5-Year Follow-up Study. J. Hum. Reprod. Sci. 2020, 13, 31–37. [Google Scholar] [CrossRef]
- Cervello, I.; Gil-Sanchis, C.; Santamaria, X.; Cabanillas, S.; Diaz, A.; Faus, A.; Pellicer, A.; Simon, C. Human CD133(+) bone marrow-derived stem cells promote endometrial proliferation in a murine model of Asherman syndrome. Fertil. Steril. 2015, 104, 1552.e1-3–1560.e1-3. [Google Scholar] [CrossRef] [PubMed]
- Domnina, A.; Novikova, P.; Obidina, J.; Fridlyanskaya, I.; Alekseenko, L.; Kozhukharova, I.; Lyublinskaya, O.; Zenin, V.; Nikolsky, N. Human mesenchymal stem cells in spheroids improve fertility in model animals with damaged endometrium. Stem Cell Res. Ther. 2018, 9, 50. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Zhang, Q.; Sun, J.; Lai, D. Human amniotic epithelial cells improve fertility in an intrauterine adhesion mouse model. Stem Cell Res. Ther. 2019, 10, 257. [Google Scholar] [CrossRef] [PubMed]
- Santamaria, X.; Cabanillas, S.; Cervello, I.; Arbona, C.; Raga, F.; Ferro, J.; Palmero, J.; Remohi, J.; Pellicer, A.; Simon, C. Autologous cell therapy with CD133+ bone marrow-derived stem cells for refractory Asherman’s syndrome and endometrial atrophy: A pilot cohort study. Hum. Reprod. 2016, 31, 1087–1096. [Google Scholar] [CrossRef]
- Lee, S.Y.; Shin, J.E.; Kwon, H.; Choi, D.H.; Kim, J.H. Effect of Autologous Adipose-Derived Stromal Vascular Fraction Transplantation on Endometrial Regeneration in Patients of Asherman’s Syndrome: A Pilot Study. Reprod. Sci. 2020, 27, 561–568. [Google Scholar] [CrossRef]
- Cao, Y.; Sun, H.; Zhu, H.; Zhu, X.; Tang, X.; Yan, G.; Wang, J.; Bai, D.; Wang, L.; Zhou, Q.; et al. Allogeneic cell therapy using umbilical cord MSCs on collagen scaffolds for patients with recurrent uterine adhesion: A phase I clinical trial. Stem Cell Res. Ther. 2018, 9, 192. [Google Scholar] [CrossRef]
- Tan, J.; Li, P.; Wang, Q.; Li, Y.; Li, X.; Zhao, D.; Xu, X.; Kong, L. Autologous menstrual blood-derived stromal cells transplantation for severe Asherman’s syndrome. Hum. Reprod. 2016, 31, 2723–2729. [Google Scholar] [CrossRef]
- Canady, J.; Karrer, S.; Fleck, M.; Bosserhoff, A.K. Fibrosing connective tissue disorders of the skin: Molecular similarities and distinctions. J. Dermatol. Sci. 2013, 70, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Christner, P.J.; Artlett, C.M.; Conway, R.F.; Jimenez, S.A. Increased numbers of microchimeric cells of fetal origin are associated with dermal fibrosis in mice following injection of vinyl chloride. Arthritis Rheum. 2000, 43, 2598–2605. [Google Scholar] [CrossRef]
- Servettaz, A.; Goulvestre, C.; Kavian, N.; Nicco, C.; Guilpain, P.; Chereau, C.; Vuiblet, V.; Guillevin, L.; Mouthon, L.; Weill, B.; et al. Selective oxidation of DNA topoisomerase 1 induces systemic sclerosis in the mouse. J. Immunol. 2009, 182, 5855–5864. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T. Animal model of systemic sclerosis. J. Dermatol. 2010, 37, 26–41. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Melichian, D.; Komura, K.; Hinchcliff, M.; Lam, A.P.; Lafyatis, R.; Gottardi, C.J.; MacDougald, O.A.; Varga, J. Canonical Wnt signaling induces skin fibrosis and subcutaneous lipoatrophy: A novel mouse model for scleroderma? Arthritis. Rheum. 2011, 63, 1707–1717. [Google Scholar] [CrossRef] [PubMed]
- Medsger, T.A., Jr. Epidemiology of systemic sclerosis. Clin. Dermatol. 1994, 12, 207–216. [Google Scholar] [CrossRef]
- Gilliam, A.C. Scleroderma. Curr. Dir. Autoimmun. 2008, 10, 258–279. [Google Scholar]
- Gurtner, G.C.; Werner, S.; Barrandon, Y.; Longaker, M.T. Wound repair and regeneration. Nature 2008, 453, 314–321. [Google Scholar] [CrossRef]
- Lorenz, H.P.; Adzick, N.S. Scarless Skin Wound Repair in the Fetus. West. J. Med. 1993, 159, 350–355. [Google Scholar]
- Merkel, J.R.; DiPaolo, B.R.; Hallock, G.G.; Rice, D.C. Type I and type III collagen content of healing wounds in fetal and adult rats. Proc. Soc. Exp. Biol Med. 1988, 187, 493–497. [Google Scholar] [CrossRef] [PubMed]
- Limandjaja, G.C.; Niessen, F.B.; Scheper, R.J.; Gibbs, S. Hypertrophic scars and keloids: Overview of the evidence and practical guide for differentiating between these abnormal scars. Exp. Dermatol. 2021, 30, 146–161. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, K.; Chen, C.; Wang, D.; Xu, X.; Qu, C.; Yamaza, T.; Cai, T.; Chen, W.; Sun, L.; Shi, S. Mesenchymal-stem-cell-induced immunoregulation involves FAS-ligand-/FAS-mediated T cell apoptosis. Cell Stem Cell 2012, 10, 544–555. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Wang, D.; Moshaverinia, A.; Liu, D.; Kou, X.; Yu, W.; Yang, R.; Sun, L.; Shi, S. Mesenchymal stem cell transplantation in tight-skin mice identifies miR-151-5p as a therapeutic target for systemic sclerosis. Cell Res. 2017, 27, 559–577. [Google Scholar] [CrossRef]
- Maria, A.T.; Toupet, K.; Bony, C.; Pirot, N.; Vozenin, M.C.; Petit, B.; Roger, P.; Batteux, F.; le Quellec, A.; Jorgensen, C.; et al. Antifibrotic, Antioxidant, and Immunomodulatory Effects of Mesenchymal Stem Cells in HOCl-Induced Systemic Sclerosis. Arthritis Rheumatol. 2016, 68, 1013–1025. [Google Scholar] [CrossRef]
- Okamura, A.; Matsushita, T.; Komuro, A.; Kobayashi, T.; Maeda, S.; Hamaguchi, Y.; Takehara, K. Adipose-derived stromal/stem cells successfully attenuate the fibrosis of scleroderma mouse models. Int. J. Rheum. Dis. 2020, 23, 216–225. [Google Scholar] [CrossRef]
- Rubio, G.A.; Elliot, S.J.; Wikramanayake, T.C.; Xia, X.; Pereira-Simon, S.; Thaller, S.R.; Glinos, G.D.; Jozic, I.; Hirt, P.; Pastar, I.; et al. Mesenchymal stromal cells prevent bleomycin-induced lung and skin fibrosis in aged mice and restore wound healing. J. Cell Physiol. 2018, 233, 5503–5512. [Google Scholar] [CrossRef]
- Domergue, S.; Bony, C.; Maumus, M.; Toupet, K.; Frouin, E.; Rigau, V.; Vozenin, M.C.; Magalon, G.; Jorgensen, C.; Noel, D. Comparison between Stromal Vascular Fraction and Adipose Mesenchymal Stem Cells in Remodeling Hypertrophic Scars. PLoS ONE 2016, 11, e0156161. [Google Scholar] [CrossRef]
- Liu, J.; Ren, J.; Su, L.; Cheng, S.; Zhou, J.; Ye, X.; Dong, Y.; Sun, S.; Qi, F.; Liu, Z.; et al. Human adipose tissue-derived stem cells inhibit the activity of keloid fibroblasts and fibrosis in a keloid model by paracrine signaling. Burns 2018, 44, 370–385. [Google Scholar] [CrossRef]
- Henrique-Neto, A.; Vasconcelos, M.Y.K.; Dias, J.B.E.; de Moraes, D.A.; Goncalves, M.S.; Zanin-Silva, D.C.; Zucoloto, T.G.; de Oliveira, M.F.C.; Dotoli, G.M.; Weffort, L.F.; et al. Hematopoietic stem cell transplantation for systemic sclerosis: Brazilian experience. Adv. Rheumatol. 2021, 61, 9. [Google Scholar] [CrossRef]
- Burt, R.K.; Shah, S.J.; Dill, K.; Grant, T.; Gheorghiade, M.; Schroeder, J.; Craig, R.; Hirano, I.; Marshall, K.; Ruderman, E.; et al. Autologous non-myeloablative haemopoietic stem-cell transplantation compared with pulse cyclophosphamide once per month for systemic sclerosis (ASSIST): An open-label, randomised phase 2 trial. Lancet 2011, 378, 498–506. [Google Scholar] [CrossRef]
- van Laar, J.M.; Farge, D.; Sont, J.K.; Naraghi, K.; Marjanovic, Z.; Larghero, J.; Schuerwegh, A.J.; Marijt, E.W.; Vonk, M.C.; Schattenberg, A.V.; et al. Autologous hematopoietic stem cell transplantation vs intravenous pulse cyclophosphamide in diffuse cutaneous systemic sclerosis: A randomized clinical trial. JAMA 2014, 311, 2490–2498. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, K.M.; Goldmuntz, E.A.; Keyes-Elstein, L.; McSweeney, P.A.; Pinckney, A.; Welch, B.; Mayes, M.D.; Nash, R.A.; Crofford, L.J.; Eggleston, B.; et al. Myeloablative Autologous Stem-Cell Transplantation for Severe Scleroderma. N. Engl. J. Med. 2018, 378, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Scuderi, N.; Ceccarelli, S.; Onesti, M.G.; Fioramonti, P.; Guidi, C.; Romano, F.; Frati, L.; Angeloni, A.; Marchese, C. Human adipose-derived stromal cells for cell-based therapies in the treatment of systemic sclerosis. Cell Transpl. 2013, 22, 779–795. [Google Scholar] [CrossRef] [PubMed]
- Granel, B.; Daumas, A.; Jouve, E.; Harle, J.R.; Nguyen, P.S.; Chabannon, C.; Colavolpe, N.; Reynier, J.C.; Truillet, R.; Mallet, S.; et al. Safety, tolerability and potential efficacy of injection of autologous adipose-derived stromal vascular fraction in the fingers of patients with systemic sclerosis: An open-label phase I trial. Ann. Rheum Dis. 2015, 74, 2175–2182. [Google Scholar] [CrossRef]
- Almadori, A.; Griffin, M.; Ryan, C.M.; Hunt, D.F.; Hansen, E.; Kumar, R.; Abraham, D.J.; Denton, C.P.; Butler, P.E.M. Stem cell enriched lipotransfer reverses the effects of fibrosis in systemic sclerosis. PLoS ONE 2019, 14, e0218068. [Google Scholar] [CrossRef]
- Del Papa, N.; Caviggioli, F.; Sambataro, D.; Zaccara, E.; Vinci, V.; di Luca, G.; Parafioriti, A.; Armiraglio, E.; Maglione, W.; Polosa, R.; et al. Autologous fat grafting in the treatment of fibrotic perioral changes in patients with systemic sclerosis. Cell Transpl. 2015, 24, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Nussbaum, J.; Minami, E.; Laflamme, M.A.; Virag, J.A.; Ware, C.B.; Masino, A.; Muskheli, V.; Pabon, L.; Reinecke, H.; Murry, C.E. Transplantation of undifferentiated murine embryonic stem cells in the heart: Teratoma formation and immune response. FASEB J. 2007, 21, 1345–1357. [Google Scholar] [CrossRef]
- Murry, C.E.; Keller, G. Differentiation of embryonic stem cells to clinically relevant populations: Lessons from embryonic development. Cell 2008, 132, 661–680. [Google Scholar] [CrossRef] [PubMed]
- Kroon, E.; Martinson, L.A.; Kadoya, K.; Bang, A.G.; Kelly, O.G.; Eliazer, S.; Young, H.; Richardson, M.; Smart, N.G.; Cunningham, J.; et al. Pancreatic endoderm derived from human embryonic stem cells generates glucose-responsive insulin-secreting cells in vivo. Nat. Biotechnol. 2008, 26, 443–452. [Google Scholar] [CrossRef]
- Prokhorova, T.A.; Harkness, L.M.; Frandsen, U.; Ditzel, N.; Schroder, H.D.; Burns, J.S.; Kassem, M. Teratoma formation by human embryonic stem cells is site dependent and enhanced by the presence of Matrigel. Stem Cells Dev. 2009, 18, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Laflamme, M.A.; Gold, J.; Xu, C.; Hassanipour, M.; Rosler, E.; Police, S.; Muskheli, V.; Murry, C.E. Formation of human myocardium in the rat heart from human embryonic stem cells. Am. J. Pathol. 2005, 167, 663–671. [Google Scholar] [CrossRef]
- Meyer, J.R. The significance of induced pluripotent stem cells for basic research and clinical therapy. J. Med. Ethics 2008, 34, 849–851. [Google Scholar] [CrossRef]
- Kiskinis, E.; Eggan, K. Progress toward the clinical application of patient-specific pluripotent stem cells. J. Clin. Investig. 2010, 120, 51–59. [Google Scholar] [CrossRef]
- Breitbach, M.; Bostani, T.; Roell, W.; Xia, Y.; Dewald, O.; Nygren, J.M.; Fries, J.W.; Tiemann, K.; Bohlen, H.; Hescheler, J.; et al. Potential risks of bone marrow cell transplantation into infarcted hearts. Blood 2007, 110, 1362–1369. [Google Scholar] [CrossRef]
- Yoon, Y.S.; Park, J.S.; Tkebuchava, T.; Luedeman, C.; Losordo, D.W. Unexpected severe calcification after transplantation of bone marrow cells in acute myocardial infarction. Circulation 2004, 109, 3154–3157. [Google Scholar] [CrossRef]
- Portalska, K.J.; Leferink, A.; Groen, N.; Fernandes, H.; Moroni, L.; van Blitterswijk, C.; de Boer, J. Endothelial differentiation of mesenchymal stromal cells. PLoS ONE 2012, 7, e46842. [Google Scholar]
- Oswald, J.; Boxberger, S.; Jorgensen, B.; Feldmann, S.; Ehninger, G.; Bornhauser, M.; Werner, C. Mesenchymal stem cells can be differentiated into endothelial cells in vitro. Stem Cells 2004, 22, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Marriott, S.; Baskir, R.S.; Gaskill, C.; Menon, S.; Carrier, E.J.; Williams, J.; Talati, M.; Helm, K.; Alford, C.E.; Kropski, J.A.; et al. ABCG2pos lung mesenchymal stem cells are a novel pericyte subpopulation that contributes to fibrotic remodeling. Am. J. Physiol. Cell Physiol. 2014, 307, C684–C698. [Google Scholar] [CrossRef]
- Schneider, R.K.; Mullally, A.; Dugourd, A.; Peisker, F.; Hoogenboezem, R.; van Strien, P.M.H.; Bindels, E.M.; Heckl, D.; Busche, G.; Fleck, D.; et al. Gli1(+) Mesenchymal Stromal Cells Are a Key Driver of Bone Marrow Fibrosis and an Important Cellular Therapeutic Target. Cell Stem Cell 2017, 20, 785–800.e8. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.O.; Han, J.W.; Kim, J.M.; Cho, H.J.; Park, C.; Lee, N.; Kim, D.W.; Yoon, Y.S. Malignant tumor formation after transplantation of short-term cultured bone marrow mesenchymal stem cells in experimental myocardial infarction and diabetic neuropathy. Circ. Res. 2011, 108, 1340–1347. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).