
Alpha-Synuclein and Cognitive Decline in Parkinson Disease


{kind=link}
{kind=link}
Abstract
1. Introduction
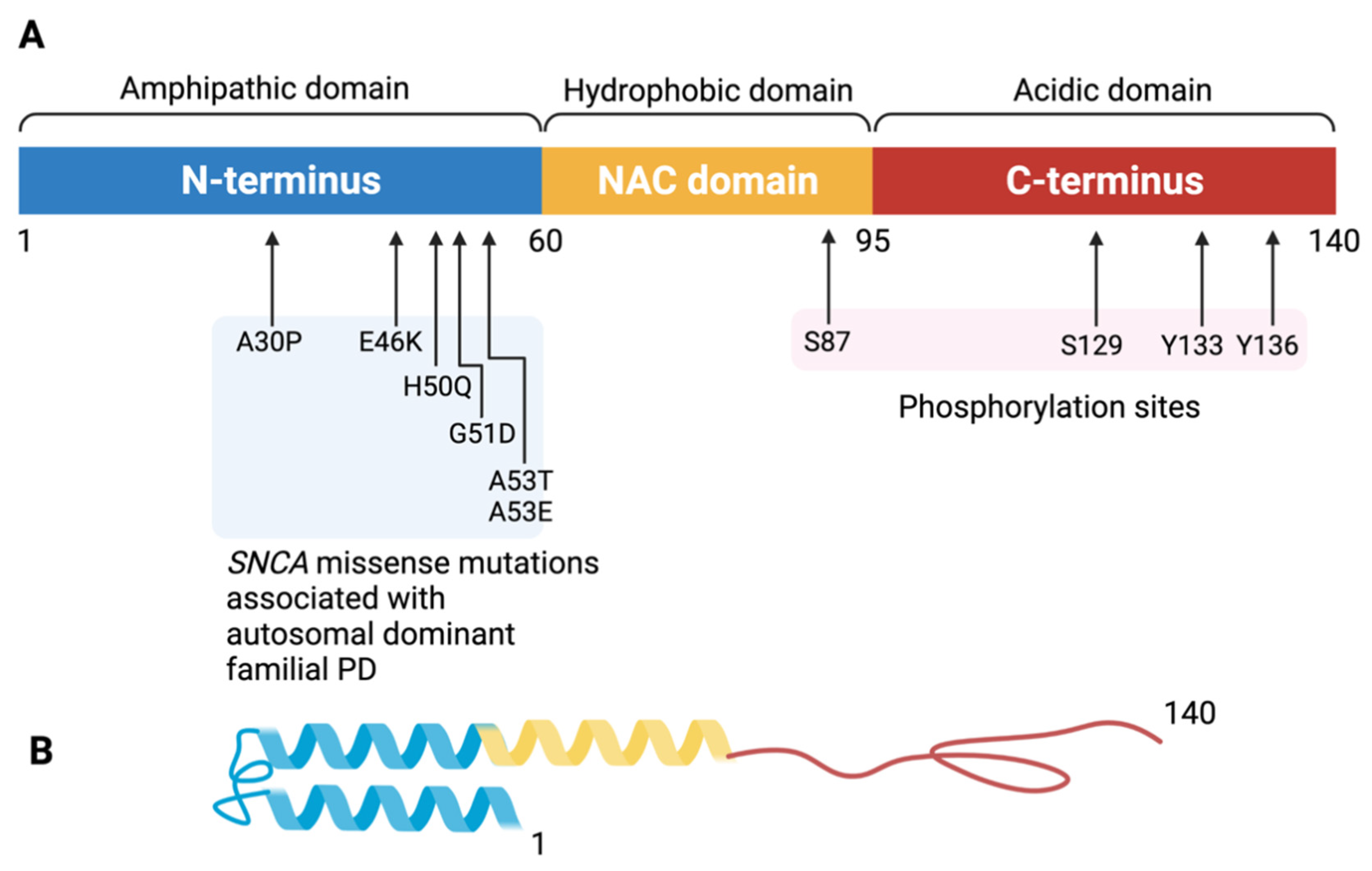
2. Alpha-Synuclein, Lewy Body, and Dementia
3. Physiological Function and Potential Toxicity of Alpha-Synuclein
4. Parkinson Disease and Cognitive Decline: Genetic Contribution, Alpha-Synuclein Propagation, and Protein–Protein Interaction
4.1. Genetic Contribution
4.1.1. PD-Related Genes
- SNCA
- LRRK2
- GBA
- Parkin/PINK1
4.1.2. Non-PD Related Genes
- APOE
- MAPT
4.1.3. Polygenic Risk Score (PRS) in PDD
4.2. Alpha-Synuclein Propagation
4.3. Protein–Protein Interaction
5. Alpha-Synuclein as a Biomarker of PDD
5.1. αS, Amyloid-β (Aβ), and Tau Pathology in PDD
5.2. MicroRNAs as Biomarkers for PD/PDD
5.3. EV Proteins as Biomarkers for PD/PDD
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Postuma, R.B.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C.W.; Oertel, W.; Obeso, J.; Marek, K.; Litvan, I.; Lang, A.E.; et al. MDS clinical diagnostic criteria for Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2015, 30, 1591–1601. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, K.R.; Healy, D.G.; Schapira, A.H.V. Non-motor symptoms of Parkinson’s disease: Diagnosis and management. Lancet Neurol. 2006, 5, 235–245. [Google Scholar] [CrossRef]
- Sauerbier, A.; Jenner, P.; Todorova, A.; Chaudhuri, K.R. Non motor subtypes and Parkinson’s disease. Parkinsonism Relat. Disord. 2016, 22 (Suppl. 1), S41–S46. [Google Scholar] [CrossRef]
- Muslimovic, D.; Post, B.; Speelman, J.D.; Schmand, B. Cognitive profile of patients with newly diagnosed Parkinson disease. Neurology 2005, 65, 1239–1245. [Google Scholar] [CrossRef]
- Szeto, J.Y.Y.; Walton, C.C.; Rizos, A.; Martinez-Martin, P.; Halliday, G.M.; Naismith, S.L.; Chaudhuri, K.R.; Lewis, S.J.G. Dementia in long-term Parkinson’s disease patients: A multicentre retrospective study. NPJ Parkinsons Dis. 2020, 6, 2. [Google Scholar] [CrossRef]
- Foltynie, T.; Brayne, C.E.; Robbins, T.W.; Barker, R.A. The cognitive ability of an incident cohort of Parkinson’s patients in the UK. The CamPaIGN study. Brain 2004, 127, 550–560. [Google Scholar] [CrossRef]
- Wojtala, J.; Heber, I.A.; Neuser, P.; Heller, J.; Kalbe, E.; Rehberg, S.P.; Storch, A.; Linse, K.; Schneider, C.; Gräber, S.; et al. Cognitive decline in Parkinson’s disease: The impact of the motor phenotype on cognition. J. Neurol. Neurosurg. Psychiatry 2019, 90, 171–179. [Google Scholar] [CrossRef]
- Goldman, J.G.; Litvan, I. Mild cognitive impairment in Parkinson’s disease. Minerva Med. 2011, 102, 441–459. [Google Scholar] [PubMed]
- Aarsland, D.; Andersen, K.; Larsen, J.P.; Lolk, A.; Nielsen, H.; Kragh-Sorensen, P. Risk of dementia in Parkinson’s disease: A community-based, prospective study. Neurology 2001, 56, 730–736. [Google Scholar] [CrossRef] [PubMed]
- Padovani, A.; Costanzi, C.; Gilberti, N.; Borroni, B. Parkinson’s disease and dementia. Neurol. Sci. 2006, 27 (Suppl. 1), S40–S43. [Google Scholar] [CrossRef]
- Hely, M.A.; Reid, W.G.; Adena, M.A.; Halliday, G.M.; Morris, J.G. The Sydney multicenter study of Parkinson’s disease: The inevitability of dementia at 20 years. Mov. Disord. Off. J. Mov. Disord. Soc. 2008, 23, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Aarsland, D.; Kurz, M.W. The epidemiology of dementia associated with Parkinson disease. J. Neurol. Sci. 2010, 289, 18–22. [Google Scholar] [CrossRef]
- Churchyard, A.; Lees, A.J. The relationship between dementia and direct involvement of the hippocampus and amygdala in Parkinson’s disease. Neurology 1997, 49, 1570–1576. [Google Scholar] [CrossRef] [PubMed]
- Hurtig, H.I.; Trojanowski, J.Q.; Galvin, J.; Ewbank, D.; Schmidt, M.L.; Lee, V.M.; Clark, C.M.; Glosser, G.; Stern, M.B.; Gollomp, S.M.; et al. Alpha-synuclein cortical Lewy bodies correlate with dementia in Parkinson’s disease. Neurology 2000, 54, 1916–1921. [Google Scholar] [CrossRef]
- Bertrand, E.; Lechowicz, W.; Szpak, G.M.; Lewandowska, E.; Dymecki, J.; Wierzba-Bobrowicz, T. Limbic neuropathology in idiopathic Parkinson’s disease with concomitant dementia. Folia Neuropathol. 2004, 42, 141–150. [Google Scholar] [PubMed]
- Mattila, P.M.; Rinne, J.O.; Helenius, H.; Dickson, D.W.; Röyttä, M. Alpha-synuclein-immunoreactive cortical Lewy bodies are associated with cognitive impairment in Parkinson’s disease. Acta Neuropathol. 2000, 100, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Kövari, E.; Gold, G.; Herrmann, F.R.; Canuto, A.; Hof, P.R.; Bouras, C.; Giannakopoulos, P. Lewy body densities in the entorhinal and anterior cingulate cortex predict cognitive deficits in Parkinson’s disease. Acta Neuropathol. 2003, 106, 83–88. [Google Scholar] [CrossRef]
- Lo, R.Y.; Tanner, C.M.; Albers, K.B.; Leimpeter, A.D.; Fross, R.D.; Bernstein, A.L.; McGuire, V.; Quesenberry, C.P.; Nelson, L.M.; Van Den Eeden, S.K. Clinical features in early Parkinson disease and survival. Arch. Neurol. 2009, 66, 1353–1358. [Google Scholar] [CrossRef]
- Rosenthal, E.; Brennan, L.; Xie, S.; Hurtig, H.; Milber, J.; Weintraub, D.; Karlawish, J.; Siderowf, A. Association between cognition and function in patients with Parkinson disease with and without dementia. Mov. Disord. Off. J. Mov. Disord. Soc. 2010, 25, 1170–1176. [Google Scholar] [CrossRef] [PubMed]
- Jenner, P.; Morris, H.R.; Robbins, T.W.; Goedert, M.; Hardy, J.; Ben-Shlomo, Y.; Bolam, P.; Burn, D.; Hindle, J.V.; Brooks, D. Parkinson’s disease--the debate on the clinical phenomenology, aetiology, pathology and pathogenesis. J. Parkinsons Dis. 2013, 3, 1–11. [Google Scholar] [CrossRef]
- Coon, E.A.; Mandrekar, J.N.; Berini, S.E.; Benarroch, E.E.; Sandroni, P.; Low, P.A.; Singer, W. Predicting phenoconversion in pure autonomic failure. Neurology 2020, 95, e889–e897. [Google Scholar] [CrossRef] [PubMed]
- Boeve, B.F.; Silber, M.H.; Ferman, T.J.; Lucas, J.A.; Parisi, J.E. Association of REM sleep behavior disorder and neurodegenerative disease may reflect an underlying synucleinopathy. Mov. Disord. Off. J. Mov. Disord. Soc. 2001, 16, 622–630. [Google Scholar] [CrossRef] [PubMed]
- Postuma, R.B.; Iranzo, A.; Hu, M.; Högl, B.; Boeve, B.F.; Manni, R.; Oertel, W.H.; Arnulf, I.; Ferini-Strambi, L.; Puligheddu, M.; et al. Risk and predictors of dementia and parkinsonism in idiopathic REM sleep behaviour disorder: A multicentre study. Brain 2019, 142, 744–759. [Google Scholar] [CrossRef]
- Galvin, J.E.; Uryu, K.; Lee, V.M.-Y.; Trojanowski, J.Q. Axon pathology in Parkinson’s disease and Lewy body dementia hippocampus contains α-, β-, and γ-synuclein. Proc. Natl. Acad. Sci. USA 1999, 96, 13450–13455. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.-Y.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. α-Synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Cairns, N.J.; Lantos, P.L.; Goedert, M. Filamentous α-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci. Lett. 1998, 251, 205–208. [Google Scholar] [CrossRef]
- Beyer, K.; Domingo-Sàbat, M.; Ariza, A. Molecular pathology of Lewy body diseases. Int. J. Mol. Sci. 2009, 10, 724–745. [Google Scholar] [CrossRef] [PubMed]
- Kosaka, K. Latest concept of Lewy body disease. Psychiatry Clin. Neurosci. 2014, 68, 391–394. [Google Scholar] [CrossRef]
- Emre, M.; Aarsland, D.; Brown, R.; Burn, D.J.; Duyckaerts, C.; Mizuno, Y.; Broe, G.A.; Cummings, J.; Dickson, D.W.; Gauthier, S.; et al. Clinical diagnostic criteria for dementia associated with Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2007, 22, 1689–1707. [Google Scholar] [CrossRef]
- McKeith, I.G.; Boeve, B.F.; Dickson, D.W.; Halliday, G.; Taylor, J.-P.; Weintraub, D.; Aarsland, D.; Galvin, J.; Attems, J.; Ballard, C.G.; et al. Diagnosis and management of dementia with Lewy bodies: Fourth consensus report of the DLB Consortium. Neurology 2017, 89, 88–100. [Google Scholar] [CrossRef]
- Compta, Y.; Parkkinen, L.; O’Sullivan, S.S.; Vandrovcova, J.; Holton, J.L.; Collins, C.; Lashley, T.; Kallis, C.; Williams, D.R.; de Silva, R.; et al. Lewy- and Alzheimer-type pathologies in Parkinson’s disease dementia: Which is more important? Brain 2011, 134, 1493–1505. [Google Scholar] [CrossRef]
- Bohnen, N.I.; Kaufer, D.I.; Ivanco, L.S.; Lopresti, B.; Koeppe, R.A.; Davis, J.G.; Mathis, C.A.; Moore, R.Y.; DeKosky, S.T. Cortical cholinergic function is more severely affected in parkinsonian dementia than in Alzheimer disease: An in vivo positron emission tomographic study. Arch. Neurol. 2003, 60, 1745–1748. [Google Scholar] [CrossRef]
- Cersosimo, M.G. Propagation of alpha-synuclein pathology from the olfactory bulb: Possible role in the pathogenesis of dementia with Lewy bodies. Cell Tissue Res. 2018, 373, 233–243. [Google Scholar] [CrossRef]
- Smirnov, D.S.; Galasko, D.; Edland, S.D.; Filoteo, J.V.; Hansen, L.A.; Salmon, D.P. Cognitive decline profiles differ in Parkinson disease dementia and dementia with Lewy bodies. Neurology 2020, 94, e2076–e2087. [Google Scholar] [CrossRef] [PubMed]
- Schirinzi, T.; Canevelli, M.; Suppa, A.; Bologna, M.; Marsili, L. The continuum between neurodegeneration, brain plasticity, and movement: A critical appraisal. Rev. Neurosci. 2020, 31, 723–742. [Google Scholar] [CrossRef]
- Chan, F.; Lax, N.Z.; Davies, C.H.; Turnbull, D.M.; Cunningham, M.O. Neuronal oscillations: A physiological correlate for targeting mitochondrial dysfunction in neurodegenerative diseases? Neuropharmacology 2016, 102, 48–58. [Google Scholar] [CrossRef]
- Maroteaux, L.; Campanelli, J.T.; Scheller, R.H. Synuclein: A neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 1988, 8, 2804–2815. [Google Scholar] [CrossRef]
- Bartels, T.; Ahlstrom, L.S.; Leftin, A.; Kamp, F.; Haass, C.; Brown, M.F.; Beyer, K. The N-terminus of the intrinsically disordered protein α-synuclein triggers membrane binding and helix folding. Biophys. J. 2010, 99, 2116–2124. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Ivanova, M.I.; Sawaya, M.R.; Cascio, D.; Reyes, F.E.; Shi, D.; Sangwan, S.; Guenther, E.L.; Johnson, L.M.; Zhang, M.; et al. Structure of the toxic core of α-synuclein from invisible crystals. Nature 2015, 525, 486–490. [Google Scholar] [CrossRef]
- Souza, J.M.; Giasson, B.I.; Lee, V.M.; Ischiropoulos, H. Chaperone-like activity of synucleins. FEBS Lett. 2000, 474, 116–119. [Google Scholar] [CrossRef]
- Sorrentino, Z.A.; Giasson, B.I. The emerging role of α-synuclein truncation in aggregation and disease. J. Biol. Chem. 2020, 295, 10224–10244. [Google Scholar] [CrossRef]
- Burré, J.; Sharma, M.; Südhof, T.C. Definition of a molecular pathway mediating α-synuclein neurotoxicity. J. Neurosci. Off. J. Soc. Neurosci. 2015, 35, 5221–5232. [Google Scholar] [CrossRef]
- Nakamura, K.; Nemani, V.M.; Azarbal, F.; Skibinski, G.; Levy, J.M.; Egami, K.; Munishkina, L.; Zhang, J.; Gardner, B.; Wakabayashi, J.; et al. Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein alpha-synuclein. J. Biol. Chem. 2011, 286, 20710–20726. [Google Scholar] [CrossRef] [PubMed]
- Bridi, J.C.; Hirth, F. Mechanisms of α-Synuclein Induced Synaptopathy in Parkinson’s Disease. Front. Neurosci. 2018, 12, 80. [Google Scholar] [CrossRef]
- Burré, J. The Synaptic Function of α-Synuclein. J. Parkinsons Dis. 2015, 5, 699–713. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, K.; Tanji, K.; Mori, F.; Takahashi, H. The Lewy body in Parkinson’s disease: Molecules implicated in the formation and degradation of alpha-synuclein aggregates. Neuropathology 2007, 27, 494–506. [Google Scholar] [CrossRef]
- Shahmoradian, S.H.; Lewis, A.J.; Genoud, C.; Hench, J.; Moors, T.E.; Navarro, P.P.; Castaño-Díez, D.; Schweighauser, G.; Graff-Meyer, A.; Goldie, K.N.; et al. Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat. Neurosci. 2019, 22, 1099–1109. [Google Scholar] [CrossRef]
- Dettmer, U.; Newman, A.J.; Soldner, F.; Luth, E.S.; Kim, N.C.; Von Saucken, V.E.; Sanderson, J.B.; Jaenisch, R.; Bartels, T.; Selkoe, D. Parkinson-causing α-synuclein missense mutations shift native tetramers to monomers as a mechanism for disease initiation. Nat. Commun. 2015, 6, 1–16. [Google Scholar] [CrossRef]
- Ferreon, A.C.M.; Gambin, Y.; Lemke, E.A.; Deniz, A.A. Interplay of α-synuclein binding and conformational switching probed by single-molecule fluorescence. Proc. Natl. Acad. Sci. USA 2009, 106, 5645–5650. [Google Scholar] [CrossRef] [PubMed]
- Frimpong, A.K.; Abzalimov, R.R.; Uversky, V.N.; Kaltashov, I.A. Characterization of intrinsically disordered proteins with electrospray ionization mass spectrometry: Conformational heterogeneity of α-synuclein. Proteins Struct. Funct. Bioinform. 2010, 78, 714–722. [Google Scholar] [CrossRef]
- Rokad, D.; Ghaisas, S.; Harischandra, D.S.; Jin, H.; Anantharam, V.; Kanthasamy, A.; Kanthasamy, A.G. Role of neurotoxicants and traumatic brain injury in α-synuclein protein misfolding and aggregation. Brain Res. Bull. 2017, 133, 60–70. [Google Scholar] [CrossRef]
- Guhathakurta, S.; Bok, E.; Evangelista, B.A.; Kim, Y.-S. Deregulation of α-synuclein in Parkinson’s disease: Insight from epigenetic structure and transcriptional regulation of SNCA. Prog. Neurobiol. 2017, 154, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Alam, P.; Bousset, L.; Melki, R.; Otzen, D.E. α-synuclein oligomers and fibrils: A spectrum of species, a spectrum of toxicities. J. Neurochem. 2019, 150, 522–534. [Google Scholar] [CrossRef] [PubMed]
- Cremades, N.; Chen, S.; Dobson, C. Structural characteristics of α-synuclein oligomers. Int. Rev. Cell Mol. Biol. 2017, 329, 79–143. [Google Scholar] [PubMed]
- Peelaerts, W.; Bousset, L.; Van der Perren, A.; Moskalyuk, A.; Pulizzi, R.; Giugliano, M.; Van den Haute, C.; Melki, R.; Baekelandt, V. α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 2015, 522, 340–344. [Google Scholar] [CrossRef]
- Chen, S.W.; Drakulic, S.; Deas, E.; Ouberai, M.; Aprile, F.A.; Arranz, R.; Ness, S.; Roodveldt, C.; Guilliams, T.; De-Genst, E.J. Structural characterization of toxic oligomers that are kinetically trapped during α-synuclein fibril formation. Proc. Natl. Acad. Sci. USA 2015, 112, E1994–E2003. [Google Scholar] [CrossRef]
- Cremades, N.; Cohen, S.I.; Deas, E.; Abramov, A.Y.; Chen, A.Y.; Orte, A.; Sandal, M.; Clarke, R.W.; Dunne, P.; Aprile, F.A. Direct observation of the interconversion of normal and toxic forms of α-synuclein. Cell 2012, 149, 1048–1059. [Google Scholar] [CrossRef]
- Fusco, G.; Chen, S.W.; Williamson, P.T.; Cascella, R.; Perni, M.; Jarvis, J.A.; Cecchi, C.; Vendruscolo, M.; Chiti, F.; Cremades, N. Structural basis of membrane disruption and cellular toxicity by α-synuclein oligomers. Science 2017, 358, 1440–1443. [Google Scholar] [CrossRef]
- Gath, J.; Bousset, L.; Habenstein, B.; Melki, R.; Böckmann, A.; Meier, B.H. Unlike twins: An NMR comparison of two α-synuclein polymorphs featuring different toxicity. PLoS ONE 2014, 9, e90659. [Google Scholar] [CrossRef]
- Monsellier, E.; Bousset, L.; Melki, R. α-Synuclein and huntingtin exon 1 amyloid fibrils bind laterally to the cellular membrane. Sci. Rep. 2016, 6, 19180. [Google Scholar] [CrossRef]
- Pieri, L.; Madiona, K.; Bousset, L.; Melki, R. Fibrillar α-synuclein and huntingtin exon 1 assemblies are toxic to the cells. Biophys. J. 2012, 102, 2894–2905. [Google Scholar] [CrossRef]
- Paslawski, W.; Andreasen, M.; Nielsen, S.B.; Lorenzen, N.; Thomsen, K.; Kaspersen, J.D.; Pedersen, J.S.; Otzen, D.E. High stability and cooperative unfolding of α-synuclein oligomers. Biochemistry 2014, 53, 6252–6263. [Google Scholar] [CrossRef] [PubMed]
- Mahul-Mellier, A.-L.; Burtscher, J.; Maharjan, N.; Weerens, L.; Croisier, M.; Kuttler, F.; Leleu, M.; Knott, G.W.; Lashuel, H.A. The process of Lewy body formation, rather than simply α-synuclein fibrillization, is one of the major drivers of neurodegeneration. Proc. Natl. Acad. Sci. USA 2020, 117, 4971–4982. [Google Scholar] [CrossRef] [PubMed]
- Boutros, S.W.; Raber, J.; Unni, V.K. Effects of Alpha-Synuclein Targeted Antisense Oligonucleotides on Lewy Body-Like Pathology and Behavioral Disturbances Induced by Injections of Pre-Formed Fibrils in the Mouse Motor Cortex. J. Parkinsons Dis. 2021, 11, 1091–1115. [Google Scholar] [CrossRef]
- Bernal-Conde, L.D.; Ramos-Acevedo, R.; Reyes-Hernández, M.A.; Balbuena-Olvera, A.J.; Morales-Moreno, I.D.; Argüero-Sánchez, R.; Schüle, B.; Guerra-Crespo, M. Alpha-Synuclein Physiology and Pathology: A Perspective on Cellular Structures and Organelles. Front. Neurosci. 2020, 13, 1399. [Google Scholar] [CrossRef]
- Payton, J.E.; Perrin, R.J.; Woods, W.S.; George, J.M. Structural determinants of PLD2 inhibition by α-synuclein. J. Mol. Biol. 2004, 337, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Dalfó, E.; Ferrer, I. α-Synuclein binding to rab3a in multiple system atrophy. Neurosci. Lett. 2005, 380, 170–175. [Google Scholar] [CrossRef]
- Burré, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Südhof, T.C. α-Synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef]
- Zhu, M.; Li, J.; Fink, A.L. The association of α-synuclein with membranes affects bilayer structure, stability, and fibril formation. J. Biol. Chem. 2003, 278, 40186–40197. [Google Scholar] [CrossRef]
- Volles, M.J.; Lansbury, P.T. Vesicle permeabilization by protofibrillar α-synuclein is sensitive to Parkinson’s disease-linked mutations and occurs by a pore-like mechanism. Biochemistry 2002, 41, 4595–4602. [Google Scholar] [CrossRef]
- Kayed, R.; Sokolov, Y.; Edmonds, B.; McIntire, T.M.; Milton, S.C.; Hall, J.E.; Glabe, C.G. Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J. Biol. Chem. 2004, 279, 46363–46366. [Google Scholar] [CrossRef] [PubMed]
- Cecchi, C.; Stefani, M. The amyloid-cell membrane system. The interplay between the biophysical features of oligomers/fibrils and cell membrane defines amyloid toxicity. Biophys. Chem. 2013, 182, 30–43. [Google Scholar] [CrossRef]
- Iyer, A.; Schilderink, N.; Claessens, M.M.; Subramaniam, V. Membrane-bound alpha synuclein clusters induce impaired lipid diffusion and increased lipid packing. Biophys. J. 2016, 111, 2440–2449. [Google Scholar] [CrossRef]
- Logan, T.; Bendor, J.; Toupin, C.; Thorn, K.; Edwards, R.H. α-Synuclein promotes dilation of the exocytotic fusion pore. Nat. Neurosci. 2017, 20, 681–689. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.E.; Brender, J.R.; Ramamoorthy, A. Induction of negative curvature as a mechanism of cell toxicity by amyloidogenic peptides: The case of islet amyloid polypeptide. J. Am. Chem. Soc. 2009, 131, 4470–4478. [Google Scholar] [CrossRef] [PubMed]
- Soll, L.G.; Eisen, J.N.; Vargas, K.J.; Medeiros, A.T.; Hammar, K.M.; Morgan, J.R. α-Synuclein-112 impairs synaptic vesicle recycling consistent with its enhanced membrane binding properties. Front. Cell Dev. Biol. 2020, 8, 405. [Google Scholar] [CrossRef]
- Cooper, A.A.; Gitler, A.D.; Cashikar, A.; Haynes, C.M.; Hill, K.J.; Bhullar, B.; Liu, K.; Xu, K.; Strathearn, K.E.; Liu, F. α-Synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 2006, 313, 324–328. [Google Scholar] [CrossRef]
- Gosavi, N.; Lee, H.-J.; Lee, J.S.; Patel, S.; Lee, S.-J. Golgi fragmentation occurs in the cells with prefibrillar α-synuclein aggregates and precedes the formation of fibrillar inclusion. J. Biol. Chem. 2002, 277, 48984–48992. [Google Scholar] [CrossRef]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial import and accumulation of α-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar] [CrossRef]
- Nakamura, K. α-Synuclein and mitochondria: Partners in crime? Neurotherapeutics 2013, 10, 391–399. [Google Scholar] [CrossRef]
- Gao, G.; Wang, Z.; Lu, L.; Duan, C.; Wang, X.; Yang, H. Morphological analysis of mitochondria for evaluating the toxicity of α-synuclein in transgenic mice and isolated preparations by atomic force microscopy. Biomed. Pharmacother. 2017, 96, 1380–1388. [Google Scholar] [CrossRef] [PubMed]
- Stefanis, L.; Larsen, K.E.; Rideout, H.J.; Sulzer, D.; Greene, L.A. Expression of A53T mutant but not wild-type α-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J. Neurosci. 2001, 21, 9549–9560. [Google Scholar] [CrossRef]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired degradation of mutant α-synuclein by chaperone-mediated autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef] [PubMed]
- Xilouri, M.; Brekk, O.R.; Stefanis, L. Alpha-synuclein and protein degradation systems: A reciprocal relationship. Mol. Neurobiol. 2013, 47, 537–551. [Google Scholar] [CrossRef] [PubMed]
- Cookson, M.R. Alpha-Synuclein and neuronal cell death. Mol. Neurodegener. 2009, 4, 9. [Google Scholar] [CrossRef]
- Rocha, E.M.; De Miranda, B.; Sanders, L.H. Alpha-synuclein: Pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol. Dis. 2018, 109, 249–257. [Google Scholar] [CrossRef]
- Mullin, S.; Schapira, A. Alpha-Synuclein and mitochondrial dysfunction in Parkinson’s disease. Mol. Neurobiol. 2013, 47, 587–597. [Google Scholar] [CrossRef]
- Osterberg, V.R.; Spinelli, K.J.; Weston, L.J.; Luk, K.C.; Woltjer, R.L.; Unni, V.K. Progressive aggregation of alpha-synuclein and selective degeneration of lewy inclusion-bearing neurons in a mouse model of parkinsonism. Cell Rep. 2015, 10, 1252–1260. [Google Scholar] [CrossRef]
- Volpicelli-Daley, L.A.; Luk, K.C.; Patel, T.P.; Tanik, S.A.; Riddle, D.M.; Stieber, A.; Meaney, D.F.; Trojanowski, J.Q.; Lee, V.M.-Y. Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 2011, 72, 57–71. [Google Scholar] [CrossRef]
- Cavaliere, F.; Cerf, L.; Dehay, B.; Ramos-Gonzalez, P.; De Giorgi, F.; Bourdenx, M.; Bessede, A.; Obeso, J.A.; Matute, C.; Ichas, F. In vitro α-synuclein neurotoxicity and spreading among neurons and astrocytes using Lewy body extracts from Parkinson disease brains. Neurobiol. Dis. 2017, 103, 101–112. [Google Scholar] [CrossRef]
- Mezias, C.; Rey, N.; Brundin, P.; Raj, A. Neural connectivity predicts spreading of alpha-synuclein pathology in fibril-injected mouse models: Involvement of retrograde and anterograde axonal propagation. Neurobiol. Dis. 2020, 134, 104623. [Google Scholar] [CrossRef]
- Hijaz, B.A.; Volpicelli-Daley, L.A. Initiation and propagation of alpha-synuclein aggregation in the nervous system. Mol. Neurodegener. 2020, 15, 19. [Google Scholar] [CrossRef] [PubMed]
- Recasens, A.; Dehay, B.; Bové, J.; Carballo-Carbajal, I.; Dovero, S.; Pérez-Villalba, A.; Fernagut, P.O.; Blesa, J.; Parent, A.; Perier, C.; et al. Lewy body extracts from Parkinson disease brains trigger α-synuclein pathology and neurodegeneration in mice and monkeys. Ann. Neurol. 2014, 75, 351–362. [Google Scholar] [CrossRef] [PubMed]
- Howitt, J.; Hill, A.F. Exosomes in the Pathology of Neurodegenerative Diseases. J. Biol. Chem. 2016, 291, 26589–26597. [Google Scholar] [CrossRef]
- Gousset, K.; Schiff, E.; Langevin, C.; Marijanovic, Z.; Caputo, A.; Browman, D.T.; Chenouard, N.; de Chaumont, F.; Martino, A.; Enninga, J.; et al. Prions hijack tunnelling nanotubes for intercellular spread. Nat. Cell Biol. 2009, 11, 328–336. [Google Scholar] [CrossRef]
- Lee, H.J.; Patel, S.; Lee, S.J. Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J. Neurosci. 2005, 25, 6016–6024. [Google Scholar] [CrossRef]
- Ghiglieri, V.; Calabrese, V.; Calabresi, P. Alpha-Synuclein: From Early Synaptic Dysfunction to Neurodegeneration. Front. Neurol. 2018, 9, 295. [Google Scholar] [CrossRef]
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of α-synuclein: From structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 2013, 14, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Roman-Vendrell, C.; Medeiros, A.T.; Sanderson, J.B.; Jiang, H.; Bartels, T.; Morgan, J.R. Effects of Excess Brain-Derived Human alpha-Synuclein on Synaptic Vesicle Trafficking. Front. Neurosci. 2021, 15, 639414. [Google Scholar] [CrossRef]
- Wu, Q.; Takano, H.; Riddle, D.M.; Trojanowski, J.Q.; Coulter, D.A.; Lee, V.M. Alpha-Synuclein (alphaSyn) Preformed Fibrils Induce Endogenous alphaSyn Aggregation, Compromise Synaptic Activity and Enhance Synapse Loss in Cultured Excitatory Hippocampal Neurons. J. Neurosci. 2019, 39, 5080–5094. [Google Scholar] [CrossRef]
- Cascella, R.; Chen, S.W.; Bigi, A.; Camino, J.D.; Xu, C.K.; Dobson, C.M.; Chiti, F.; Cremades, N.; Cecchi, C. The release of toxic oligomers from α-synuclein fibrils induces dysfunction in neuronal cells. Nat. Commun. 2021, 12, 1814. [Google Scholar] [CrossRef] [PubMed]
- Alza, N.P.; Iglesias Gonzalez, P.A.; Conde, M.A.; Uranga, R.M.; Salvador, G.A. Lipids at the Crossroad of alpha-Synuclein Function and Dysfunction: Biological and Pathological Implications. Front. Cell Neurosci. 2019, 13, 175. [Google Scholar] [CrossRef]
- O’Leary, E.I.; Lee, J.C. Interplay between α-synuclein amyloid formation and membrane structure. Biochim. Et Biophys. Acta (BBA)-Proteins Proteom. 2019, 1867, 483–491. [Google Scholar] [CrossRef] [PubMed]
- Gelders, G.; Baekelandt, V.; Van der Perren, A. Linking Neuroinflammation and Neurodegeneration in Parkinson’s Disease. J. Immunol. Res. 2018, 2018, 4784268. [Google Scholar] [CrossRef]
- Rosborough, K.; Patel, N.; Kalia, L.V. α-Synuclein and Parkinsonism: Updates and Future Perspectives. Curr. Neurol. Neurosci. Rep. 2017, 17, 31. [Google Scholar] [CrossRef]
- Kasten, M.; Klein, C. The many faces of alpha-synuclein mutations. Mov. Disord. 2013, 28, 697–701. [Google Scholar] [CrossRef] [PubMed]
- Zarranz, J.J.; Alegre, J.; Gomez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atares, B.; et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef]
- Carmona-Abellan, M.; Gabilondo, I.; Murueta-Goyena, A.; Khurana, V.; Tijero, B.; Luquin, M.R.; Acera, M.; Del Pino, R.; Gardeazabal, J.; Martínez-Valbuena, I.; et al. Small fiber neuropathy and phosphorylated alpha-synuclein in the skin of E46K-SNCA mutation carriers. Parkinsonism Relat. Disord. 2019, 65, 139–145. [Google Scholar] [CrossRef]
- Tokutake, T.; Ishikawa, A.; Yoshimura, N.; Miyashita, A.; Kuwano, R.; Nishizawa, M.; Ikeuchi, T. Clinical and neuroimaging features of patient with early-onset Parkinson’s disease with dementia carrying SNCA p.G51D mutation. Parkinsonism Relat. Disord. 2014, 20, 262–264. [Google Scholar] [CrossRef] [PubMed]
- Wittke, C.; Petkovic, S.; Dobricic, V.; Schaake, S.; Respondek, G.; Weissbach, A.; Madoev, H.; Trinh, J.; Vollstedt, E.J.; Kuhnke, N.; et al. Genotype-Phenotype Relations for the Atypical Parkinsonism Genes: MDSGene Systematic Review. Mov. Disord. Off. J. Mov. Disord. Soc. 2021, 36, 1499–1510. [Google Scholar] [CrossRef]
- Marsili, L.; Vizcarra, J.A.; Sturchio, A.; Dwivedi, A.K.; Keeling, E.G.; Patel, D.; Mishra, M.; Farooqi, A.; Merola, A.; Fasano, A.; et al. When does postural instability appear in monogenic parkinsonisms? An individual-patient meta-analysis. J. Neurol. 2020, 268, 3203–3211. [Google Scholar] [CrossRef] [PubMed]
- Allan, L.M.; Ballard, C.G.; Allen, J.; Murray, A.; Davidson, A.W.; McKeith, I.G.; Kenny, R.A. Autonomic dysfunction in dementia. J. Neurol. Neurosurg. Psychiatry 2007, 78, 671–677. [Google Scholar] [CrossRef]
- Schneider, S.A.; Alcalay, R.N. Neuropathology of genetic synucleinopathies with parkinsonism: Review of the literature. Mov. Disord. Off. J. Mov. Disord. Soc. 2017, 32, 1504–1523. [Google Scholar] [CrossRef] [PubMed]
- Kalia, L.V.; Lang, A.E.; Hazrati, L.N.; Fujioka, S.; Wszolek, Z.K.; Dickson, D.W.; Ross, O.A.; Van Deerlin, V.M.; Trojanowski, J.Q.; Hurtig, H.I.; et al. Clinical correlations with Lewy body pathology in LRRK2-related Parkinson disease. JAMA Neurol. 2015, 72, 100–105. [Google Scholar] [CrossRef]
- Junqueira, V.B.; Barros, S.B.; Chan, S.S.; Rodrigues, L.; Giavarotti, L.; Abud, R.L.; Deucher, G.P. Aging and oxidative stress. Mol. Asp. Med. 2004, 25, 5–16. [Google Scholar] [CrossRef]
- Lindner, A.B.; Demarez, A. Protein aggregation as a paradigm of aging. Biochim. Biophys. Acta 2009, 1790, 980–996. [Google Scholar] [CrossRef]
- Tong, Y.; Yamaguchi, H.; Giaime, E.; Boyle, S.; Kopan, R.; Kelleher, R.J., 3rd; Shen, J. Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of alpha-synuclein, and apoptotic cell death in aged mice. Proc. Natl. Acad. Sci. USA 2010, 107, 9879–9884. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.; Giaime, E.; Yamaguchi, H.; Ichimura, T.; Liu, Y.; Si, H.; Cai, H.; Bonventre, J.V.; Shen, J. Loss of leucine-rich repeat kinase 2 causes age-dependent bi-phasic alterations of the autophagy pathway. Mol. Neurodegener. 2012, 7, 2. [Google Scholar] [CrossRef]
- Bieri, G.; Brahic, M.; Bousset, L.; Couthouis, J.; Kramer, N.J.; Ma, R.; Nakayama, L.; Monbureau, M.; Defensor, E.; Schüle, B.; et al. LRRK2 modifies α-syn pathology and spread in mouse models and human neurons. Acta Neuropathol. 2019, 137, 961–980. [Google Scholar] [CrossRef]
- Weston, L.J.; Stackhouse, T.L.; Spinelli, K.J.; Boutros, S.W.; Rose, E.P.; Osterberg, V.R.; Luk, K.C.; Raber, J.; Weissman, T.A.; Unni, V.K. Genetic deletion of Polo-like kinase 2 reduces alpha-synuclein serine-129 phosphorylation in presynaptic terminals but not Lewy bodies. J. Biol. Chem. 2021, 296, 100273. [Google Scholar] [CrossRef]
- Oueslati, A. Implication of Alpha-Synuclein Phosphorylation at S129 in Synucleinopathies: What Have We Learned in the Last Decade? J. Parkinsons Dis. 2016, 6, 39–51. [Google Scholar] [CrossRef]
- Steger, M.; Diez, F.; Dhekne, H.S.; Lis, P.; Nirujogi, R.S.; Karayel, O.; Tonelli, F.; Martinez, T.N.; Lorentzen, E.; Pfeffer, S.R.; et al. Systematic proteomic analysis of LRRK2-mediated Rab GTPase phosphorylation establishes a connection to ciliogenesis. Elife 2017, 6, e31012. [Google Scholar] [CrossRef]
- Jeong, G.R.; Jang, E.H.; Bae, J.R.; Jun, S.; Kang, H.C.; Park, C.H.; Shin, J.H.; Yamamoto, Y.; Tanaka-Yamamoto, K.; Dawson, V.L.; et al. Dysregulated phosphorylation of Rab GTPases by LRRK2 induces neurodegeneration. Mol. Neurodegener. 2018, 13, 8. [Google Scholar] [CrossRef] [PubMed]
- Bae, E.-J.; Kim, D.-K.; Kim, C.; Mante, M.; Adame, A.; Rockenstein, E.; Ulusoy, A.; Klinkenberg, M.; Jeong, G.R.; Bae, J.R.; et al. LRRK2 kinase regulates α-synuclein propagation via RAB35 phosphorylation. Nat. Commun. 2018, 9, 3465. [Google Scholar] [CrossRef]
- Bonet-Ponce, L.; Cookson, M.R. The role of Rab GTPases in the pathobiology of Parkinson’ disease. Curr. Opin. Cell Biol. 2019, 59, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Sidransky, E.; Nalls, M.A.; Aasly, J.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef]
- Tayebi, N.; Callahan, M.; Madike, V.; Stubblefield, B.K.; Orvisky, E.; Krasnewich, D.; Fillano, J.J.; Sidransky, E. Gaucher disease and parkinsonism: A phenotypic and genotypic characterization. Mol. Genet. Metab. 2001, 73, 313–321. [Google Scholar] [CrossRef]
- Mitsui, J.; Mizuta, I.; Toyoda, A.; Ashida, R.; Takahashi, Y.; Goto, J.; Fukuda, Y.; Date, H.; Iwata, A.; Yamamoto, M.; et al. Mutations for Gaucher disease confer high susceptibility to Parkinson disease. Arch. Neurol. 2009, 66, 571–576. [Google Scholar] [CrossRef]
- Cullen, V.; Sardi, S.P.; Ng, J.; Xu, Y.H.; Sun, Y.; Tomlinson, J.J.; Kolodziej, P.; Kahn, I.; Saftig, P.; Woulfe, J.; et al. Acid beta-glucosidase mutants linked to Gaucher disease, Parkinson disease, and Lewy body dementia alter alpha-synuclein processing. Ann. Neurol. 2011, 69, 940–953. [Google Scholar] [CrossRef]
- Tsuang, D.; Leverenz, J.B.; Lopez, O.L.; Hamilton, R.L.; Bennett, D.A.; Schneider, J.A.; Buchman, A.S.; Larson, E.B.; Crane, P.K.; Kaye, J.A.; et al. GBA mutations increase risk for Lewy body disease with and without Alzheimer disease pathology. Neurology 2012, 79, 1944–1950. [Google Scholar] [CrossRef] [PubMed]
- Cilia, R.; Tunesi, S.; Marotta, G.; Cereda, E.; Siri, C.; Tesei, S.; Zecchinelli, A.L.; Canesi, M.; Mariani, C.B.; Meucci, N.; et al. Survival and dementia in GBA-associated Parkinson’s disease: The mutation matters. Ann. Neurol. 2016, 80, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Neumann, J.; Bras, J.; Deas, E.; O’Sullivan, S.S.; Parkkinen, L.; Lachmann, R.H.; Li, A.; Holton, J.; Guerreiro, R.; Paudel, R.; et al. Glucocerebrosidase mutations in clinical and pathologically proven Parkinson’s disease. Brain 2009, 132, 1783–1794. [Google Scholar] [CrossRef]
- Mazzulli, J.R.; Xu, Y.H.; Sun, Y.; Knight, A.L.; McLean, P.J.; Caldwell, G.A.; Sidransky, E.; Grabowski, G.A.; Krainc, D. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011, 146, 37–52. [Google Scholar] [CrossRef]
- Kitada, T.; Asakawa, S.; Hattori, N.; Matsumine, H.; Yamamura, Y.; Minoshima, S.; Yokochi, M.; Mizuno, Y.; Shimizu, N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998, 392, 605–608. [Google Scholar] [CrossRef]
- Bonifati, V.; Rizzu, P.; van Baren, M.J.; Schaap, O.; Breedveld, G.J.; Krieger, E.; Dekker, M.C.; Squitieri, F.; Ibanez, P.; Joosse, M.; et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003, 299, 256–259. [Google Scholar] [CrossRef]
- Valente, E.M.; Abou-Sleiman, P.M.; Caputo, V.; Muqit, M.M.; Harvey, K.; Gispert, S.; Ali, Z.; Del Turco, D.; Bentivoglio, A.R.; Healy, D.G.; et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 2004, 304, 1158–1160. [Google Scholar] [CrossRef] [PubMed]
- Petrucelli, L.; O’Farrell, C.; Lockhart, P.J.; Baptista, M.; Kehoe, K.; Vink, L.; Choi, P.; Wolozin, B.; Farrer, M.; Hardy, J.; et al. Parkin protects against the toxicity associated with mutant alpha-synuclein: Proteasome dysfunction selectively affects catecholaminergic neurons. Neuron 2002, 36, 1007–1019. [Google Scholar] [CrossRef]
- Haywood, A.F.; Staveley, B.E. Parkin counteracts symptoms in a Drosophila model of Parkinson’s disease. BMC Neurosci. 2004, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Khandelwal, P.J.; Dumanis, S.B.; Feng, L.R.; Maguire-Zeiss, K.; Rebeck, G.W.; Lashuel, H.A.; Moussa, C.E.H. Parkinson-related parkin reduces α-Synuclein phosphorylation in a gene transfer model. Mol. Neurodegener. 2010, 5, 47. [Google Scholar] [CrossRef]
- Miyakawa, S.; Ogino, M.; Funabe, S.; Uchino, A.; Shimo, Y.; Hattori, N.; Ichinoe, M.; Mikami, T.; Saegusa, M.; Nishiyama, K.; et al. Lewy body pathology in a patient with a homozygous parkin deletion. Mov. Disord. Off. J. Mov. Disord. Soc. 2013, 28, 388–391. [Google Scholar] [CrossRef]
- Sasaki, S.; Shirata, A.; Yamane, K.; Iwata, M. Parkin-positive autosomal recessive juvenile Parkinsonism with alpha-synuclein-positive inclusions. Neurology 2004, 63, 678–682. [Google Scholar] [CrossRef] [PubMed]
- Farrer, M.; Chan, P.; Chen, R.; Tan, L.; Lincoln, S.; Hernandez, D.; Forno, L.; Gwinn-Hardy, K.; Petrucelli, L.; Hussey, J.; et al. Lewy bodies and parkinsonism in families with parkin mutations. Ann. Neurol. 2001, 50, 293–300. [Google Scholar] [CrossRef] [PubMed]
- Alcalay, R.N.; Caccappolo, E.; Mejia-Santana, H.; Tang, M.X.; Rosado, L.; Orbe Reilly, M.; Ruiz, D.; Louis, E.D.; Comella, C.L.; Nance, M.A.; et al. Cognitive and motor function in long-duration PARKIN-associated Parkinson disease. JAMA Neurol. 2014, 71, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tomiyama, H.; Sato, K.; Hatano, Y.; Yoshino, H.; Atsumi, M.; Kitaguchi, M.; Sasaki, S.; Kawaguchi, S.; Miyajima, H.; et al. Clinicogenetic study of PINK1 mutations in autosomal recessive early-onset parkinsonism. Neurology 2005, 64, 1955–1957. [Google Scholar] [CrossRef] [PubMed]
- Tunold, J.-A.; Geut, H.; Rozemuller, J.M.A.; Henriksen, S.P.; Toft, M.; van de Berg, W.D.J.; Pihlstrøm, L. APOE and MAPT Are Associated With Dementia in Neuropathologically Confirmed Parkinson’s Disease. Front. Neurol. 2021, 12, 52. [Google Scholar] [CrossRef]
- Bras, J.; Guerreiro, R.; Darwent, L.; Parkkinen, L.; Ansorge, O.; Escott-Price, V.; Hernandez, D.G.; Nalls, M.A.; Clark, L.N.; Honig, L.S.; et al. Genetic analysis implicates APOE, SNCA and suggests lysosomal dysfunction in the etiology of dementia with Lewy bodies. Hum. Mol. Genet. 2014, 23, 6139–6146. [Google Scholar] [CrossRef]
- Guerreiro, R.; Ross, O.A.; Kun-Rodrigues, C.; Hernandez, D.G.; Orme, T.; Eicher, J.D.; Shepherd, C.E.; Parkkinen, L.; Darwent, L.; Heckman, M.G.; et al. Investigating the genetic architecture of dementia with Lewy bodies: A two-stage genome-wide association study. Lancet Neurol. 2018, 17, 64–74. [Google Scholar] [CrossRef]
- Tsuang, D.; Leverenz, J.B.; Lopez, O.L.; Hamilton, R.L.; Bennett, D.A.; Schneider, J.A.; Buchman, A.S.; Larson, E.B.; Crane, P.K.; Kaye, J.A.; et al. APOE ε4 increases risk for dementia in pure synucleinopathies. JAMA Neurol. 2013, 70, 223–228. [Google Scholar] [CrossRef]
- Huang, X.; Chen, P.; Kaufer, D.I.; Troster, A.I.; Poole, C. Apolipoprotein E and dementia in Parkinson disease: A meta-analysis. Arch. Neurol. 2006, 63, 189–193. [Google Scholar] [CrossRef] [PubMed]
- Dickson, D.W.; Heckman, M.G.; Murray, M.E.; Soto, A.I.; Walton, R.L.; Diehl, N.N.; van Gerpen, J.A.; Uitti, R.J.; Wszolek, Z.K.; Ertekin-Taner, N.; et al. APOE ε4 is associated with severity of Lewy body pathology independent of Alzheimer pathology. Neurology 2018, 91, e1182–e1195. [Google Scholar] [CrossRef]
- Tan, M.M.X.; Lawton, M.A.; Jabbari, E.; Reynolds, R.H.; Iwaki, H.; Blauwendraat, C.; Kanavou, S.; Pollard, M.I.; Hubbard, L.; Malek, N.; et al. Genome-Wide Association Studies of Cognitive and Motor Progression in Parkinson’s Disease. Mov. Disord. 2021, 36, 424–433. [Google Scholar] [CrossRef]
- Davis, A.A.; Inman, C.E.; Wargel, Z.M.; Dube, U.; Freeberg, B.M.; Galluppi, A.; Haines, J.N.; Dhavale, D.D.; Miller, R.; Choudhury, F.A.; et al. APOE genotype regulates pathology and disease progression in synucleinopathy. Sci. Transl. Med. 2020, 12, eaay3069. [Google Scholar] [CrossRef]
- Zhao, N.; Attrebi, O.N.; Ren, Y.; Qiao, W.; Sonustun, B.; Martens, Y.A.; Meneses, A.D.; Li, F.; Shue, F.; Zheng, J.; et al. APOE4 exacerbates α-synuclein pathology and related toxicity independent of amyloid. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Pascale, E.; Di Battista, M.E.; Rubino, A.; Purcaro, C.; Valente, M.; Fattapposta, F.; Ferraguti, G.; Meco, G. Genetic Architecture of MAPT Gene Region in Parkinson Disease Subtypes. Front. Cell Neurosci. 2016, 10, 96. [Google Scholar] [CrossRef]
- Giasson, B.I.; Forman, M.S.; Higuchi, M.; Golbe, L.I.; Graves, C.L.; Kotzbauer, P.T.; Trojanowski, J.Q.; Lee, V.M. Initiation and synergistic fibrillization of tau and alpha-synuclein. Science 2003, 300, 636–640. [Google Scholar] [CrossRef] [PubMed]
- Seto-Salvia, N.; Clarimon, J.; Pagonabarraga, J.; Pascual-Sedano, B.; Campolongo, A.; Combarros, O.; Mateo, J.I.; Regana, D.; Martinez-Corral, M.; Marquie, M.; et al. Dementia risk in Parkinson disease: Disentangling the role of MAPT haplotypes. Arch. Neurol. 2011, 68, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Williams-Gray, C.H.; Evans, J.R.; Goris, A.; Foltynie, T.; Ban, M.; Robbins, T.W.; Brayne, C.; Kolachana, B.S.; Weinberger, D.R.; Sawcer, S.J.; et al. The distinct cognitive syndromes of Parkinson’s disease: 5 year follow-up of the CamPaIGN cohort. Brain 2009, 132, 2958–2969. [Google Scholar] [CrossRef] [PubMed]
- Goris, A.; Williams-Gray, C.H.; Clark, G.R.; Foltynie, T.; Lewis, S.J.; Brown, J.; Ban, M.; Spillantini, M.G.; Compston, A.; Burn, D.J.; et al. Tau and alpha-synuclein in susceptibility to, and dementia in, Parkinson’s disease. Ann. Neurol. 2007, 62, 145–153. [Google Scholar] [CrossRef]
- Mamah, C.E.; Lesnick, T.G.; Lincoln, S.J.; Strain, K.J.; de Andrade, M.; Bower, J.H.; Ahlskog, J.E.; Rocca, W.A.; Farrer, M.J.; Maraganore, D.M. Interaction of alpha-synuclein and tau genotypes in Parkinson’s disease. Ann. Neurol. 2005, 57, 439–443. [Google Scholar] [CrossRef]
- Botta-Orfila, T.; Ezquerra, M.; Rios, J.; Fernandez-Santiago, R.; Cervantes, S.; Samaranch, L.; Pastor, P.; Marti, M.J.; Munoz, E.; Valldeoriola, F.; et al. Lack of interaction of SNCA and MAPT genotypes in Parkinson’s disease. Eur. J. Neurol. 2011, 18, e32. [Google Scholar] [CrossRef]
- Elbaz, A.; Ross, O.A.; Ioannidis, J.P.; Soto-Ortolaza, A.I.; Moisan, F.; Aasly, J.; Annesi, G.; Bozi, M.; Brighina, L.; Chartier-Harlin, M.C.; et al. Independent and joint effects of the MAPT and SNCA genes in Parkinson disease. Ann. Neurol. 2011, 69, 778–792. [Google Scholar] [CrossRef]
- Biernacka, J.M.; Armasu, S.M.; Cunningham, J.M.; Ahlskog, J.E.; Chung, S.J.; Maraganore, D.M. Do interactions between SNCA, MAPT, and LRRK2 genes contribute to Parkinson’s disease susceptibility? Parkinsonism Relat. Disord. 2011, 17, 730–736. [Google Scholar] [CrossRef][Green Version]
- Dudbridge, F. Power and predictive accuracy of polygenic risk scores. PLoS Genet. 2013, 9, e1003348. [Google Scholar] [CrossRef]
- Foo, J.N.; Chew, E.G.Y.; Chung, S.J.; Peng, R.; Blauwendraat, C.; Nalls, M.A.; Mok, K.Y.; Satake, W.; Toda, T.; Chao, Y.; et al. Identification of Risk Loci for Parkinson Disease in Asians and Comparison of Risk Between Asians and Europeans: A Genome-Wide Association Study. JAMA Neurol. 2020, 77, 746–754. [Google Scholar] [CrossRef]
- Paul, K.C.; Schulz, J.; Bronstein, J.M.; Lill, C.M.; Ritz, B.R. Association of Polygenic Risk Score With Cognitive Decline and Motor Progression in Parkinson Disease. JAMA Neurol. 2018, 75, 360. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Del Tredici, K.; Rub, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Braak, H.; Ghebremedhin, E.; Rub, U.; Bratzke, H.; Del Tredici, K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004, 318, 121–134. [Google Scholar] [CrossRef]
- Beach, T.G.; Adler, C.H.; Lue, L.; Sue, L.I.; Bachalakuri, J.; Henry-Watson, J.; Sasse, J.; Boyer, S.; Shirohi, S.; Brooks, R.; et al. Unified staging system for Lewy body disorders: Correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol. 2009, 117, 613–634. [Google Scholar] [CrossRef]
- Rey, N.L.; Wesson, D.W.; Brundin, P. The olfactory bulb as the entry site for prion-like propagation in neurodegenerative diseases. Neurobiol. Dis. 2018, 109, 226–248. [Google Scholar] [CrossRef]
- Borghammer, P.; Van Den Berge, N. Brain-First versus Gut-First Parkinson’s Disease: A Hypothesis. J. Parkinsons Dis. 2019, 9, S281–S295. [Google Scholar] [CrossRef] [PubMed]
- Giguère, N.; Burke Nanni, S.; Trudeau, L.-E. On Cell Loss and Selective Vulnerability of Neuronal Populations in Parkinson’s Disease. Front. Neurol. 2018, 9, 455. [Google Scholar] [CrossRef] [PubMed]
- Double, K.L. Neuronal vulnerability in Parkinson’s disease. Parkinsonism Relat. Disord. 2012, 18, S52–S54. [Google Scholar] [CrossRef]
- Bloch, A.; Probst, A.; Bissig, H.; Adams, H.; Tolnay, M. Alpha-synuclein pathology of the spinal and peripheral autonomic nervous system in neurologically unimpaired elderly subjects. Neuropathol. Appl. Neurobiol. 2006, 32, 284–295. [Google Scholar] [CrossRef]
- Borghammer, P. The α-Synuclein Origin and Connectome Model (SOC Model) of Parkinson’s Disease: Explaining Motor Asymmetry, Non-Motor Phenotypes, and Cognitive Decline. J. Parkinsons Dis. 2021, 11, 455–474. [Google Scholar] [CrossRef] [PubMed]
- Aarsland, D.; Perry, R.; Brown, A.; Larsen, J.P.; Ballard, C. Neuropathology of dementia in Parkinson’s disease: A prospective, community-based study. Ann. Neurol. 2005, 58, 773–776. [Google Scholar] [CrossRef]
- Ballard, C.; Ziabreva, I.; Perry, R.; Larsen, J.P.; O’Brien, J.; McKeith, I.; Perry, E.; Aarsland, D. Differences in neuropathologic characteristics across the Lewy body dementia spectrum. Neurology 2006, 67, 1931–1934. [Google Scholar] [CrossRef]
- Hernandez, S.M.; Tikhonova, E.B.; Karamyshev, A.L. Protein-Protein Interactions in Alpha-Synuclein Biogenesis: New Potential Targets in Parkinson’s Disease. Front. Aging Neurosci. 2020, 12, 72. [Google Scholar] [CrossRef]
- Auluck, P.K.; Chan, H.Y.; Trojanowski, J.Q.; Lee, V.M.; Bonini, N.M. Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson’s disease. Science 2002, 295, 865–868. [Google Scholar] [CrossRef]
- Sot, B.; Rubio-Muñoz, A.; Leal-Quintero, A.; Martínez-Sabando, J.; Marcilla, M.; Roodveldt, C.; Valpuesta, J.M. The chaperonin CCT inhibits assembly of α-synuclein amyloid fibrils by a specific, conformation-dependent interaction. Sci. Rep. 2017, 7, 40859. [Google Scholar] [CrossRef] [PubMed]
- Giasson, B.I.; Duda, J.E.; Murray, I.V.J.; Chen, Q.; Souza, J.M.; Hurtig, H.I.; Ischiropoulos, H.; Trojanowski, J.Q.; Lee, V.M.-Y. Oxidative Damage Linked to Neurodegeneration by Selective alpha-Synuclein Nitration in Synucleinopathy Lesions. Science 2000, 290, 985–989. [Google Scholar] [CrossRef]
- Shimura, H.; Schlossmacher, M.G.; Hattori, N.; Frosch, M.P.; Trockenbacher, A.; Schneider, R.; Mizuno, Y.; Kosik, K.S.; Selkoe, D.J. Ubiquitination of a New Form of alpha-Synuclein by Parkin from Human Brain: Implications for Parkinson’s Disease. Science 2001, 293, 263–269. [Google Scholar] [CrossRef]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. α-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef]
- Li, W.; West, N.; Colla, E.; Pletnikova, O.; Troncoso, J.C.; Marsh, L.; Dawson, T.M.; Jäkälä, P.; Hartmann, T.; Price, D.L. Aggregation promoting C-terminal truncation of α-synuclein is a normal cellular process and is enhanced by the familial Parkinson’s disease-linked mutations. Proc. Natl. Acad. Sci. USA 2005, 102, 2162–2167. [Google Scholar] [CrossRef] [PubMed]
- Dorval, V.; Fraser, P.E. Small ubiquitin-like modifier (SUMO) modification of natively unfolded proteins tau and alpha-synuclein. J. Biol. Chem. 2006, 281, 9919–9924. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.P.; Walker, D.E.; Goldstein, J.M.; de Laat, R.; Banducci, K.; Caccavello, R.J.; Barbour, R.; Huang, J.; Kling, K.; Lee, M.; et al. Phosphorylation of Ser-129 Is the Dominant Pathological Modification of a-Synuclein in Familial and Sporadic Lewy Body Disease. J. Biol. Chem. 2006, 281, 29739–29752. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.G.; Lue, L.-F.; Adler, C.H.; Shill, H.A.; Caviness, J.N.; Sabbagh, M.N.; Akiyama, H.; Serrano, G.E.; Sue, L.I.; Beach, T.G. Changes in properties of serine 129 phosphorylated α-synuclein with progression of Lewy-type histopathology in human brains. Exp. Neurol. 2013, 240, 190–204. [Google Scholar] [CrossRef] [PubMed]
- McFarland, M.A.; Ellis, C.E.; Markey, S.P.; Nussbaum, R.L. Proteomics analysis identifies phosphorylation-dependent alpha-synuclein protein interactions. Mol. Cell Proteom. 2008, 7, 2123–2137. [Google Scholar] [CrossRef]
- Leverenz, J.B.; Umar, I.; Wang, Q.; Montine, T.J.; McMillan, P.J.; Tsuang, D.W.; Jin, J.; Pan, C.; Shin, J.; Zhu, D.; et al. Proteomic identification of novel proteins in cortical lewy bodies. Brain Pathol. 2007, 17, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Gorbatyuk, O.S.; Li, S.; Sullivan, L.F.; Chen, W.; Kondrikova, G.; Manfredsson, F.P.; Mandel, R.J.; Muzyczka, N. The phosphorylation state of Ser-129 in human alpha-synuclein determines neurodegeneration in a rat model of Parkinson disease. Proc. Natl. Acad. Sci. USA 2008, 105, 763–768. [Google Scholar] [CrossRef]
- Febbraro, F.; Sahin, G.; Farran, A.; Soares, S.; Jensen, P.H.; Kirik, D.; Romero-Ramos, M. Ser129D mutant alpha-synuclein induces earlier motor dysfunction while S129A results in distinctive pathology in a rat model of Parkinson’s disease. Neurobiol. Dis. 2013, 56, 47–58. [Google Scholar] [CrossRef]
- Lee, H.J.; Suk, J.E.; Bae, E.J.; Lee, J.H.; Paik, S.R.; Lee, S.J. Assembly-dependent endocytosis and clearance of extracellular alpha-synuclein. Int. J. Biochem. Cell Biol. 2008, 40, 1835–1849. [Google Scholar] [CrossRef]
- Lee, H.J.; Suk, J.E.; Bae, E.J.; Lee, S.J. Clearance and deposition of extracellular alpha-synuclein aggregates in microglia. Biochem. Biophys. Res. Commun. 2008, 372, 423–428. [Google Scholar] [CrossRef]
- Giasson, B.I.; Lee, V.M.Y. Are Ubiquitination Pathways Central to Parkinson’s Disease? Cell 2003, 114, 1–8. [Google Scholar] [CrossRef]
- Tofaris, G.K.; Layfield, R.; Spillantini, M.G. Alpha-synuclein metabolism and aggregation is linked to ubiquitin-independent degradation by the proteasome. FEBS Lett. 2001, 509, 22–26. [Google Scholar] [CrossRef]
- Vogiatzi, T.; Xilouri, M.; Vekrellis, K.; Stefanis, L. Wild Type α-Synuclein Is Degraded by Chaperone-mediated Autophagy and Macroautophagy in Neuronal Cells. J. Biol. Chem. 2008, 283, 23542–23556. [Google Scholar] [CrossRef]
- Lee, V.M.; Giasson, B.I.; Trojanowski, J.Q. More than just two peas in a pod: Common amyloidogenic properties of tau and alpha-synuclein in neurodegenerative diseases. Trends Neurosci. 2004, 27, 129–134. [Google Scholar] [CrossRef]
- Badiola, N.; De Oliveira, R.M.; Herrera, F.; Guardia-Laguarta, C.; Gonçalves, S.A.; Pera, M.; Suárez-Calvet, M.; Clarimon, J.; Outeiro, T.F.; Lleó, A. Tau Enhances α-Synuclein Aggregation and Toxicity in Cellular Models of Synucleinopathy. PLoS ONE 2011, 6, e26609. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Rockenstein, E.; Veinbergs, I.; Sagara, Y.; Mallory, M.; Hashimoto, M.; Mucke, L. β-Amyloid peptides enhance α-synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer’s disease and Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2001, 98, 12245–12250. [Google Scholar] [CrossRef]
- Clinton, L.K.; Blurton-Jones, M.; Myczek, K.; Trojanowski, J.Q.; LaFerla, F.M. Synergistic Interactions between Abeta, tau, and alpha-synuclein: Acceleration of neuropathology and cognitive decline. J. Neurosci. 2010, 30, 7281–7289. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.; Malek, N.; Grosset, K.; Cullen, B.; Gentleman, S.; Grosset, D.G. Neuropathology of dementia in patients with Parkinson’s disease: A systematic review of autopsy studies. J. Neurol. Neurosurg. Psychiatry 2019, 90, 1234–1243. [Google Scholar] [CrossRef]
- Irwin, D.J.; Lee, V.M.Y.; Trojanowski, J.Q. Parkinson’s disease dementia: Convergence of α-synuclein, tau and amyloid-β pathologies. Nat. Rev. Neurosci. 2013, 14, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Gomperts, S.N.; Locascio, J.J.; Makaretz, S.J.; Schultz, A.; Caso, C.; Vasdev, N.; Sperling, R.; Growdon, J.H.; Dickerson, B.C.; Johnson, K. Tau Positron Emission Tomographic Imaging in the Lewy Body Diseases. JAMA Neurol. 2016, 73, 1334–1341. [Google Scholar] [CrossRef] [PubMed]
- Bassil, F.; Meymand, E.S.; Brown, H.J.; Xu, H.; Cox, T.O.; Pattabhiraman, S.; Maghames, C.M.; Wu, Q.; Zhang, B.; Trojanowski, J.Q.; et al. α-Synuclein modulates tau spreading in mouse brains. J. Exp. Med. 2021, 218, e20192193. [Google Scholar] [CrossRef]
- Hong, Z.; Shi, M.; Chung, K.A.; Quinn, J.F.; Peskind, E.R.; Galasko, D.; Jankovic, J.; Zabetian, C.P.; Leverenz, J.B.; Baird, G.; et al. DJ-1 and alpha-synuclein in human cerebrospinal fluid as biomarkers of Parkinson’s disease. Brain 2010, 133, 713–726. [Google Scholar] [CrossRef] [PubMed]
- Mollenhauer, B.; Cullen, V.; Kahn, I.; Krastins, B.; Outeiro, T.F.; Pepivani, I.; Ng, J.; Schulz-Schaeffer, W.; Kretzschmar, H.A.; McLean, P.J.; et al. Direct quantification of CSF alpha-synuclein by ELISA and first cross-sectional study in patients with neurodegeneration. Exp. Neurol. 2008, 213, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Mollenhauer, B.; Locascio, J.J.; Schulz-Schaeffer, W.; Sixel-Döring, F.; Trenkwalder, C.; Schlossmacher, M.G. α-Synuclein and tau concentrations in cerebrospinal fluid of patients presenting with parkinsonism: A cohort study. Lancet Neurol. 2011, 10, 230–240. [Google Scholar] [CrossRef]
- Tokuda, T.; Salem, S.A.; Allsop, D.; Mizuno, T.; Nakagawa, M.; Qureshi, M.M.; Locascio, J.J.; Schlossmacher, M.G.; El-Agnaf, O.M. Decreased alpha-synuclein in cerebrospinal fluid of aged individuals and subjects with Parkinson’s disease. Biochem. Biophys. Res. Commun. 2006, 349, 162–166. [Google Scholar] [CrossRef]
- Wennström, M.; Surova, Y.; Hall, S.; Nilsson, C.; Minthon, L.; Boström, F.; Hansson, O.; Nielsen, H.M. Low CSF levels of both α-synuclein and the α-synuclein cleaving enzyme neurosin in patients with synucleinopathy. PLoS ONE 2013, 8, e53250. [Google Scholar] [CrossRef]
- Stewart, T.; Liu, C.; Ginghina, C.; Cain, K.C.; Auinger, P.; Cholerton, B.; Shi, M.; Zhang, J. Cerebrospinal fluid α-synuclein predicts cognitive decline in Parkinson disease progression in the DATATOP cohort. Am. J. Pathol. 2014, 184, 966–975. [Google Scholar] [CrossRef]
- Hall, S.; Surova, Y.; Öhrfelt, A.; Zetterberg, H.; Lindqvist, D.; Hansson, O. CSF biomarkers and clinical progression of Parkinson disease. Neurology 2015, 84, 57–63. [Google Scholar] [CrossRef]
- Goldman, J.G.; Andrews, H.; Amara, A.; Naito, A.; Alcalay, R.N.; Shaw, L.M.; Taylor, P.; Xie, T.; Tuite, P.; Henchcliffe, C.; et al. Cerebrospinal fluid, plasma, and saliva in the BioFIND study: Relationships among biomarkers and Parkinson’s disease Features. Mov. Disord. 2018, 33, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-H.; Yang, S.-Y.; Horng, H.-E.; Yang, C.-C.; Chieh, J.-J.; Chen, H.-H.; Liu, B.-H.; Chiu, M.-J. Plasma α-synuclein predicts cognitive decline in Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2017, 88, 818–824. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-W.; Yang, S.-Y.; Yang, C.-C.; Chang, C.-W.; Wu, Y.-R. Plasma and Serum Alpha-Synuclein as a Biomarker of Diagnosis in Patients with Parkinson’s Disease. Front. Neurol. 2020, 10, 1388. [Google Scholar] [CrossRef]
- Maass, F.; Rikker, S.; Dambeck, V.; Warth, C.; Tatenhorst, L.; Csoti, I.; Schmitz, M.; Zerr, I.; Leha, A.; Bähr, M.; et al. Increased alpha-synuclein tear fluid levels in patients with Parkinson’s disease. Sci. Rep. 2020, 10, 8507. [Google Scholar] [CrossRef]
- Wang, Z.; Becker, K.; Donadio, V.; Siedlak, S.; Yuan, J.; Rezaee, M.; Incensi, A.; Kuzkina, A.; Orru, C.D.; Tatsuoka, C.; et al. Skin alpha-Synuclein Aggregation Seeding Activity as a Novel Biomarker for Parkinson Disease. JAMA Neurol. 2021, 78, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Donadio, V.; Incensi, A.; El-Agnaf, O.; Rizzo, G.; Vaikath, N.; Del Sorbo, F.; Scaglione, C.; Capellari, S.; Elia, A.; Stanzani Maserati, M.; et al. Skin α-synuclein deposits differ in clinical variants of synucleinopathy: An in vivo study. Sci. Rep. 2018, 8, 14246. [Google Scholar] [CrossRef]
- Mikolaenko, I.; Pletnikova, O.; Kawas, C.H.; O’Brien, R.; Resnick, S.M.; Crain, B.; Troncoso, J.C. Alpha-synuclein lesions in normal aging, Parkinson disease, and Alzheimer disease: Evidence from the Baltimore Longitudinal Study of Aging (BLSA). J. Neuropathol. Exp. Neurol. 2005, 64, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Hughes, A.J.; Daniel, S.E.; Blankson, S.; Lees, A.J. A clinicopathologic study of 100 cases of Parkinson’s disease. Arch. Neurol. 1993, 50, 140–148. [Google Scholar] [CrossRef]
- Sabbagh, M.N.; Adler, C.H.; Lahti, T.J.; Connor, D.J.; Vedders, L.; Peterson, L.K.; Caviness, J.N.; Shill, H.A.; Sue, L.I.; Ziabreva, I. Parkinson’s disease with dementia: Comparing patients with and without Alzheimer pathology. Alzheimer Dis. Assoc. Disord. 2009, 23, 295. [Google Scholar] [CrossRef]
- Lin, C.-H.; Wu, R.-M. Biomarkers of cognitive decline in Parkinson’s disease. Parkinsonism Relat. Disord. 2015, 21, 431–443. [Google Scholar] [CrossRef]
- Jellinger, K.; Seppi, K.; Wenning, G.; Poewe, W. Impact of coexistent Alzheimer pathology on the natural history of Parkinson’s disease. J. Neural Transm. 2002, 109, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.-Y.; Zuo, L.-J.; Wang, F.; Chen, Z.-J.; Hu, Y.; Wang, Y.-J.; Wang, X.-M.; Zhang, W. Potential biomarkers relating pathological proteins, neuroinflammatory factors and free radicals in PD patients with cognitive impairment: A cross-sectional study. BMC Neurol. 2014, 14, 113. [Google Scholar] [CrossRef]
- Liu, C.; Cholerton, B.; Shi, M.; Ginghina, C.; Cain, K.C.; Auinger, P.; Zhang, J. The Parkinson Study Group DATATOP Investigators. CSF tau and tau/Aβ42 predict cognitive decline in Parkinson’s disease. Parkinsonism Relat. Disord. 2015, 21, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Tang, Y.; Bai, X.; Liang, X.; Fan, Y.; Shen, Y.; Huang, F.; Wang, J. Association of the serum microRNA-29 family with cognitive impairment in Parkinson’s disease. Aging (Albany NY) 2020, 12, 13518. [Google Scholar] [CrossRef]
- Yang, T.T.; Liu, C.G.; Gao, S.C.; Zhang, Y.; Wang, P.C. The serum exosome derived MicroRNA− 135a,− 193b, and− 384 were potential Alzheimer’s disease biomarkers. Biomed. Environ. Sci. 2018, 31, 87–96. [Google Scholar]
- Zhao, L.; Wang, Z. MicroRNAs: Game changers in the regulation of α-synuclein in Parkinson’s disease. Parkinsons Dis. 2019, 2019, 1743183. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Rapp, J.; Rainone, S.; Hébert, S.S. MicroRNAs underlying memory deficits in neurodegenerative disorders. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2017, 73, 79–86. [Google Scholar] [CrossRef]
- Kanagaraj, N.; Beiping, H.; Dheen, S.; Tay, S. Downregulation of miR-124 in MPTP-treated mouse model of Parkinson’s disease and MPP iodide-treated MN9D cells modulates the expression of the calpain/cdk5 pathway proteins. Neuroscience 2014, 272, 167–179. [Google Scholar] [CrossRef]
- Wang, H.; Ye, Y.; Zhu, Z.; Mo, L.; Lin, C.; Wang, Q.; Wang, H.; Gong, X.; He, X.; Lu, G. MiR-124 Regulates Apoptosis and Autophagy Process in MPTP Model of P arkinson’s Disease by Targeting to B im. Brain Pathol. 2016, 26, 167–176. [Google Scholar] [CrossRef]
- Barbagallo, C.; Mostile, G.; Baglieri, G.; Giunta, F.; Luca, A.; Raciti, L.; Zappia, M.; Purrello, M.; Ragusa, M.; Nicoletti, A. Specific signatures of serum miRNAs as potential biomarkers to discriminate clinically similar neurodegenerative and vascular-related diseases. Cell. Mol. Neurobiol. 2020, 40, 531–546. [Google Scholar] [CrossRef]
- Dos Santos, M.C.T.; Barreto-Sanz, M.A.; Correia, B.R.S.; Bell, R.; Widnall, C.; Perez, L.T.; Berteau, C.; Schulte, C.; Scheller, D.; Berg, D. miRNA-based signatures in cerebrospinal fluid as potential diagnostic tools for early stage Parkinson’s disease. Oncotarget 2018, 9, 17455. [Google Scholar] [CrossRef] [PubMed]
- Gui, Y.; Liu, H.; Zhang, L.; Lv, W.; Hu, X. Altered microRNA profiles in cerebrospinal fluid exosome in Parkinson disease and Alzheimer disease. Oncotarget 2015, 6, 37043. [Google Scholar] [CrossRef] [PubMed]
- Rajgor, D. Macro roles for microRNAs in neurodegenerative diseases. Non-Coding RNA Res. 2018, 3, 154–159. [Google Scholar] [CrossRef]
- Sonntag, K.-C. MicroRNAs and deregulated gene expression networks in neurodegeneration. Brain Res. 2010, 1338, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Lau, P.; Bossers, K.; Janky, R.S.; Salta, E.; Frigerio, C.S.; Barbash, S.; Rothman, R.; Sierksma, A.S.; Thathiah, A.; Greenberg, D. Alteration of the micro RNA network during the progression of Alzheimer’s disease. EMBO Mol. Med. 2013, 5, 1613–1634. [Google Scholar] [CrossRef]
- Doxakis, E. Post-transcriptional regulation of α-synuclein expression by mir-7 and mir-153. J. Biol. Chem. 2010, 285, 12726–12734. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Zhu, H.; Xu, Y.; Huang, L.; Ma, C.; Deng, W.; Liu, Y.; Qin, C. MicroRNA-153 negatively regulates the expression of amyloid precursor protein and amyloid precursor-like protein 2. Brain Res. 2012, 1455, 103–113. [Google Scholar] [CrossRef]
- Lee, J.G.; Takahama, S.; Zhang, G.; Tomarev, S.I.; Ye, Y. Unconventional secretion of misfolded proteins promotes adaptation to proteasome dysfunction in mammalian cells. Nat. Cell Biol. 2016, 18, 765–776. [Google Scholar] [CrossRef]
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, 6748. [Google Scholar] [CrossRef]
- Fiandaca, M.S.; Kapogiannis, D.; Mapstone, M.; Boxer, A.; Eitan, E.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Federoff, H.J.; Miller, B.L.; et al. Identification of preclinical Alzheimer’s disease by a profile of pathogenic proteins in neurally derived blood exosomes: A case-control study. Alzheimers Dement. 2015, 11, 600–607.e1. [Google Scholar] [CrossRef]
- Serpente, M.; Fenoglio, C.; D’Anca, M.; Arcaro, M.; Sorrentino, F.; Visconte, C.; Arighi, A.; Fumagalli, G.G.; Porretti, L.; Cattaneo, A.; et al. MiRNA Profiling in Plasma Neural-Derived Small Extracellular Vesicles from Patients with Alzheimer’s Disease. Cells 2020, 9, 1443. [Google Scholar] [CrossRef]
- Stuendl, A.; Kunadt, M.; Kruse, N.; Bartels, C.; Moebius, W.; Danzer, K.M.; Mollenhauer, B.; Schneider, A. Induction of α-synuclein aggregate formation by CSF exosomes from patients with Parkinson’s disease and dementia with Lewy bodies. Brain 2015, 139, 481–494. [Google Scholar] [CrossRef]
- Jiang, C.; Hopfner, F.; Katsikoudi, A.; Hein, R.; Catli, C.; Evetts, S.; Huang, Y.; Wang, H.; Ryder, J.W.; Kuhlenbaeumer, G.; et al. Serum neuronal exosomes predict and differentiate Parkinson’s disease from atypical parkinsonism. J. Neurol. Neurosurg. Psychiatry 2020, 91, 720–729. [Google Scholar] [CrossRef]
- Chung, C.-C.; Chan, L.; Chen, J.-H.; Bamodu, O.A.; Hong, C.-T. Neurofilament light chain level in plasma extracellular vesicles and Parkinson’s disease. Ther. Adv. Neurol. Disord. 2020, 13, 1756286420975917. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.-Y.; Chan, L.; Chung, C.-C.; Chiu, J.-Y.; Hsieh, Y.-C.; Hong, C.-T. Altered insulin receptor substrate 1 phosphorylation in blood neuron-derived extracellular vesicles from patients with Parkinson’s disease. Front. Cell Dev. Biol. 2020, 8, 1490. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.-C.; Chan, L.; Chen, J.-H.; Hung, Y.-C.; Hong, C.-T. Plasma Extracellular Vesicle α-Synuclein Level in Patients with Parkinson’s Disease. Biomolecules 2021, 11, 744. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Huber, B.R.; Zhang, J. Biomarkers for cognitive impairment in Parkinson disease. Brain Pathol. 2010, 20, 660–671. [Google Scholar] [CrossRef] [PubMed]
- Emmanouilidou, E.; Melachroinou, K.; Roumeliotis, T.; Garbis, S.D.; Ntzouni, M.; Margaritis, L.H.; Stefanis, L.; Vekrellis, K. Cell-Produced α-Synuclein Is Secreted in a Calcium-Dependent Manner by Exosomes and Impacts Neuronal Survival. J. Neurosci. 2010, 30, 6838–6851. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Seow, Y.; Schapira, A.H.; Gardiner, C.; Sargent, I.L.; Wood, M.J.A.; Cooper, J.M. Lysosomal dysfunction increases exosome-mediated alpha-synuclein release and transmission. Neurobiol. Dis. 2011, 42, 360–367. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fan, T.-S.; Liu, S.C.-H.; Wu, R.-M. Alpha-Synuclein and Cognitive Decline in Parkinson Disease. Life 2021, 11, 1239. https://doi.org/10.3390/life11111239
Fan T-S, Liu SC-H, Wu R-M. Alpha-Synuclein and Cognitive Decline in Parkinson Disease. Life. 2021; 11(11):1239. https://doi.org/10.3390/life11111239
Chicago/Turabian StyleFan, Tian-Sin, Sam Chi-Hao Liu, and Ruey-Meei Wu. 2021. "Alpha-Synuclein and Cognitive Decline in Parkinson Disease" Life 11, no. 11: 1239. https://doi.org/10.3390/life11111239
APA StyleFan, T.-S., Liu, S. C.-H., & Wu, R.-M. (2021). Alpha-Synuclein and Cognitive Decline in Parkinson Disease. Life, 11(11), 1239. https://doi.org/10.3390/life11111239