Quis Custodiet Ipsos Custodes (Who Controls the Controllers)? Two Decades of Studies on HDAC9


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Class IIa Histone Deacetylases (HDACs): A Short Introduction
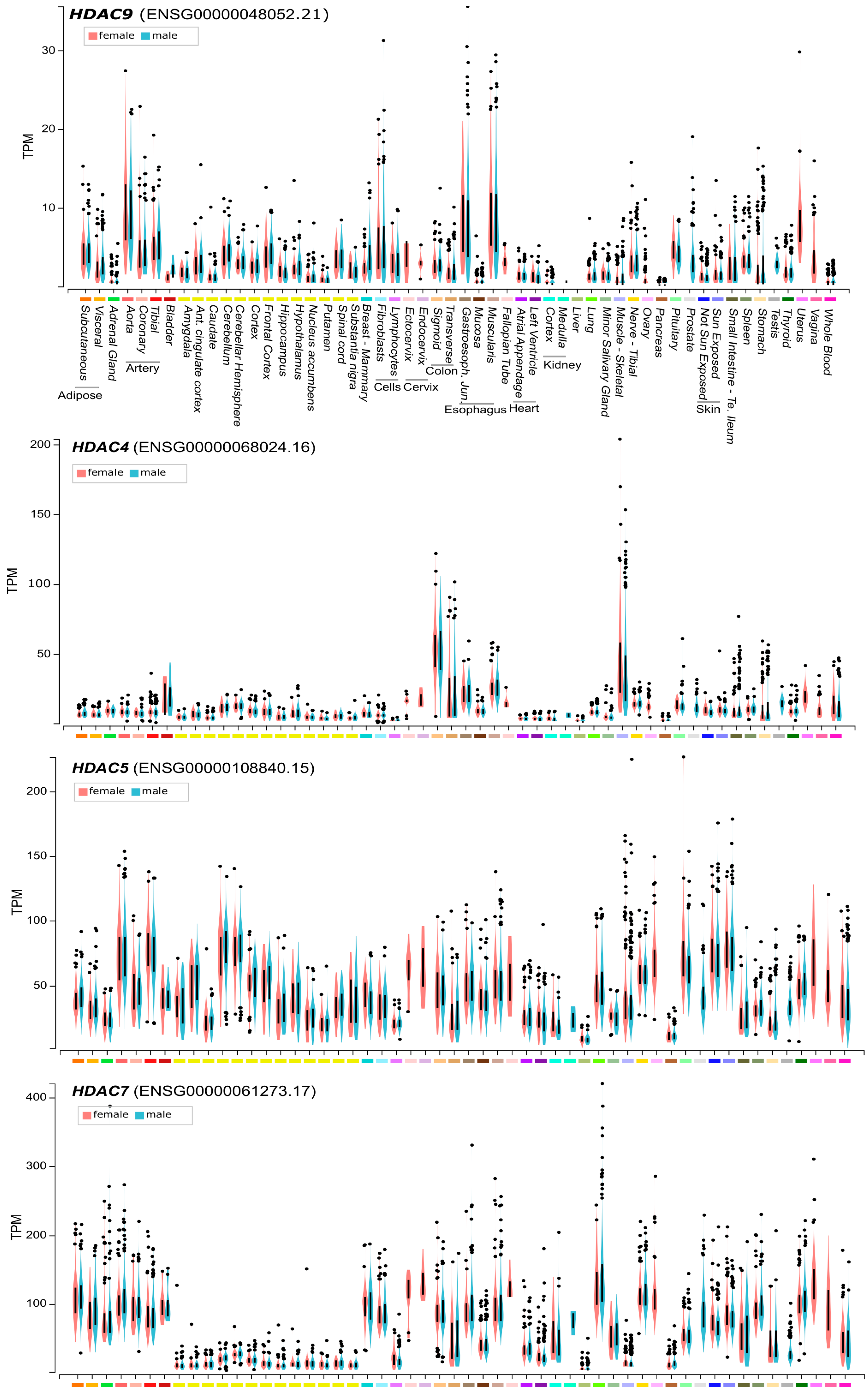
2. HDAC9: An Historical Perspective
3. Regulation of HDAC9 Expression during Differentiation: The Adipogenesis and the Risk of Diabetes
4. HDAC9 in Atherosclerotic Plaques and Cardiovascular Disease (CVD)
5. HDAC9 Expression in Inflammation and in the Immune Response
6. Regulation of HDAC9 Expression and Cancer
7. HDAC9 and DNA Damage
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Lahm, A.; Paolini, C.; Pallaoro, M.; Nardi, M.C.; Jones, P.; Neddermann, P.; Sambucini, S.; Bottomley, M.J.; Lo Surdo, P.; Carfí, A.; et al. Unraveling the hidden catalytic activity of vertebrate class IIa histone deacetylases. Proc. Natl. Acad. Sci. USA 2007, 104, 17335–17340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Giorgio, E.; Brancolini, C. Regulation of class IIa HDAC activities: It is not only matter of subcellular localization. Epigenomics 2016, 8, 251–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clocchiatti, A.; Di Giorgio, E.; Demarchi, F.; Brancolini, C. Beside the MEF2 axis: Unconventional functions of HDAC4. Cell Signal. 2013, 25, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Asfaha, Y.; Schrenk, C.; Alves Avelar, L.A.; Hamacher, A.; Pflieger, M.; Kassack, M.U.; Kurz, T. Recent advances in class IIa histone deacetylases research. Bioorg. Med. Chem. 2019, 27, 115087. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, Q.; Ding, Y.; Liu, Y.; Zhao, D.; Zhao, K.; Shen, Q.; Liu, X.; Zhu, X.; Li, N.; et al. Methyltransferase Dnmt3a upregulates HDAC9 to deacetylate the kinase TBK1 for activation of antiviral innate immunity. Nat. Immunol. 2016, 17, 806–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paroni, G.; Fontanini, A.; Cernotta, N.; Foti, C.; Gupta, M.P.; Yang, X.J.; Fasino, D.; Brancolini, C. Dephosphorylation and caspase processing generate distinct nuclear pools of histone deacetylase 4. Mol. Cell Biol. 2007, 27, 6718–6732. [Google Scholar] [CrossRef] [Green Version]
- Cernotta, N.; Clocchiatti, A.; Florean, C.; Brancolini, C. Ubiquitin-dependent degradation of HDAC4, a new regulator of random cell motility. Mol. Biol. Cell. 2011, 22, 278–289. [Google Scholar] [CrossRef]
- Lehmann, L.H.; Jebessa, Z.H.; Kreusser, M.M.; Horsch, A.; He, T.; Kronlage, M.; Dewenter, M.; Sramek, V.; Oehl, U.; Krebs-Haupenthal, J.; et al. A proteolytic fragment of histone deacetylase 4 protects the heart from failure by regulating the hexosamine biosynthetic pathway. Nat. Med. 2018, 24, 62–72. [Google Scholar] [CrossRef]
- Parra, M. Class IIa HDACs-New insights into their functions in physiology and pathology. FEBS J. 2015, 282, 1736–1744. [Google Scholar] [CrossRef]
- Parra, M.; Verdin, E. Regulatory signal transduction pathways for class IIa histone deacetylases. Curr. Opin. Pharmacol. 2010, 10, 454–460. [Google Scholar] [CrossRef]
- Paroni, G.; Cernotta, N.; Dello Russo, C.; Gallinari, P.; Pallaoro, M.; Foti, C.; Talamo, F.; Orsatti, L.; Steinkühler, C.; Brancolini, C. PP2A regulates HDAC4 nuclear import. Mol. Biol. Cell 2008, 19, 655–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sparrow, D.B.; Miska, E.A.; Langley, E.; Reynaud-Deonauth, S.; Kotecha, S.; Towers, N.; Spohr, G.; Kouzarides, T.; Mohun, T.J. MEF-2 function is modified by a novel co-repressor, MITR. EMBO J. 1999, 18, 5085–5098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Richon, V.M.; Rifkind, R.A.; Marks, P.A. Identification of a transcriptional repressor related to the noncatalytic domain of histone deacetylases 4 and 5. Proc. Natl. Acad. Sci. USA 2000, 97, 1056–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.L.; McKinsey, T.A.; Lu, J.R.; Olson, E.N. Association of COOH-terminal-binding protein (CtBP) and MEF2-interacting transcription repressor (MITR) contributes to transcriptional repression of the MEF2 transcription factor. J. Biol. Chem. 2001, 276, 35–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.L.; McKinsey, T.A.; Olson, E.N. The transcriptional corepressor MITR is a signal-responsive inhibitor of myogenesis. Proc. Natl. Acad. Sci. USA 2001, 98, 7354–7359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Giorgio, E.; Clocchiatti, A.; Piccinin, S.; Sgorbissa, A.; Viviani, G.; Peruzzo, P.; Romeo, S.; Rossi, S.; Dei Tos, A.P.; Maestro, R.; et al. MEF2 is a converging hub for histone deacetylase 4 and phosphatidylinositol 3-kinase/Akt-induced transformation. Mol. Cell Biol. 2013, 33, 4473–4491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Backs, J.; Backs, T.; Bezprozvannaya, S.; McKinsey, T.A.; Olson, E.N. Histone deacetylase 5 acquires calcium/calmodulin-dependent kinase II responsiveness by oligomerization with histone deacetylase 4. Mol. Cell Biol. 2008, 28, 3437–3445. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Marks, P.A.; Rifkind, R.A.; Richon, V.M. Cloning and characterization of a histone deacetylase, HDAC9. Proc. Natl. Acad. Sci. USA 2001, 98, 10572–10577. [Google Scholar] [CrossRef] [Green Version]
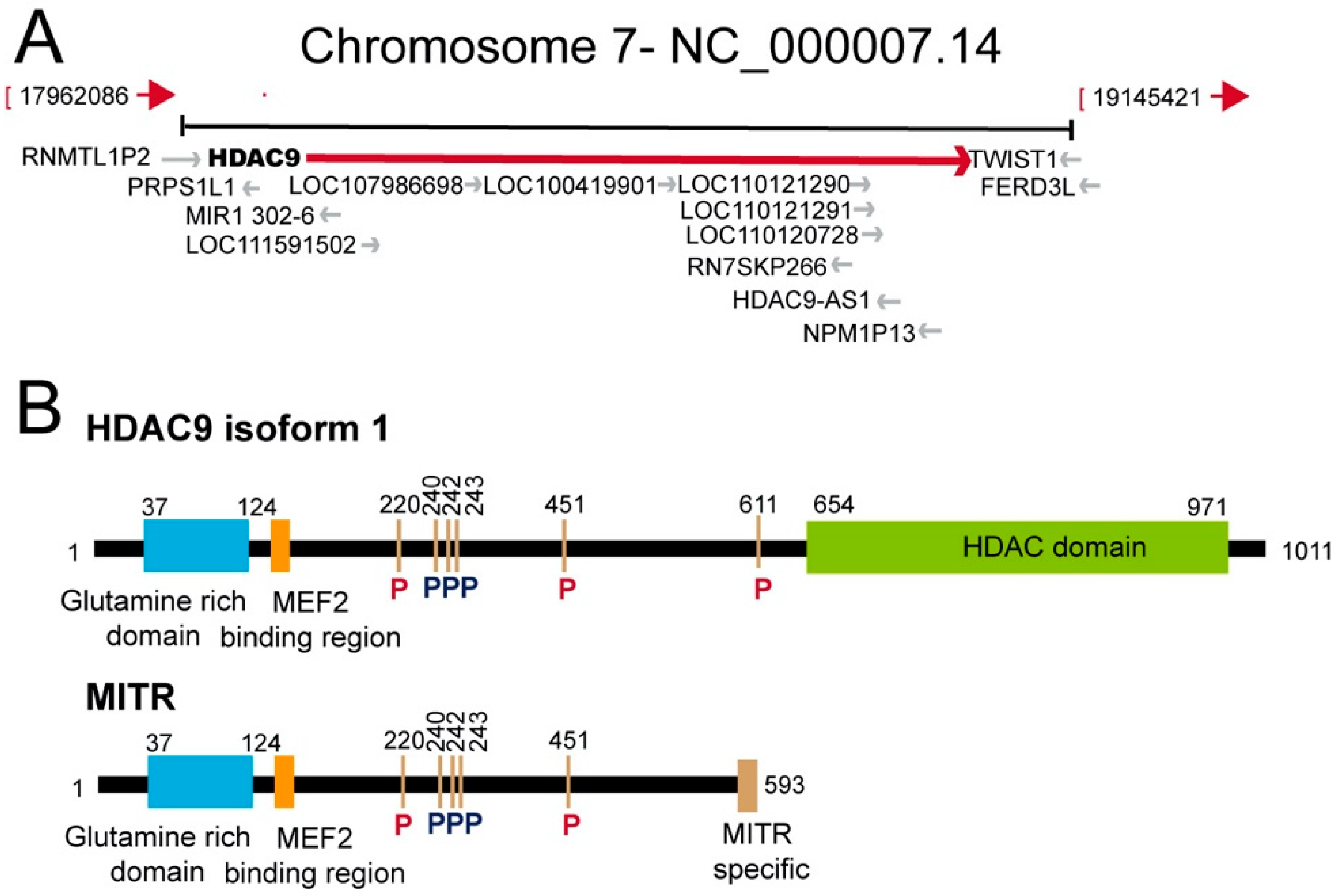
- Mahlknecht, U.; Schnittger, S.; Will, J.; Cicek, N.; Hoelzer, D. Chromosomal organization and localization of the human histone deacetylase 9 gene (HDAC9). Biochem. Biophys. Res. Commun. 2002, 293, 182–191. [Google Scholar] [CrossRef]
- Petrie, K.; Guidez, F.; Howell, L.; Healy, L.; Waxman, S.; Greaves, M.; Zelent, A. The histone deacetylase 9 gene encodes multiple protein isoforms. J. Biol. Chem. 2003, 278, 16059–16072. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.L.; McKinsey, T.A.; Olson, E.N. Association of class II histone deacetylases with heterochromatin protein 1: Potential role for histone methylation in control of muscle differentiation. Mol. Cell Biol. 2002, 22, 7302–7312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.L.; McKinsey, T.A.; Chang, S.; Antos, C.L.; Hill, J.A.; Olson, E.N. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell 2002, 110, 479–488. [Google Scholar] [CrossRef] [Green Version]
- He, T.; Huang, J.; Chen, L.; Han, G.; Stanmore, D.; Krebs-Haupenthal, J.; Avkiran, M.; Hagenmüller, M.; Backs, J. Cyclic AMP represses pathological MEF2 activation by myocyte-specific hypo-phosphorylation of HDAC5. J. Mol. Cell Cardiol. 2020, 145, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Harrison, B.C.; Huynh, K.; Lundgaard, G.L.; Helmke, S.M.; Perryman, M.B.; McKinsey, T.A. Protein kinase C-related kinase targets nuclear localization signals in a subset of class IIa histone deacetylases. FEBS Lett. 2010, 584, 1103–1110. [Google Scholar] [CrossRef] [Green Version]
- Hu, S.; Cho, E.H.; Lee, J.Y. Histone Deacetylase 9: Its Role in the Pathogenesis of Diabetes and Other Chronic Diseases. Diabetes Metab. J. 2020, 44, 234–244. [Google Scholar] [CrossRef]
- Di Giorgio, E.; Dalla, E.; Franforte, E.; Paluvai, H.; Minisini, M.; Trevisanut, M.; Picco, R.; Brancolini, C. Different class IIa HDACs repressive complexes regulate specific epigenetic responses related to cell survival in leiomyosarcoma cells. Nucleic Acids Res. 2020, 48, 646–664. [Google Scholar] [CrossRef] [Green Version]
- Clocchiatti, A.; Di Giorgio, E.; Viviani, G.; Streuli, C.; Sgorbissa, A.; Picco, R.; Cutano, V.; Brancolini, C. The MEF2-HDAC axis controls proliferation of mammary epithelial cells and acini formation in vitro. J. Cell Sci. 2015, 128, 3961–3976. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.H.; Yeh, F.L.; Yeh, S.P.; Ma, H.T.; Hung, S.C.; Hung, M.C.; Li, L.Y. Myocyte enhancer factor-2 interacting transcriptional repressor (MITR) is a switch that promotes osteogenesis and inhibits adipogenesis of mesenchymal stem cells by inactivating peroxisome proliferator-activated receptor gamma-2. J. Biol. Chem. 2011, 286, 10671–10680. [Google Scholar] [CrossRef] [Green Version]
- Jin, Z.; Wei, W.; Huynh, H.; Wan, Y. HDAC9 Inhibits Osteoclastogenesis via Mutual Suppression of PPARγ/RANKL Signaling. Mol. Endocrinol. 2015, 29, 730–738. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, T.K.; Idelman, G.; Blanco, V.; Blomkalns, A.L.; Piegore, M.G., Jr.; Weintraub, D.S.; Kumar, S.; Rajsheker, S.; Manka, D.; Rudich, S.M.; et al. Histone deacetylase 9 is a negative regulator of adipogenic differentiation. J. Biol. Chem. 2011, 286, 27836–27847. [Google Scholar] [CrossRef] [Green Version]
- Wong, R.H.; Chang, I.; Hudak, C.S.; Hyun, S.; Kwan, H.Y.; Sul, H.S. A role of DNA-PK for the metabolic gene regulation in response to insulin. Cell 2009, 136, 1056–1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.J.; Cheng, P.; Liang, M.K.; Chen, Y.S.; Lu, Q.; Wang, J.Y.; Xia, Z.Y.; Zhou, H.D.; Cao, X.; Xie, H.; et al. MicroRNA-188 regulates age-related switch between osteoblast and adipocyte differentiation. J. Clin. Investig. 2015, 125, 1509–1522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, T.K.; Basford, J.E.; Knoll, E.; Tong, W.S.; Blanco, V.; Blomkalns, A.L.; Rudich, S.; Lentsch, A.B.; Hui, D.Y.; Weintraub, N.L. HDAC9 knockout mice are protected from adipose tissue dysfunction and systemic metabolic disease during high-fat feeding. Diabetes 2014, 63, 176–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khamis, A.; Boutry, R.; Canouil, M.; Mathew, S.; Lobbens, S.; Crouch, H.; Andrew, T.; Abderrahmani, A.; Tamanini, F.; Froguel, P. Histone deacetylase 9 promoter hypomethylation associated with adipocyte dysfunction is a statin-related metabolic effect. Clin. Epigenet. 2020, 12, 68. [Google Scholar] [CrossRef]
- Lenoir, O.; Flosseau, K.; Ma, F.X.; Blondeau, B.; Mai, A.; Bassel-Duby, R.; Ravassard, P.; Olson, E.N.; Haumaitre, C.; Scharfmann, R. Specific control of pancreatic endocrine β- and δ-cell mass by class IIa histone deacetylases HDAC4, HDAC5, and HDAC9. Diabetes 2011, 60, 2861–2871. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Wang, N.; Dong, M.; Guo, M.; Zhao, Y.; Zhuo, Z.; Zhang, C.; Chi, X.; Pan, Y.; Jiang, J.; et al. The Metabolic Regulator Histone Deacetylase 9 Contributes to Glucose Homeostasis Abnormality Induced by Hepatitis C Virus Infection. Diabetes 2015, 64, 4088–4098. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Zhang, Z.; Wang, N.; Guo, M.; Chi, X.; Pan, Y.; Jiang, J.; Niu, J.; Ksimu, S.; Li, J.Z.; et al. Role of HDAC9-FoxO1 Axis in the Transcriptional Program Associated with Hepatic Gluconeogenesis. Sci. Rep. 2017, 7, 6102. [Google Scholar] [CrossRef] [Green Version]
- Spracklen, C.N.; Karaderi, T.; Yaghootkar, H.; Schurmann, C.; Fine, R.S.; Kutalik, Z.; Preuss, M.H.; Lu, Y.; Wittemans, L.B.L.; Adair, L.S.; et al. Exome-Derived Adiponectin-Associated Variants Implicate Obesity and Lipid Biology. Am. J. Hum. Genet. 2019, 105, 15–28. [Google Scholar] [CrossRef] [Green Version]
- Méjat, A.; Ramond, F.; Bassel-Duby, R.; Khochbin, S.; Olson, E.N.; Schaeffer, L. Histone deacetylase 9 couples neuronal activity to muscle chromatin acetylation and gene expression. Nat. Neurosci. 2005, 8, 313–321. [Google Scholar] [CrossRef]
- Macpherson, P.C.; Farshi, P.; Goldman, D. Dach2-Hdac9 signaling regulates reinnervation of muscle endplates. Development 2015, 142, 4038–4048. [Google Scholar] [CrossRef] [Green Version]
- Litke, C.; Bading, H.; Mauceri, D. Histone deacetylase 4 shapes neuronal morphology via a mechanism involving regulation of expression of vascular endothelial growth factor D. J. Biol. Chem. 2018, 293, 8196–8207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valzania, A.; Catale, C.; Viscomi, M.T.; Puglisi-Allegra, S.; Carola, V. Histone deacetylase 5 modulates the effects of social adversity in early life on cocaine-induced behavior. Physiol. Behav. 2017, 171, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Sando, R., 3rd; Gounko, N.; Pieraut, S.; Liao, L.; Yates, J., 3rd; Maximov, A. HDAC4 governs a transcriptional program essential for synaptic plasticity and memory. Cell 2012, 151, 821–834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi, M.; Carreira, M.B.; Cooper, Y.A.; Bobadilla, A.C.; Heinsbroek, J.A.; Koike, N.; Larson, E.B.; Balmuth, E.A.; Hughes, B.W.; Penrod, R.D.; et al. HDAC5 and Its Target Gene, Npas4, Function in the Nucleus Accumbens to Regulate Cocaine-Conditioned Behaviors. Neuron 2017, 96, 130–144.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woldemichael, B.T.; Jawaid, A.; Kremer, E.A.; Gaur, N.; Krol, J.; Marchais, A.; Mansuy, I.M. The microRNA cluster miR-183/96/182 contributes to long-term memory in a protein phosphatase 1-dependent manner. Nat. Commun. 2016, 7, 12594. [Google Scholar] [CrossRef] [Green Version]
- Rahman, M.S.; Woollard, K. Atherosclerosis. Adv. Exp. Med. Biol. 2017, 1003, 121–144. [Google Scholar] [CrossRef]
- International Stroke Genetics Consortium (ISGC); Wellcome Trust Case Control Consortium 2 (WTCCC2); Bellenguez, C.; Bevan, S.; Gschwendtner, A.; Spencer, C.C.; Burgess, A.I.; Pirinen, M.; Jackson, C.A.; Traylor, M.; et al. Genome-wide association study identifies a variant in HDAC9 associated with large vessel ischemic stroke. Nat. Genet. 2012, 44, 328–333. [Google Scholar] [CrossRef]
- Traylor, M.; Farrall, M.; Holliday, E.G.; Sudlow, C.; Hopewell, J.C.; Cheng, Y.C.; Fornage, M.; Ikram, M.A.; Malik, R.; Bevan, S.; et al. Genetic risk factors for ischaemic stroke and its subtypes (the METASTROKE collaboration): A meta-analysis of genome-wide association studies. Lancet Neurol. 2012, 11, 951–962. [Google Scholar] [CrossRef] [Green Version]
- Markus, H.S.; Mäkelä, K.M.; Bevan, S.; Raitoharju, E.; Oksala, N.; Bis, J.C.; O’Donnell, C.; Hainsworth, A.; Lehtimäki, T. Evidence HDAC9 genetic variant associated with ischemic stroke increases risk via promoting carotid atherosclerosis. Stroke 2013, 44, 1220–1225. [Google Scholar] [CrossRef] [Green Version]
- Han, Y.; Sun, W.; Wang, L.; Tao, S.; Tian, L.; Hao, Y.; Zhang, W.; Wu, S.; Li, S.; Lv, H. HDAC9 gene is associated with stroke risk in a Chinese population. Exp. Biol. Med. 2013, 238, 842–847. [Google Scholar] [CrossRef] [PubMed]
- Dichgans, M.; Malik, R.; König, I.R.; Rosand, J.; Clarke, R.; Gretarsdottir, S.; Thorleifsson, G.; Mitchell, B.D.; Assimes, T.L.; Levi, C.; et al. Shared genetic susceptibility to ischemic stroke and coronary artery disease: A genome-wide analysis of common variants. Stroke 2014, 45, 24–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azghandi, S.; Prell, C.; van der Laan, S.W.; Schneider, M.; Malik, R.; Berer, K.; Gerdes, N.; Pasterkamp, G.; Weber, C.; Haffner, C.; et al. Deficiency of the stroke relevant HDAC9 gene attenuates atherosclerosis in accord with allele-specific effects at 7p21.1. Stroke 2015, 46, 197–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.B.; Han, Y.D.; Sabina, S.; Cui, N.H.; Zhang, S.; Liu, Z.J.; Li, C.; Zheng, F. HDAC9 Variant Rs2107595 Modifies Susceptibility to Coronary Artery Disease and the Severity of Coronary Atherosclerosis in a Chinese Han Population. PLoS ONE 2016, 11, e0160449. [Google Scholar] [CrossRef] [PubMed]
- Shroff, N.; Ander, B.P.; Zhan, X.; Stamova, B.; Liu, D.; Hull, H.; Hamade, F.R.; Dykstra-Aiello, C.; Ng, K.; Sharp, F.R.; et al. HDAC9 Polymorphism Alters Blood Gene Expression in Patients with Large Vessel Atherosclerotic Stroke. Transl. Stroke Res. 2019, 10, 19–25. [Google Scholar] [CrossRef]
- Shi, W.; Wei, X.; Wang, Z.; Han, H.; Fu, Y.; Liu, J.; Zhang, Y.; Guo, J.; Dong, C.; Zhou, D.; et al. HDAC9 exacerbates endothelial injury in cerebral ischaemia/reperfusion injury. J. Cell Mol. Med. 2016, 20, 1139–1149. [Google Scholar] [CrossRef] [Green Version]
- Kuang, S.; Wang, Z.; Su, L.; Han, X.; Dong, Q. Neuroprotection of histone deacetylase inhibitor TMP269 in cerebral ischemia/reperfusion rat. Int. J. Clin. Exp. Med. 2018, 11, 3405–3413. [Google Scholar]
- Nelson, C.P.; Goel, A.; Butterworth, A.S.; Kanoni, S.; Webb, T.R.; Marouli, E.; Zeng, L.; Ntalla, I.; Lai, F.Y.; Hopewell, J.C.; et al. Association analyses based on false discovery rate implicate new loci for coronary artery disease. Nat. Genet. 2017, 49, 1385–1391. [Google Scholar] [CrossRef]
- Matsukura, M.; Ozaki, K.; Takahashi, A.; Onouchi, Y.; Morizono, T.; Komai, H.; Shigematsu, H.; Kudo, T.; Inoue, Y.; Kimura, H.; et al. Genome-Wide Association Study of Peripheral Arterial Disease in a Japanese Population. PLoS ONE 2015, 10, e0139262. [Google Scholar] [CrossRef] [Green Version]
- Klarin, D.; Lynch, J.; Aragam, K.; Chaffin, M.; Assimes, T.L.; Huang, J.; Lee, K.M.; Shao, Q.; Huffman, J.E.; Natarajan, P.; et al. Genome-wide association study of peripheral artery disease in the Million Veteran Program. Nat. Med. 2019, 25, 1274–1279. [Google Scholar] [CrossRef]
- Malhotra, R.; Mauer, A.C.; Lino Cardenas, C.L.; Guo, X.; Yao, J.; Zhang, X.; Wunderer, F.; Smith, A.V.; Wong, Q.; Pechlivanis, S.; et al. HDAC9 is implicated in atherosclerotic aortic calcification and affects vascular smooth muscle cell phenotype. Nat. Genet. 2019, 51, 1580–1587. [Google Scholar] [CrossRef]
- Prestel, M.; Prell-Schicker, C.; Webb, T.; Malik, R.; Lindner, B.; Ziesch, N.; Rex-Haffner, M.; Röh, S.; Viturawong, T.; Lehm, M.; et al. The Atherosclerosis Risk Variant rs2107595 Mediates Allele-Specific Transcriptional Regulation of HDAC9 via E2F3 and Rb1. Stroke 2019, 50, 2651–2660. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Rong, S.; Repa, J.J.; St Clair, R.; Parks, J.S.; Mishra, N. Histone deacetylase 9 represses cholesterol efflux and alternatively activated macrophages in atherosclerosis development. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1871–1879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palm, F.; Aigner, A.; Pussinen, P.J.; Urbanek, C.; Buggle, F.; Safer, A.; Becher, H.; Grau, A.J. Association of a Multigenetic Pro-Inflammatory Profile with Ischaemic Stroke. Cerebrovasc Dis. 2020, 49, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Chiou, H.Y.; Bai, C.H.; Lien, L.M.; Hu, C.J.; Jeng, J.S.; Tang, S.C.; Lin, H.J.; Hsieh, Y.C. Interactive Effects of a Combination of the HDAC3 and HDAC9 Genes with Diabetes Mellitus on the Risk of Ischemic Stroke. Thromb. Haemost. 2020. [Google Scholar] [CrossRef] [PubMed]
- Asare, Y.; Campbell-James, T.A.; Bokov, Y.; Yu, L.L.; Prestel, M.; El Bounkari, O.; Roth, S.; Megens, R.T.A.; Straub, T.; Thomas, K.; et al. Histone Deacetylase 9 Activates IKK to Regulate Atherosclerotic Plaque Vulnerability. Circ. Res. 2020, 127, 811–823. [Google Scholar] [CrossRef] [PubMed]
- Isselbacher, E.M.; Lino Cardenas, C.L.; Lindsay, M.E. Hereditary Influence in Thoracic Aortic Aneurysm and Dissection. Circulation 2016, 133, 2516–2528. [Google Scholar] [CrossRef] [Green Version]
- Lino Cardenas, C.L.; Kessinger, C.W.; Cheng, Y.; MacDonald, C.; MacGillivray, T.; Ghoshhajra, B.; Huleihel, L.; Nuri, S.; Yeri, A.S.; Jaffer, F.A. An HDAC9-MALAT1-BRG1 complex mediates smooth muscle dysfunction in thoracic aortic aneurysm. Nat. Commun. 2018, 9, 1009. [Google Scholar] [CrossRef] [Green Version]
- Lino Cardenas, C.L.; Kessinger, C.W.; Chou, E.L.; Ghoshhajra, B.; Yeri, A.S.; Das, S.; Weintraub, N.L.; Malhotra, R.; Jaffer, F.A.; Lindsay, M.E. HDAC9 complex inhibition improves smooth muscle-dependent stenotic vascular disease. JCI Insight 2019, 4, e124706. [Google Scholar] [CrossRef] [Green Version]
- Yan, K.; Cao, Q.; Reilly, C.M.; Young, N.L.; Garcia, B.A.; Mishra, N. Histone deacetylase 9 deficiency protects against effector T cell-mediated systemic autoimmunity. J. Biol. Chem. 2011, 286, 28833–28843. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.A.; Kong, E.F.; Meiller, T.F.; Jabra-Rizk, M.A. Periodontal Diseases: Bug Induced, Host Promoted. PLoS Pathog. 2015, 11, e1004952. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Liu, W.; Wang, H.; Yang, Q.; Zhang, L.; Jin, F.; Jin, Y. Mutual inhibition between HDAC9 and miR-17 regulates osteogenesis of human periodontal ligament stem cells in inflammatory conditions. Cell Death Dis. 2018, 9, 480. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Li, H.; Li, K.; Fan, X.D. HDAC9 promotes brain ischemic injury by provoking IκBα/NF-κB and MAPKs signaling pathways. Biochem. Biophys. Res. Commun. 2018, 503, 1322–1329. [Google Scholar] [CrossRef] [PubMed]
- Sanford, J.A.; Zhang, L.J.; Williams, M.R.; Gangoiti, J.A.; Huang, C.M.; Gallo, R.L. Inhibition of HDAC8 and HDAC9 by microbial short-chain fatty acids breaks immune tolerance of the epidermis to TLR ligands. Sci. Immunol. 2016, 1, eaah4609. [Google Scholar] [CrossRef]
- Sanford, J.A.; O’Neill, A.M.; Zouboulis, C.C.; Gallo, R.L. Short-Chain Fatty Acids from Cutibacterium acnes Activate Both a Canonical and Epigenetic Inflammatory Response in Human Sebocytes. J. Immunol. 2019, 202, 1767–1776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cutano, V.; Di Giorgio, E.; Minisini, M.; Picco, R.; Dalla, E.; Brancolini, C. HDAC7-mediated control of tumour microenvironment maintains proliferative and stemness competence of human mammary epithelial cells. Mol. Oncol. 2019, 13, 1651–1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terry, L.V.; Oo, Y.H. The Next Frontier of Regulatory T Cells: Promising Immunotherapy for Autoimmune Diseases and Organ Transplantations. Front. Immunol. 2020, 11, 565518. [Google Scholar] [CrossRef] [PubMed]
- Tao, R.; de Zoeten, E.F.; Ozkaynak, E.; Chen, C.; Wang, L.; Porrett, P.M.; Li, B.; Turka, L.A.; Olson, E.N.; Greene, M.I.; et al. Deacetylase inhibition promotes the generation and function of regulatory T cells. Nat. Med. 2007, 13, 1299–1307. [Google Scholar] [CrossRef] [PubMed]
- de Zoeten, E.F.; Wang, L.; Sai, H.; Dillmann, W.H.; Hancock, W.W. Inhibition of HDAC9 increases T regulatory cell function and prevents colitis in mice. Gastroenterology 2010, 138, 583–594. [Google Scholar] [CrossRef] [Green Version]
- Cao, X. Self-regulation and cross-regulation of pattern-recognition receptor signalling in health and disease. Nat. Rev. Immunol. 2016, 16, 35–50. [Google Scholar] [CrossRef]
- Mason, D.X.; Jackson, T.J.; Lin, A.W. Molecular signature of oncogenic RAS-induced senescence. Oncogene 2004, 23, 9238–9246. [Google Scholar] [CrossRef] [Green Version]
- Haberland, M.; Arnold, M.A.; McAnally, J.; Phan, D.; Kim, Y.; Olson, E.N. Regulation of HDAC9 gene expression by MEF2 establishes a negative-feedback loop in the transcriptional circuitry of muscle differentiation. Mol. Cell Biol. 2007, 27, 518–525. [Google Scholar] [CrossRef] [Green Version]
- Di Giorgio, E.; Franforte, E.; Cefalù, S.; Rossi, S.; Dei Tos, A.P.; Brenca, M.; Polano, M.; Maestro, R.; Paluvai, H.; Picco, R.; et al. The co-existence of transcriptional activator and transcriptional repressor MEF2 complexes influences tumor aggressiveness. PLoS Genet. 2017, 13, e1006752. [Google Scholar] [CrossRef] [Green Version]
- Di Giorgio, E.; Hancock, W.W.; Brancolini, C. MEF2 and the tumorigenic process, hic sunt leones. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 261–273. [Google Scholar] [CrossRef]
- Kahali, B.; Gramling, S.J.; Marquez, S.B.; Thompson, K.; Lu, L.; Reisman, D. Identifying targets for the restoration and reactivation of BRM. Oncogene 2014, 33, 653–664. [Google Scholar] [CrossRef] [Green Version]
- Kahali, B.; Yu, J.; Marquez, S.B.; Thompson, K.W.; Liang, S.Y.; Lu, L.; Reisman, D. The silencing of the SWI/SNF subunit and anticancer gene BRM in Rhabdoid tumors. Oncotarget 2014, 5, 3316–3332. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Zhang, L.; Zhou, Y.; Li, L.; Zhao, J.; Qin, W.; Jin, Z.; Liu, W. Increase in HDAC9 suppresses myoblast differentiation via epigenetic regulation of autophagy in hypoxia. Cell Death Dis. 2019, 10, 552. [Google Scholar] [CrossRef] [Green Version]
- Di Giorgio, E.; Gagliostro, E.; Brancolini, C. Selective class IIa HDAC inhibitors: Myth or reality. Cell Mol. Life Sci. 2015, 72, 73–86. [Google Scholar] [CrossRef]
- Porter, N.J.; Christianson, D.W. Structure, mechanism, and inhibition of the zinc-dependent histone deacetylases. Curr. Opin. Struct. Biol. 2019, 59, 9–18. [Google Scholar] [CrossRef]
- Gore, J.; Craven, K.E.; Wilson, J.L.; Cote, G.A.; Cheng, M.; Nguyen, H.V.; Cramer, H.M.; Sherman, S.; Korc, M. TCGA data and patient-derived orthotopic xenografts highlight pancreatic cancer-associated angiogenesis. Oncotarget 2015, 6, 7504–7521. [Google Scholar] [CrossRef] [Green Version]
- Milde, T.; Oehme, I.; Korshunov, A.; Kopp-Schneider, A.; Remke, M.; Northcott, P.; Deubzer, H.E.; Lodrini, M.; Taylor, M.D.; von Deimling, A.; et al. HDAC5 and HDAC9 in medulloblastoma: Novel markers for risk stratification and role in tumor cell growth. Clin. Cancer Res. 2010, 16, 3240–3252. [Google Scholar] [CrossRef] [Green Version]
- Yang, R.; Wu, Y.; Wang, M.; Sun, Z.; Zou, J.; Zhang, Y.; Cui, H. HDAC9 promotes glioblastoma growth via TAZ-mediated EGFR pathway activation. Oncotarget 2015, 6, 7644–7656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gil, V.S.; Bhagat, G.; Howell, L.; Zhang, J.; Kim, C.H.; Stengel, S.; Vega, F.; Zelent, A.; Petrie, K. Deregulated expression of HDAC9 in B cells promotes development of lymphoproliferative disease and lymphoma in mice. Dis. Models Mech. 2016, 9, 1483–1495. [Google Scholar] [CrossRef] [Green Version]
- Freese, K.; Seitz, T.; Dietrich, P.; Lee, S.M.L.; Thasler, W.E.; Bosserhoff, A.; Hellerbrand, C. Histone Deacetylase Expressions in Hepatocellular Carcinoma and Functional Effects of Histone Deacetylase Inhibitors on Liver Cancer Cells In Vitro. Cancers 2019, 11, 1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanki, K.; Watanabe, R.; Nguyen Thai, L.; Zhao, C.H.; Naito, K. HDAC9 Is Preferentially Expressed in Dedifferentiated Hepatocellular Carcinoma Cells and Is Involved in an Anchorage-Independent Growth. Cancers 2020, 12, 2734. [Google Scholar] [CrossRef]
- Paluvai, H.; Di Giorgio, E.; Brancolini, C. Unscheduled HDAC4 repressive activity in human fibroblasts triggers TP53-dependent senescence and favors cell transformation. Mol. Oncol. 2018, 12, 2165–2181. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.W.; Wang, R.; Cai, Q.Q.; Qi, B.; Wu, W.; Zhang, Y.H.; Wu, X.Z. Sulfatide epigenetically regulates miR-223 and promotes the migration of human hepatocellular carcinoma cells. J. Hepatol. 2014, 60, 792–801. [Google Scholar] [CrossRef] [PubMed]
- Lian, B.; Pei, Y.C.; Jiang, Y.Z.; Xue, M.Z.; Li, D.Q.; Li, X.G.; Zheng, Y.Z.; Liu, X.Y.; Qiao, F.; Sun, W.L.; et al. Truncated HDAC9 identified by integrated genome-wide screen as the key modulator for paclitaxel resistance in triple-negative breast cancer. Theranostics 2020, 10, 11092–11109. [Google Scholar] [CrossRef]
- Linares, A.; Assou, S.; Lapierre, M.; Thouennon, E.; Duraffourd, C.; Fromaget, C.; Boulahtouf, A.; Tian, G.; Ji, J.; Sahin, O.; et al. Increased expression of the HDAC9 gene is associated with antiestrogen resistance of breast cancers. Mol. Oncol. 2019, 13, 1534–1547. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Feng, B.; Niu, Y.; Wu, J.; Yang, Y.; Shen, S.; Guo, Y.; Liang, J.; Guo, W.; Dong, Z. A novel long noncoding RNA, LOC440173, promotes the progression of esophageal squamous cell carcinoma by modulating the miR-30d-5p/HDAC9 axis and the epithelial-mesenchymal transition. Mol. Carcinog. 2020, 59, 1392–1408. [Google Scholar] [CrossRef]
- Fleming, J.L.; Dworkin, A.M.; Allain, D.C.; Fernandez, S.; Wei, L.; Peters, S.B.; Iwenofu, O.H.; Ridd, K.; Bastian, B.C.; Toland, A.E. Allele-specific imbalance mapping identifies HDAC9 as a candidate gene for cutaneous squamous cell carcinoma. Int. J. Cancer 2014, 134, 244–248. [Google Scholar] [CrossRef] [Green Version]
- Siekmann, T.E.; Gerber, M.M.; Toland, A.E. Variants in an Hdac9 intronic enhancer plasmid impact Twist1 expression in vitro. Mamm. Genome 2016, 27, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, N.; Eshel, R.; Bar Yaacov, R.; Shahar, T.; Shmulevich, F.; Dahan, I.; Levaot, N.; Kaplan, T.; Lupiáñez, D.G.; Birnbaum, R.Y. Unraveling the transcriptional regulation of TWIST1 in limb development. PLoS Genet. 2018, 14, e1007738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kai, X.; Hejun, Z.; Yang, D.; Jie, T.; Shigang, D. Identification of HDAC9 as a viable therapeutic target for the treatment of gastric cancer. Exp. Mol. Med. 2019, 51, 100. [Google Scholar] [CrossRef] [Green Version]
- Peruzzo, P.; Comelli, M.; Di Giorgio, E.; Franforte, E.; Mavelli, I.; Brancolini, C. Transformation by different oncogenes relies on specific metabolic adaptations. Cell Cycle 2016, 15, 2656–2668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Giorgio, E.; Wang, L.; Xiong, Y.; Akimova, T.; Christensen, L.M.; Han, R.; Samanta, A.; Trevisanut, M.; Bhatti, T.R.; Beier, U.H.; et al. MEF2D sustains activation of effector Foxp3+ Tregs during transplant survival and anticancer immunity. J. Clin. Investig. 2020, 130, 6242–6260. [Google Scholar] [CrossRef]
- Ning, Y.; Ding, J.; Sun, X.; Xie, Y.; Su, M.; Ma, C.; Pan, J.; Chen, J.; Jiang, H.; Qi, C. HDAC9 deficiency promotes tumor progression by decreasing the CD8 + dendritic cell infiltration of the tumor microenvironment. J. Immunother. Cancer 2020, 8, e000529. [Google Scholar] [CrossRef]
- Van, H.T.; Santos, M.A. Histone modifications and the DNA double-strand break response. Cell Cycle 2018, 17, 2399–2410. [Google Scholar] [CrossRef] [Green Version]
- Paluvai, H.; Di Giorgio, E.; Brancolini, C. The Histone Code of Senescence. Cells 2020, 9, 466. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Palmbos, P.L.; Wang, L.; Kim, E.H.; Ney, G.M.; Liu, C.; Prasad, J.; Misek, D.E.; Yu, X.; Ljungman, M.; et al. ATDC (Ataxia Telangiectasia Group D Complementing) Promotes Radioresistance through an Interaction with the RNF8 Ubiquitin Ligase. J. Biol. Chem. 2015, 290, 27146–27157. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Z.; Peng, L.; Radhakrishnan, R.; Seto, E. Histone deacetylase 9 (HDAC9) regulates the functions of the ATDC (TRIM29) protein. J. Biol. Chem. 2010, 285, 39329–39338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotian, S.; Liyanarachchi, S.; Zelent, A.; Parvin, J.D. Histone deacetylases 9 and 10 are required for homologous recombination. J. Biol. Chem. 2011, 286, 7722–7726. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brancolini, C.; Di Giorgio, E.; Formisano, L.; Gagliano, T. Quis Custodiet Ipsos Custodes (Who Controls the Controllers)? Two Decades of Studies on HDAC9. Life 2021, 11, 90. https://doi.org/10.3390/life11020090
Brancolini C, Di Giorgio E, Formisano L, Gagliano T. Quis Custodiet Ipsos Custodes (Who Controls the Controllers)? Two Decades of Studies on HDAC9. Life. 2021; 11(2):90. https://doi.org/10.3390/life11020090
Chicago/Turabian StyleBrancolini, Claudio, Eros Di Giorgio, Luigi Formisano, and Teresa Gagliano. 2021. "Quis Custodiet Ipsos Custodes (Who Controls the Controllers)? Two Decades of Studies on HDAC9" Life 11, no. 2: 90. https://doi.org/10.3390/life11020090