
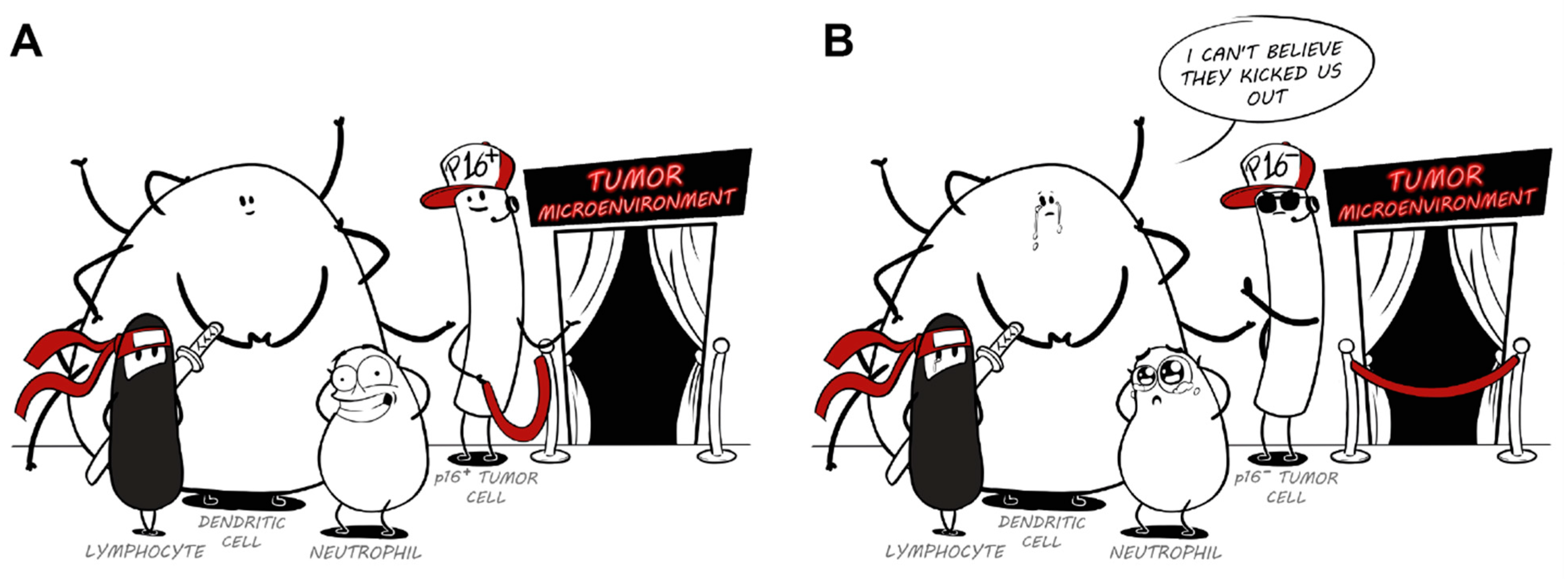
Loss of p16: A Bouncer of the Immunological Surveillance?


{kind=link}
Abstract
:1. Introduction
2. p16 in Cancer and Senescence
3. Role of SASP in Senescence and Cancer
4. p16 Regulation of Tumor Immunity
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Decker, W.K.; da Silva, R.F.; Sanabria, M.H.; Angelo, L.S.; Guimaraes, F.; Burt, B.M.; Kheradmand, F.; Paust, S. Cancer Immunotherapy: Historical Perspective of a Clinical Revolution and Emerging Preclinical Animal Models. Front. Immunol. 2017, 8, 829. [Google Scholar] [CrossRef] [Green Version]
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of Indications of FDA-Approved Immune Checkpoint Inhibitors per NCCN Guidelines with the Level of Evidence. Cancers 2020, 12, 738. [Google Scholar] [CrossRef] [Green Version]
- Aird, K.M.; Zhang, R. Nucleotide metabolism, oncogene-induced senescence and cancer. Cancer Lett. 2015, 356, 204–210. [Google Scholar] [CrossRef] [Green Version]
- Kuilman, T.; Peeper, D.S. Senescence-messaging secretome: SMS-ing cellular stress. Nat. Rev. Cancer 2009, 9, 81–94. [Google Scholar] [CrossRef]
- Coppe, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Munoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008, 6, 2853–2868. [Google Scholar] [CrossRef]
- Zhang, H.; Pan, K.H.; Cohen, S.N. Senescence-specific gene expression fingerprints reveal cell-type-dependent physical clustering of up-regulated chromosomal loci. Proc. Natl. Acad. Sci. USA 2003, 100, 3251–3256. [Google Scholar] [CrossRef] [Green Version]
- Bavik, C.; Coleman, I.; Dean, J.P.; Knudsen, B.; Plymate, S.; Nelson, P.S. The gene expression program of prostate fibroblast senescence modulates neoplastic epithelial cell proliferation through paracrine mechanisms. Cancer Res. 2006, 66, 794–802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wajapeyee, N.; Serra, R.W.; Zhu, X.; Mahalingam, M.; Green, M.R. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell 2008, 132, 363–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuilman, T.; Michaloglou, C.; Vredeveld, L.C.; Douma, S.; van Doorn, R.; Desmet, C.J.; Aarden, L.A.; Mooi, W.J.; Peeper, D.S. Oncogene-induced senescence relayed by an interleukin-dependent inflammatory network. Cell 2008, 133, 1019–1031. [Google Scholar] [CrossRef] [Green Version]
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; Da Costa, M.; Brown, C.; Popov, N.; et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 2008, 133, 1006–1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, S.; Serrano, M. A new mechanism of inactivation of the INK4/ARF locus. Cell Cycle 2006, 5, 1382–1384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharpless, N.E.; Sherr, C.J. Forging a signature of in vivo senescence. Nat. Rev. Cancer 2015, 15, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Balli, D.; Rech, A.J.; Stanger, B.Z.; Vonderheide, R.H. Immune Cytolytic Activity Stratifies Molecular Subsets of Human Pancreatic Cancer. Clin. Cancer Res. 2017, 23, 3129–3138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wartenberg, M.; Cibin, S.; Zlobec, I.; Vassella, E.; Eppenberger-Castori, S.; Terracciano, L.; Eichmann, M.D.; Worni, M.; Gloor, B.; Perren, A.; et al. Integrated Genomic and Immunophenotypic Classification of Pancreatic Cancer Reveals Three Distinct Subtypes with Prognostic/Predictive Significance. Clin. Cancer Res. 2018, 24, 4444–4454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, C.; Pabla, S.; Conroy, J.M.; Nesline, M.K.; Glenn, S.T.; Dressman, D.; Papanicolau-Sengos, A.; Burgher, B.; Andreas, J.; Giamo, V.; et al. Predicting response to checkpoint inhibitors in melanoma beyond PD-L1 and mutational burden. J. Immunother. Cancer 2018, 6, 32. [Google Scholar] [CrossRef]
- Chikenji, T.S.; Saito, Y.; Konari, N.; Nakano, M.; Mizue, Y.; Otani, M.; Fujimiya, M. p16(INK4A)-expressing mesenchymal stromal cells restore the senescence-clearance-regeneration sequence that is impaired in chronic muscle inflammation. EBioMedicine 2019, 44, 86–97. [Google Scholar] [CrossRef] [Green Version]
- Buj, R.; Leon, K.E.; Anguelov, M.A.; Aird, K.M. Suppression of p16 alleviates the senescence-associated secretory phenotype. Aging 2021, 13, 3290. [Google Scholar] [CrossRef]
- Ozenne, P.; Eymin, B.; Brambilla, E.; Gazzeri, S. The ARF tumor suppressor: Structure, functions and status in cancer. Int. J. Cancer 2010, 127, 2239–2247. [Google Scholar] [CrossRef]
- Sherr, C.J. The INK4a/ARF network in tumour suppression. Nat. Rev. Mol. Cell Biol. 2001, 2, 731–737. [Google Scholar] [CrossRef]
- Esteller, M.; Corn, P.G.; Baylin, S.B.; Herman, J.G. A gene hypermethylation profile of human cancer. Cancer Res. 2001, 61, 3225–3229. [Google Scholar] [PubMed]
- Ruas, M.; Brookes, S.; McDonald, N.Q.; Peters, G. Functional evaluation of tumour-specific variants of p16INK4a/CDKN2A: Correlation with protein structure information. Oncogene 1999, 18, 5423–5434. [Google Scholar] [CrossRef] [Green Version]
- Inoue, K.; Fry, E.A. Aberrant expression of p16(INK4a) in human cancers—A new biomarker? Cancer Rep. Rev. 2018, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quelle, D.E.; Cheng, M.; Ashmun, R.A.; Sherr, C.J. Cancer-associated mutations at the INK4a locus cancel cell cycle arrest by p16INK4a but not by the alternative reading frame protein p19ARF. Proc. Natl. Acad. Sci. USA 1997, 94, 669–673. [Google Scholar] [CrossRef] [Green Version]
- Barnum, K.J.; O’Connell, M.J. Cell cycle regulation by checkpoints. Methods Mol. Biol. 2014, 1170, 29–40. [Google Scholar] [CrossRef] [Green Version]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.Y.; Souroullas, G.P.; Diekman, B.O.; Krishnamurthy, J.; Hall, B.M.; Sorrentino, J.A.; Parker, J.S.; Sessions, G.A.; Gudkov, A.V.; Sharpless, N.E. Cells exhibiting strong p16 (INK4a) promoter activation in vivo display features of senescence. Proc. Natl. Acad. Sci. USA 2019, 116, 2603–2611. [Google Scholar] [CrossRef] [Green Version]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef] [Green Version]
- Gil, J.; Peters, G. Regulation of the INK4b-ARF-INK4a tumour suppressor locus: All for one or one for all. Nat. Rev. Mol. Cell Biol. 2006, 7, 667–677. [Google Scholar] [CrossRef]
- Itahana, K.; Campisi, J.; Dimri, G.P. Mechanisms of cellular senescence in human and mouse cells. Biogerontology 2004, 5, 1–10. [Google Scholar] [CrossRef]
- Bulavin, D.V.; Phillips, C.; Nannenga, B.; Timofeev, O.; Donehower, L.A.; Anderson, C.W.; Appella, E.; Fornace, A.J., Jr. Inactivation of the Wip1 phosphatase inhibits mammary tumorigenesis through p38 MAPK-mediated activation of the p16(Ink4a)-p19(Arf) pathway. Nat. Genet. 2004, 36, 343–350. [Google Scholar] [CrossRef]
- Deng, Q.; Liao, R.; Wu, B.L.; Sun, P. High intensity ras signaling induces premature senescence by activating p38 pathway in primary human fibroblasts. J. Biol. Chem. 2004, 279, 1050–1059. [Google Scholar] [CrossRef] [Green Version]
- Lin, A.W.; Barradas, M.; Stone, J.C.; van Aelst, L.; Serrano, M.; Lowe, S.W. Premature senescence involving p53 and p16 is activated in response to constitutive MEK/MAPK mitogenic signaling. Genes Dev. 1998, 12, 3008–3019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Chen, J.X.; Liao, R.; Deng, Q.; Zhou, J.J.; Huang, S.; Sun, P. Sequential activation of the MEK-extracellular signal-regulated kinase and MKK3/6-p38 mitogen-activated protein kinase pathways mediates oncogenic ras-induced premature senescence. Mol. Cell. Biol. 2002, 22, 3389–3403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bracken, A.P.; Kleine-Kohlbrecher, D.; Dietrich, N.; Pasini, D.; Gargiulo, G.; Beekman, C.; Theilgaard-Monch, K.; Minucci, S.; Porse, B.T.; Marine, J.C.; et al. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev. 2007, 21, 525–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dietrich, N.; Bracken, A.P.; Trinh, E.; Schjerling, C.K.; Koseki, H.; Rappsilber, J.; Helin, K.; Hansen, K.H. Bypass of senescence by the polycomb group protein CBX8 through direct binding to the INK4A-ARF locus. EMBO J. 2007, 26, 1637–1648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, T.; Teo, Y.V.; Evans, S.A.; Neretti, N.; Sedivy, J.M. Regulation of Cellular Senescence by Polycomb Chromatin Modifiers through Distinct DNA Damage- and Histone Methylation-Dependent Pathways. Cell Rep. 2018, 22, 3480–3492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakade, K.; Pan, J.; Yamasaki, T.; Murata, T.; Wasylyk, B.; Yokoyama, K.K. JDP2 (Jun Dimerization Protein 2)-deficient mouse embryonic fibroblasts are resistant to replicative senescence. J. Biol. Chem. 2009, 284, 10808–10817. [Google Scholar] [CrossRef] [Green Version]
- Ohtani, N.; Zebedee, Z.; Huot, T.J.; Stinson, J.A.; Sugimoto, M.; Ohashi, Y.; Sharrocks, A.D.; Peters, G.; Hara, E. Opposing effects of Ets and Id proteins on p16INK4a expression during cellular senescence. Nature 2001, 409, 1067–1070. [Google Scholar] [CrossRef] [PubMed]
- Passegue, E.; Wagner, E.F. JunB suppresses cell proliferation by transcriptional activation of p16(INK4a) expression. EMBO J. 2000, 19, 2969–2979. [Google Scholar] [CrossRef] [Green Version]
- Swarbrick, A.; Roy, E.; Allen, T.; Bishop, J.M. Id1 cooperates with oncogenic Ras to induce metastatic mammary carcinoma by subversion of the cellular senescence response. Proc. Natl. Acad. Sci. USA 2008, 105, 5402–5407. [Google Scholar] [CrossRef] [Green Version]
- Buj, R.; Chen, C.W.; Dahl, E.S.; Leon, K.E.; Kuskovsky, R.; Maglakelidze, N.; Navaratnarajah, M.; Zhang, G.; Doan, M.T.; Jiang, H.; et al. Suppression of p16 Induces mTORC1-Mediated Nucleotide Metabolic Reprogramming. Cell Rep. 2019, 28, 1971–1980.e1978. [Google Scholar] [CrossRef] [Green Version]
- Damsky, W.; Micevic, G.; Meeth, K.; Muthusamy, V.; Curley, D.P.; Santhanakrishnan, M.; Erdelyi, I.; Platt, J.T.; Huang, L.; Theodosakis, N.; et al. mTORC1 activation blocks BrafV600E-induced growth arrest but is insufficient for melanoma formation. Cancer Cell 2015, 27, 41–56. [Google Scholar] [CrossRef] [Green Version]
- Dankort, D.; Filenova, E.; Collado, M.; Serrano, M.; Jones, K.; McMahon, M. A new mouse model to explore the initiation, progression, and therapy of BRAFV600E-induced lung tumors. Genes Dev. 2007, 21, 379–384. [Google Scholar] [CrossRef] [Green Version]
- Goel, V.K.; Ibrahim, N.; Jiang, G.; Singhal, M.; Fee, S.; Flotte, T.; Westmoreland, S.; Haluska, F.S.; Hinds, P.W.; Haluska, F.G. Melanocytic nevus-like hyperplasia and melanoma in transgenic BRAFV600E mice. Oncogene 2009, 28, 2289–2298. [Google Scholar] [CrossRef] [Green Version]
- Haferkamp, S.; Becker, T.M.; Scurr, L.L.; Kefford, R.F.; Rizos, H. p16INK4a-induced senescence is disabled by melanoma-associated mutations. Aging Cell 2008, 7, 733–745. [Google Scholar] [CrossRef] [Green Version]
- Buj, R.; Aird, K.M. p16: Cycling off the beaten path. Mol. Cell. Oncol. 2019, 6, e1677140. [Google Scholar] [CrossRef] [Green Version]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Meljem, J.M.; Apps, J.R.; Fraser, H.C.; Martinez-Barbera, J.P. Paracrine roles of cellular senescence in promoting tumourigenesis. Br. J. Cancer 2018, 118, 1283–1288. [Google Scholar] [CrossRef] [PubMed]
- Coppe, J.P.; Rodier, F.; Patil, C.K.; Freund, A.; Desprez, P.Y.; Campisi, J. Tumor suppressor and aging biomarker p16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype. J. Biol. Chem. 2011, 286, 36396–36403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, C.; Xu, Q.; Martin, T.D.; Li, M.Z.; Demaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 2015, 349, aaa5612. [Google Scholar] [CrossRef] [Green Version]
- Malaquin, N.; Olivier, M.A.; Martinez, A.; Nadeau, S.; Sawchyn, C.; Coppe, J.P.; Cardin, G.; Mallette, F.A.; Campisi, J.; Rodier, F. Non-canonical ATM/MRN activities temporally define the senescence secretory program. EMBO Rep. 2020, 21, e50718. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Coppe, J.P.; Patil, C.K.; Hoeijmakers, W.A.; Munoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell. Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Panda, A.C.; Abdelmohsen, K.; Gorospe, M. SASP regulation by noncoding RNA. Mech. Ageing Dev. 2017, 168, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Herranz, N.; Gallage, S.; Mellone, M.; Wuestefeld, T.; Klotz, S.; Hanley, C.J.; Raguz, S.; Acosta, J.C.; Innes, A.J.; Banito, A.; et al. mTOR regulates MAPKAPK2 translation to control the senescence-associated secretory phenotype. Nat. Cell. Biol. 2015, 17, 1205–1217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laberge, R.M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell. Biol. 2015, 17, 1049–1061. [Google Scholar] [CrossRef]
- Chandra, T.; Ewels, P.A.; Schoenfelder, S.; Furlan-Magaril, M.; Wingett, S.W.; Kirschner, K.; Thuret, J.Y.; Andrews, S.; Fraser, P.; Reik, W. Global reorganization of the nuclear landscape in senescent cells. Cell Rep. 2015, 10, 471–483. [Google Scholar] [CrossRef]
- Criscione, S.W.; De Cecco, M.; Siranosian, B.; Zhang, Y.; Kreiling, J.A.; Sedivy, J.M.; Neretti, N. Reorganization of chromosome architecture in replicative cellular senescence. Sci. Adv. 2016, 2, e1500882. [Google Scholar] [CrossRef] [Green Version]
- Narita, M.; Nunez, S.; Heard, E.; Narita, M.; Lin, A.W.; Hearn, S.A.; Spector, D.L.; Hannon, G.J.; Lowe, S.W. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003, 113, 703–716. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Poustovoitov, M.V.; Ye, X.; Santos, H.A.; Chen, W.; Daganzo, S.M.; Erzberger, J.P.; Serebriiskii, I.G.; Canutescu, A.A.; Dunbrack, R.L.; et al. Formation of MacroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev. Cell 2005, 8, 19–30. [Google Scholar] [CrossRef] [Green Version]
- Sakaki, M.; Ebihara, Y.; Okamura, K.; Nakabayashi, K.; Igarashi, A.; Matsumoto, K.; Hata, K.; Kobayashi, Y.; Maehara, K. Potential roles of DNA methylation in the initiation and establishment of replicative senescence revealed by array-based methylome and transcriptome analyses. PLoS ONE 2017, 12, e0171431. [Google Scholar] [CrossRef]
- Zhang, R.; Chen, W.; Adams, P.D. Molecular dissection of formation of senescence-associated heterochromatin foci. Mol. Cell. Biol. 2007, 27, 2343–2358. [Google Scholar] [CrossRef] [Green Version]
- Cruickshanks, H.A.; McBryan, T.; Nelson, D.M.; Vanderkraats, N.D.; Shah, P.P.; van Tuyn, J.; Singh Rai, T.; Brock, C.; Donahue, G.; Dunican, D.S.; et al. Senescent cells harbour features of the cancer epigenome. Nat. Cell. Biol. 2013, 15, 1495–1506. [Google Scholar] [CrossRef]
- Parry, A.J.; Hoare, M.; Bihary, D.; Hansel-Hertsch, R.; Smith, S.; Tomimatsu, K.; Mannion, E.; Smith, A.; D’Santos, P.; Russell, I.A.; et al. NOTCH-mediated non-cell autonomous regulation of chromatin structure during senescence. Nat. Commun. 2018, 9, 1840. [Google Scholar] [CrossRef] [Green Version]
- Aird, K.M.; Iwasaki, O.; Kossenkov, A.V.; Tanizawa, H.; Fatkhutdinov, N.; Bitler, B.G.; Le, L.; Alicea, G.; Yang, T.L.; Johnson, F.B.; et al. HMGB2 orchestrates the chromatin landscape of senescence-associated secretory phenotype gene loci. J. Cell Biol. 2016, 215, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Tasdemir, N.; Banito, A.; Roe, J.S.; Alonso-Curbelo, D.; Camiolo, M.; Tschaharganeh, D.F.; Huang, C.H.; Aksoy, O.; Bolden, J.E.; Chen, C.C.; et al. BRD4 Connects Enhancer Remodeling to Senescence Immune Surveillance. Cancer Discov. 2016, 6, 612–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capell, B.C.; Drake, A.M.; Zhu, J.; Shah, P.P.; Dou, Z.; Dorsey, J.; Simola, D.F.; Donahue, G.; Sammons, M.; Rai, T.S.; et al. MLL1 is essential for the senescence-associated secretory phenotype. Genes Dev. 2016, 30, 321–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Ruiz, P.D.; McKimpson, W.M.; Novikov, L.; Kitsis, R.N.; Gamble, M.J. MacroH2A1 and ATM Play Opposing Roles in Paracrine Senescence and the Senescence-Associated Secretory Phenotype. Mol. Cell 2015, 59, 719–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayakawa, T.; Iwai, M.; Aoki, S.; Takimoto, K.; Maruyama, M.; Maruyama, W.; Motoyama, N. SIRT1 suppresses the senescence-associated secretory phenotype through epigenetic gene regulation. PLoS ONE 2015, 10, e0116480. [Google Scholar] [CrossRef]
- Leon, K.E.; Buj, R.; Lesko, E.; Dahl, E.S.; Chen, C.-W.; Imamura, Y.; Kossenkov, A.V.; Hobbs, R.P.; Arid, K.M. DOT1L modulates the senescence-associated secretory phenotype through epigenetic regulation of IL1A. bioRxiv Prepr. Serv. Biol. 2020. [Google Scholar] [CrossRef]
- Campisi, J. Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol. 2001, 11, S27–S31. [Google Scholar] [CrossRef] [Green Version]
- Dimri, G.P. What has senescence got to do with cancer? Cancer Cell 2005, 7, 505–512. [Google Scholar] [CrossRef] [Green Version]
- Krtolica, A.; Parrinello, S.; Lockett, S.; Desprez, P.Y.; Campisi, J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: A link between cancer and aging. Proc. Natl. Acad. Sci. USA 2001, 98, 12072–12077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatia, B.; Multani, A.S.; Patrawala, L.; Chen, X.; Calhoun-Davis, T.; Zhou, J.; Schroeder, L.; Schneider-Broussard, R.; Shen, J.; Pathak, S.; et al. Evidence that senescent human prostate epithelial cells enhance tumorigenicity: Cell fusion as a potential mechanism and inhibition by p16INK4a and hTERT. Int. J. Cancer 2008, 122, 1483–1495. [Google Scholar] [CrossRef] [Green Version]
- Coppe, J.P.; Kauser, K.; Campisi, J.; Beausejour, C.M. Secretion of vascular endothelial growth factor by primary human fibroblasts at senescence. J. Biol. Chem. 2006, 281, 29568–29574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoare, M.; Ito, Y.; Kang, T.W.; Weekes, M.P.; Matheson, N.J.; Patten, D.A.; Shetty, S.; Parry, A.J.; Menon, S.; Salama, R.; et al. NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat. Cell Biol. 2016, 18, 979–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basisty, N.; Kale, A.; Jeon, O.H.; Kuehnemann, C.; Payne, T.; Rao, C.; Holtz, A.; Shah, S.; Sharma, V.; Ferrucci, L.; et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol. 2020, 18, e3000599. [Google Scholar] [CrossRef] [Green Version]
- Rao, S.G.; Jackson, J.G. SASP: Tumor Suppressor or Promoter? Yes! Trends Cancer 2016, 2, 676–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez-Segura, A.; de Jong, T.V.; Melov, S.; Guryev, V.; Campisi, J.; Demaria, M. Unmasking Transcriptional Heterogeneity in Senescent Cells. Curr. Biol. 2017, 27, 2652–2660.e2654. [Google Scholar] [CrossRef] [Green Version]
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of activated stellate cells limits liver fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef] [Green Version]
- Jun, J.I.; Lau, L.F. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat. Cell Biol. 2010, 12, 676–685. [Google Scholar] [CrossRef]
- Pitiyage, G.N.; Slijepcevic, P.; Gabrani, A.; Chianea, Y.G.; Lim, K.P.; Prime, S.S.; Tilakaratne, W.M.; Fortune, F.; Parkinson, E.K. Senescent mesenchymal cells accumulate in human fibrosis by a telomere-independent mechanism and ameliorate fibrosis through matrix metalloproteinases. J. Pathol. 2011, 223, 604–617. [Google Scholar] [CrossRef] [PubMed]
- Kang, T.W.; Yevsa, T.; Woller, N.; Hoenicke, L.; Wuestefeld, T.; Dauch, D.; Hohmeyer, A.; Gereke, M.; Rudalska, R.; Potapova, A.; et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature 2011, 479, 547–551. [Google Scholar] [CrossRef]
- Georgilis, A.; Gil, J. Controlling secretion to limit chemoresistance. Genes Dev. 2016, 30, 1791–1792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ruhland, M.K.; Loza, A.J.; Capietto, A.H.; Luo, X.; Knolhoff, B.L.; Flanagan, K.C.; Belt, B.A.; Alspach, E.; Leahy, K.; Luo, J.; et al. Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat. Commun. 2016, 7, 11762. [Google Scholar] [CrossRef] [PubMed]
- Toso, A.; Revandkar, A.; Di Mitri, D.; Guccini, I.; Proietti, M.; Sarti, M.; Pinton, S.; Zhang, J.; Kalathur, M.; Civenni, G.; et al. Enhancing chemotherapy efficacy in Pten-deficient prostate tumors by activating the senescence-associated antitumor immunity. Cell Rep. 2014, 9, 75–89. [Google Scholar] [CrossRef]
- Teo, Y.V.; Rattanavirotkul, N.; Olova, N.; Salzano, A.; Quintanilla, A.; Tarrats, N.; Kiourtis, C.; Muller, M.; Green, A.R.; Adams, P.D.; et al. Notch Signaling Mediates Secondary Senescence. Cell Rep. 2019, 27, 997–1007.e1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zelenay, S.; van der Veen, A.G.; Bottcher, J.P.; Snelgrove, K.J.; Rogers, N.; Acton, S.E.; Chakravarty, P.; Girotti, M.R.; Marais, R.; Quezada, S.A.; et al. Cyclooxygenase-Dependent Tumor Growth through Evasion of Immunity. Cell 2015, 162, 1257–1270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef] [Green Version]
- DeLeon, T.T.; Almquist, D.R.; Kipp, B.R.; Langlais, B.T.; Mangold, A.; Winters, J.L.; Kosiorek, H.E.; Joseph, R.W.; Dronca, R.S.; Block, M.S.; et al. Assessment of clinical outcomes with immune checkpoint inhibitor therapy in melanoma patients with CDKN2A and TP53 pathogenic mutations. PLoS ONE 2020, 15, e0230306. [Google Scholar] [CrossRef] [Green Version]
- Helgadottir, H.; Ghiorzo, P.; van Doorn, R.; Puig, S.; Levin, M.; Kefford, R.; Lauss, M.; Queirolo, P.; Pastorino, L.; Kapiteijn, E.; et al. Efficacy of novel immunotherapy regimens in patients with metastatic melanoma with germline CDKN2A mutations. J. Med. Genet. 2020, 57, 316–321. [Google Scholar] [CrossRef] [Green Version]
- Lv, F.; Wu, J.; Miao, D.; An, W.; Wang, Y. p16 deficiency promotes nonalcoholic steatohepatitis via regulation of hepatic oxidative stress. Biochem. Biophys. Res. Commun. 2017, 486, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Chin, L.; Pomerantz, J.; Polsky, D.; Jacobson, M.; Cohen, C.; Cordon-Cardo, C.; Horner, J.W., 2nd; DePinho, R.A. Cooperative effects of INK4a and ras in melanoma susceptibility in vivo. Genes Dev. 1997, 11, 2822–2834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krimpenfort, P.; Quon, K.C.; Mooi, W.J.; Loonstra, A.; Berns, A. Loss of p16Ink4a confers susceptibility to metastatic melanoma in mice. Nature 2001, 413, 83–86. [Google Scholar] [CrossRef] [PubMed]
- Che, H.; Li, J.; Li, Y.; Ma, C.; Liu, H.; Qin, J.; Dong, J.; Zhang, Z.; Xian, C.J.; Miao, D.; et al. p16 deficiency attenuates intervertebral disc degeneration by adjusting oxidative stress and nucleus pulposus cell cycle. eLife 2020, 9, e52570. [Google Scholar] [CrossRef] [PubMed]
- Kyritsi, K.; Francis, H.; Zhou, T.; Ceci, L.; Wu, N.; Yang, Z.; Meng, F.; Chen, L.; Baiocchi, L.; Kundu, D.; et al. Downregulation of p16 Decreases Biliary Damage and Liver Fibrosis in the Mdr2(/) Mouse Model of Primary Sclerosing Cholangitis. Gene Expr. 2020, 20, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Faget, D.V.; Ren, Q.; Stewart, S.A. Unmasking senescence: Context-dependent effects of SASP in cancer. Nat. Rev. Cancer 2019, 19, 439–453. [Google Scholar] [CrossRef]
- Zhang, H.; Chen, Z.H.; Savarese, T.M. Codeletion of the genes for p16INK4, methylthioadenosine phosphorylase, interferon-alpha1, interferon-beta1, and other 9p21 markers in human malignant cell lines. Cancer Genet. Cytogenet. 1996, 86, 22–28. [Google Scholar] [CrossRef]
- Roy, D.M.; Walsh, L.A.; Desrichard, A.; Huse, J.T.; Wu, W.; Gao, J.; Bose, P.; Lee, W.; Chan, T.A. Integrated Genomics for Pinpointing Survival Loci within Arm-Level Somatic Copy Number Alterations. Cancer Cell 2016, 29, 737–750. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Kim, H.; Feng, Y.; Li, Y.; Tamiya, H.; Tocci, S.; Ronai, Z.A. PRMT5 control of cGAS/STING and NLRC5 pathways defines melanoma response to antitumor immunity. Sci. Transl. Med. 2020, 12, eaaz5683. [Google Scholar] [CrossRef] [PubMed]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z.; et al. Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 2017, 550, 402–406. [Google Scholar] [CrossRef] [Green Version]
- Ivanov, A.; Pawlikowski, J.; Manoharan, I.; van Tuyn, J.; Nelson, D.M.; Rai, T.S.; Shah, P.P.; Hewitt, G.; Korolchuk, V.I.; Passos, J.F.; et al. Lysosome-mediated processing of chromatin in senescence. J. Cell Biol. 2013, 202, 129–143. [Google Scholar] [CrossRef] [PubMed]
- Gluck, S.; Guey, B.; Gulen, M.F.; Wolter, K.; Kang, T.W.; Schmacke, N.A.; Bridgeman, A.; Rehwinkel, J.; Zender, L.; Ablasser, A. Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat. Cell Biol. 2017, 19, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Linsley, P.S.; Speake, C.; Whalen, E.; Chaussabel, D. Copy number loss of the interferon gene cluster in melanomas is linked to reduced T cell infiltrate and poor patient prognosis. PLoS ONE 2014, 9, e109760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, W.; Zender, L.; Miething, C.; Dickins, R.A.; Hernando, E.; Krizhanovsky, V.; Cordon-Cardo, C.; Lowe, S.W. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007, 445, 656–660. [Google Scholar] [CrossRef] [Green Version]
- Braumuller, H.; Wieder, T.; Brenner, E.; Assmann, S.; Hahn, M.; Alkhaled, M.; Schilbach, K.; Essmann, F.; Kneilling, M.; Griessinger, C.; et al. T-helper-1-cell cytokines drive cancer into senescence. Nature 2013, 494, 361–365. [Google Scholar] [CrossRef] [Green Version]
- Brenner, E.; Schorg, B.F.; Ahmetlic, F.; Wieder, T.; Hilke, F.J.; Simon, N.; Schroeder, C.; Demidov, G.; Riedel, T.; Fehrenbacher, B.; et al. Cancer immune control needs senescence induction by interferon-dependent cell cycle regulator pathways in tumours. Nat. Commun. 2020, 11, 1335. [Google Scholar] [CrossRef] [Green Version]
- Hilke, F.J.; Sinnberg, T.; Gschwind, A.; Niessner, H.; Demidov, G.; Amaral, T.; Ossowski, S.; Bonzheim, I.; Rocken, M.; Riess, O.; et al. Distinct Mutation Patterns Reveal Melanoma Subtypes and Influence Immunotherapy Response in Advanced Melanoma Patients. Cancers 2020, 12, 2359. [Google Scholar] [CrossRef]
- Stein, G.H.; Drullinger, L.F.; Soulard, A.; Dulic, V. Differential roles for cyclin-dependent kinase inhibitors p21 and p16 in the mechanisms of senescence and differentiation in human fibroblasts. Mol. Cell. Biol. 1999, 19, 2109–2117. [Google Scholar] [CrossRef] [Green Version]
- Novais, E.J.; Diekman, B.O.; Shapiro, I.M.; Risbud, M.V. p16(Ink4a) deletion in cells of the intervertebral disc affects their matrix homeostasis and senescence associated secretory phenotype without altering onset of senescence. Matrix Biol. 2019, 82, 54–70. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leon, K.E.; Tangudu, N.K.; Aird, K.M.; Buj, R. Loss of p16: A Bouncer of the Immunological Surveillance? Life 2021, 11, 309. https://doi.org/10.3390/life11040309
Leon KE, Tangudu NK, Aird KM, Buj R. Loss of p16: A Bouncer of the Immunological Surveillance? Life. 2021; 11(4):309. https://doi.org/10.3390/life11040309
Chicago/Turabian StyleLeon, Kelly E., Naveen Kumar Tangudu, Katherine M. Aird, and Raquel Buj. 2021. "Loss of p16: A Bouncer of the Immunological Surveillance?" Life 11, no. 4: 309. https://doi.org/10.3390/life11040309
APA StyleLeon, K. E., Tangudu, N. K., Aird, K. M., & Buj, R. (2021). Loss of p16: A Bouncer of the Immunological Surveillance? Life, 11(4), 309. https://doi.org/10.3390/life11040309