
Organization of the Respiratory Supercomplexes in Cells with Defective Complex III: Structural Features and Metabolic Consequences




Abstract
:1. Introduction
2. Mitochondrial Proteins Are Encoded by Two Genomes
3. Both Genomes Contribute to the Onset of Mitochondrial Diseases
4. The OXPHOS System
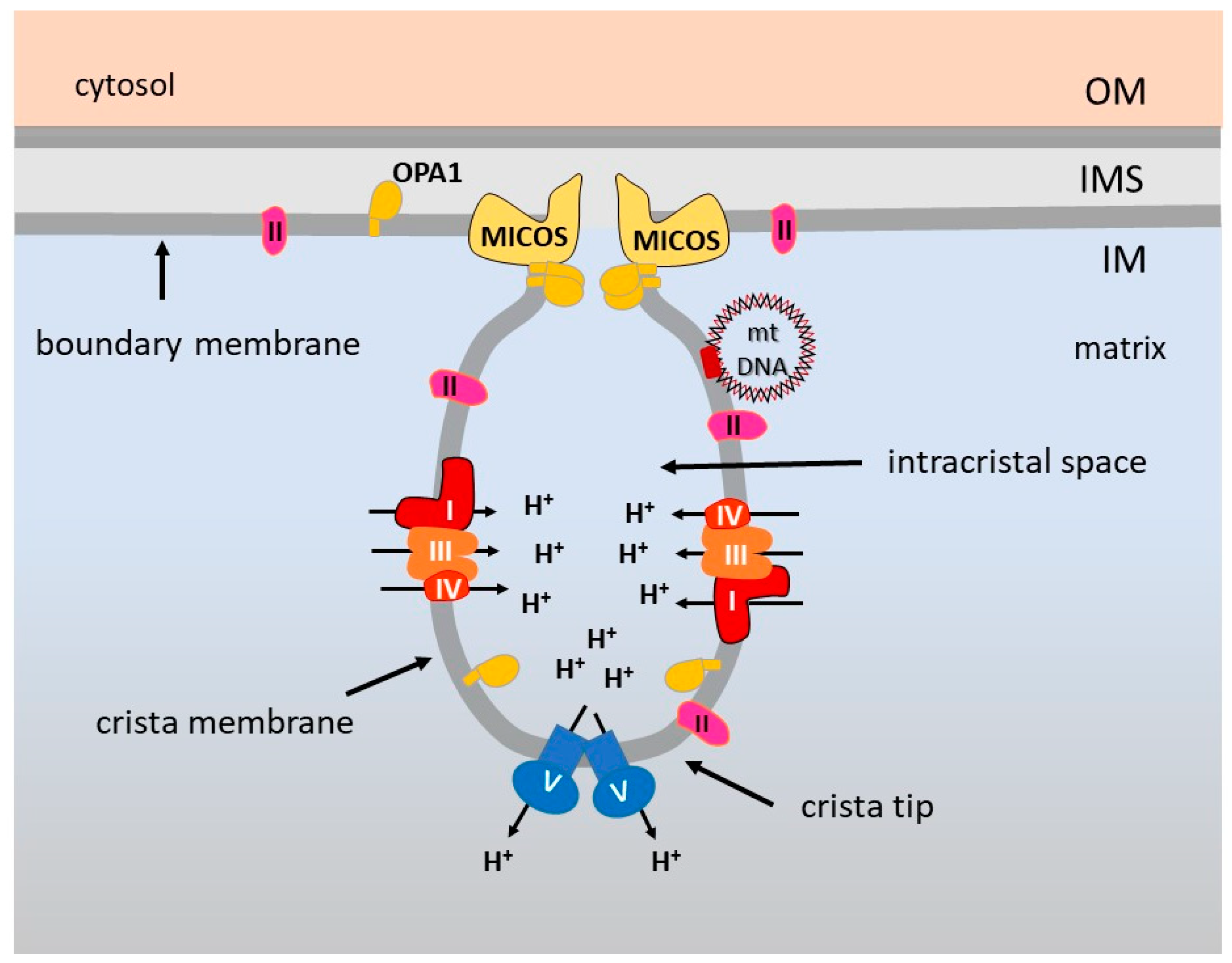
5. OXPHOS Optimization by the Inner Membrane Architecture
6. Supramolecular Organization of the Respiratory Complexes
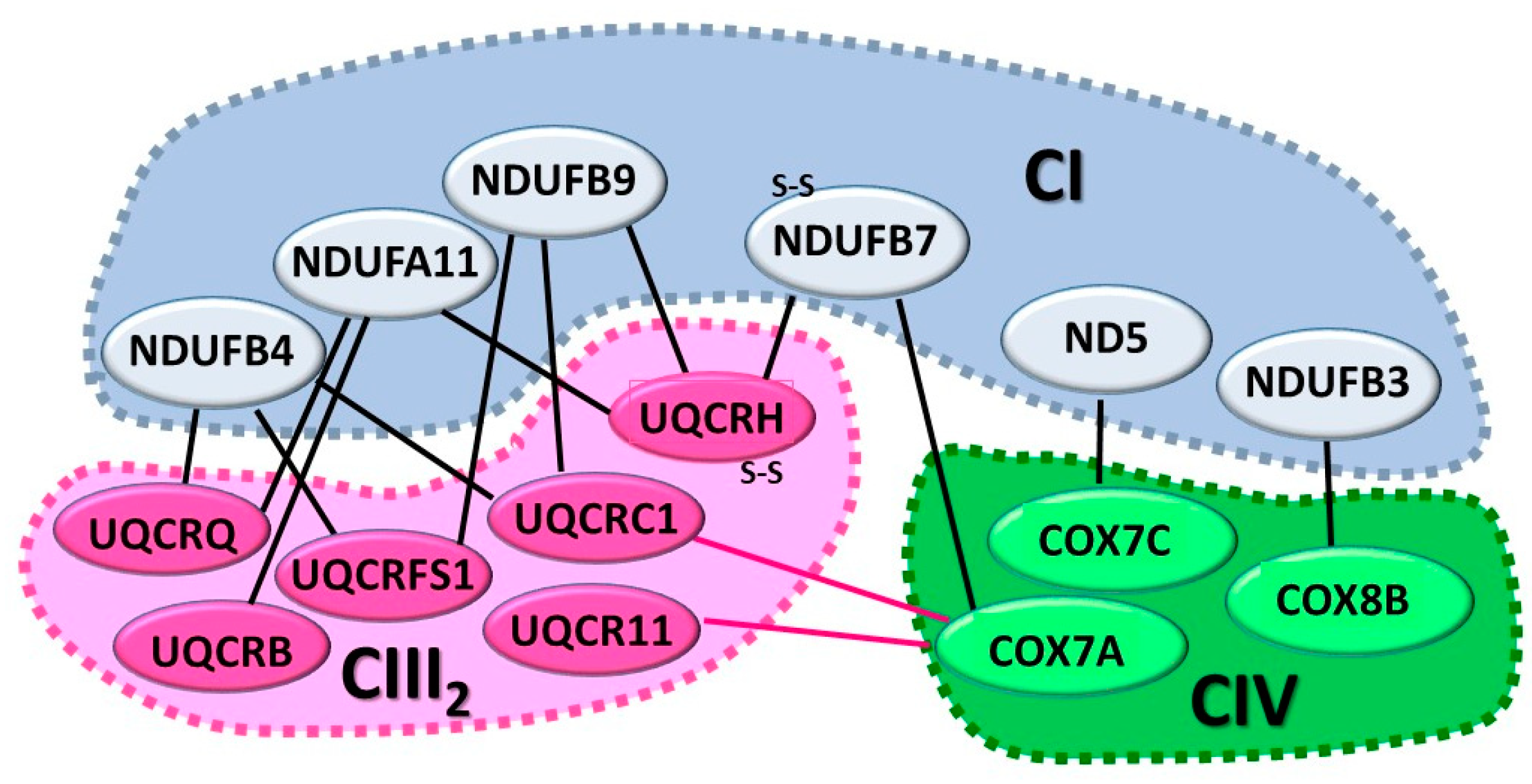
6.1. The CI+CIII2+CIV SC or Respirasome
6.2. The CI+CIII2 SC
6.3. The CIII2+CIV SC
7. SCs Assembly Factors
8. Models of SC Organization
9. Functional Roles of the SCs and CIII Involvement
10. SCs Biogenesis and Role of CIII
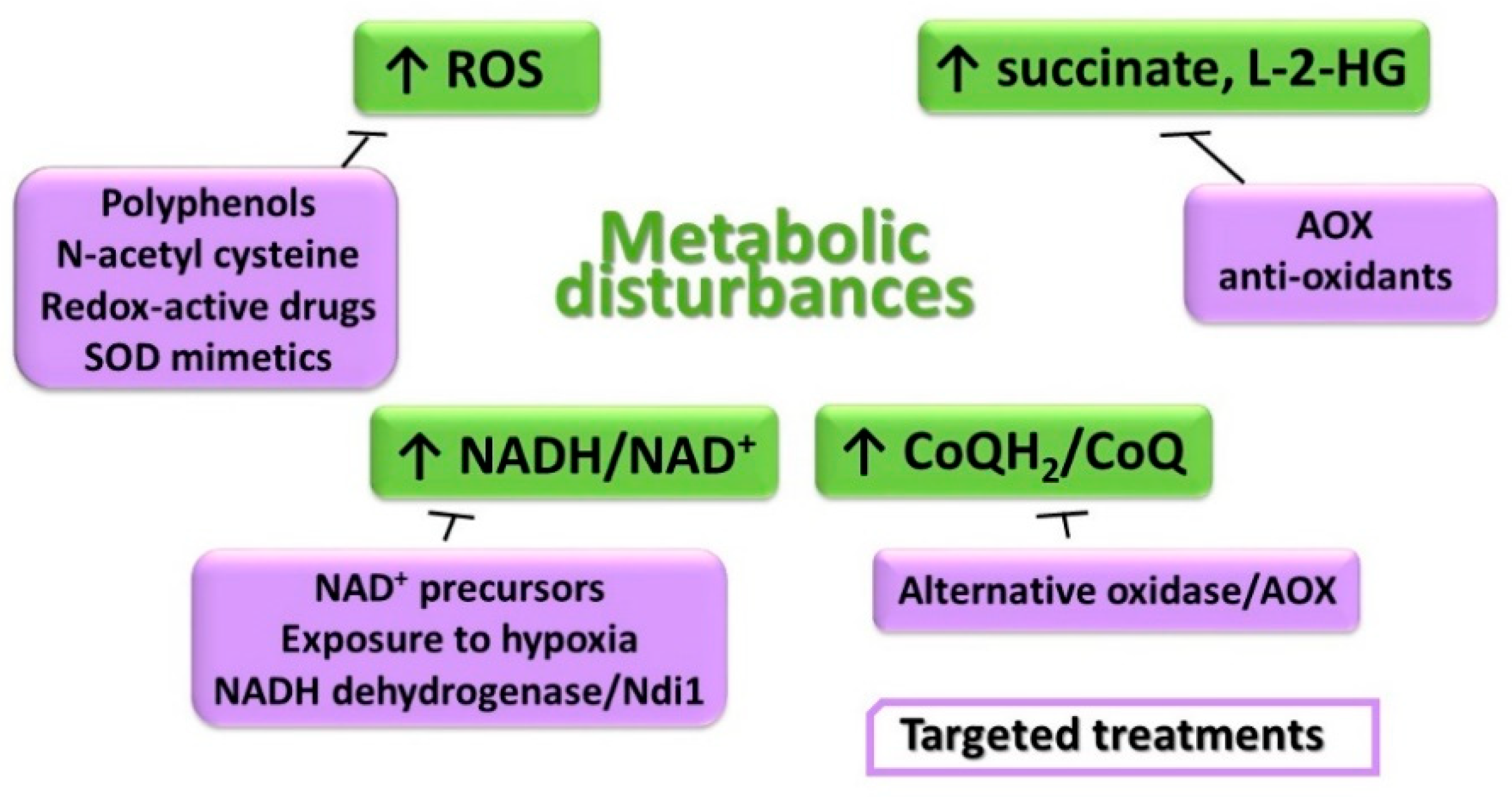
11. Metabolic Disturbances and Treatment Options
11.1. Unbalanced Intracellular Redox Homeostasis
11.2. Accumulation of the Reduced Form of Pyridine Nucleotides and CoQ
11.3. Elevation of Succinate and Effects on Gene Expression Regulation
12. Conclusions and Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Raimundo, N.; Baysal, B.E.; Shadel, G.S. Revisiting the TCA cycle: Signaling to tumor formation. Trends Mol. Med. 2011, 17, 641–649. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Reyes, I.; Diebold, L.P.; Kong, H.; Schieber, M.; Huang, H.; Hensley, C.T.; Mehta, M.M.; Wang, T.; Santos, J.H.; Woychik, R.; et al. TCA Cycle and Mitochondrial Membrane Potential Are Necessary for Diverse Biological Functions. Mol. Cell 2016, 61, 199–209. [Google Scholar] [CrossRef] [Green Version]
- Kastaniotis, A.J.; Autio, K.J.; Kerätär, J.M.; Monteuuis, G.; Mäkelä, A.M.; Nair, R.R.; Pietikäinen, L.P.; Shvetsova, A.; Chen, Z.; Hiltunen, J.K. Mitochondrial fatty acid synthesis, fatty acids and mitochondrial physiology. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 39–48. [Google Scholar] [CrossRef]
- Benador, I.Y.; Veliova, M.; Liesa, M.; Shirihai, O.S. Mitochondria Bound to Lipid Droplets: Where Mitochondrial Dynamics Regulate Lipid Storage and Utilization. Cell Metab. 2019, 29, 827–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lill, R.; Broderick, J.B.; Dean, D.R. Special issue on iron-sulfur proteins: Structure, function, biogenesis and diseases. Biochim. Biophys. Acta Bioenerg. 2015, 1853, 1251–1252. [Google Scholar] [CrossRef] [Green Version]
- Minton, D.R.; Nam, M.; McLaughlin, D.J.; Shin, J.; Bayraktar, E.C.; Alvarez, S.W.; Sviderskiy, V.O.; Papagiannakopoulos, T.; Sabatini, D.M.; Birsoy, K.; et al. Serine Catabolism by SHMT2 Is Required for Proper Mitochondrial Translation Initiation and Maintenance of Formylmethionyl-tRNAs. Mol. Cell 2018, 69, 610–621.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gherardi, G.; Monticelli, H.; Rizzuto, R.; Mammucari, C. The Mitochondrial Ca2+ Uptake and the Fine-Tuning of Aerobic Metabolism. Front. Physiol. 2020, 11, 554904. [Google Scholar] [CrossRef] [PubMed]
- Sena, L.A.; Chandel, N.S. Physiological Roles of Mitochondrial Reactive Oxygen Species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bock, F.J.; Tait, S.W.G. Mitochondria as multifaceted regulators of cell death. Nat. Rev. Mol. Cell Biol. 2020, 21, 85–100. [Google Scholar] [CrossRef]
- Liesa, M.; Shirihai, O.S. Mitochondrial Dynamics in the Regulation of Nutrient Utilization and Energy Expenditure. Cell Metab. 2013, 17, 491–506. [Google Scholar] [CrossRef] [Green Version]
- Morgenstern, M.; Stiller, S.B.; Lübbert, P.; Peikert, C.D.; Dannenmaier, S.; Drepper, F.; Weill, U.; Höß, P.; Feuerstein, R.; Gebert, M.; et al. Definition of a High-Confidence Mitochondrial Proteome at Quantitative Scale. Cell Rep. 2017, 19, 2836–2852. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulak, N.A.; Pichler, G.; Paron, I.; Nagaraj, N.; Mann, M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods 2014, 11, 319–324. [Google Scholar] [CrossRef]
- Monteuuis, G.; Miścicka, A.; Świrski, M.; Zenad, L.; Niemitalo, O.; Wrobel, L.; Alam, J.; Chacinska, A.; Kastaniotis, A.J.; Kufel, J. Non-canonical translation initiation in yeast generates a cryptic pool of mitochondrial proteins. Nucleic Acids Res. 2019, 47, 5777–5791. [Google Scholar] [CrossRef] [Green Version]
- Harding, J.W.; Pyeritz, E.A.; Copeland, E.S.; White, H.B. Role of glycerol 3-phosphate dehydrogenase in glyceride metabolism. Effect of diet on enzyme activities in chicken liver. Biochem. J. 1975, 146, 223–229. [Google Scholar] [CrossRef] [Green Version]
- Watmough, N.J.; Frerman, F.E. The electron transfer flavoprotein: Ubiquinone oxidoreductases. Biochim. Biophys. Acta Bioenerg. 2010, 1797, 1910–1916. [Google Scholar] [CrossRef]
- Alcázar-Fabra, M.; Navas, P.; Brea-Calvo, G. Coenzyme Q biosynthesis and its role in the respiratory chain structure. Biochim. Biophys. Acta Bioenerg. 2016, 1857, 1073–1078. [Google Scholar] [CrossRef] [PubMed]
- Henriques, B.J.; Olsen, R.K.J.; Gomes, C.M.; Bross, P. Electron transfer flavoprotein and its role in mitochondrial energy metabolism in health and disease. Gene 2021, 776, 145407. [Google Scholar] [CrossRef]
- Evans, D.R.; Guy, H.I. Mammalian Pyrimidine Biosynthesis: Fresh Insights into an Ancient Pathway. J. Biol. Chem. 2004, 279, 33035–33038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvi, F.; Gadda, G. Human choline dehydrogenase: Medical promises and biochemical challenges. Arch. Biochem. Biophys. 2013, 537, 243–252. [Google Scholar] [CrossRef]
- Hildebrandt, T.M.; Grieshaber, M.K. Three enzymatic activities catalyze the oxidation of sulfide to thiosulfate in mammalian and invertebrate mitochondria. FEBS J. 2008, 275, 3352–3361. [Google Scholar] [CrossRef]
- Hancock, C.N.; Liu, W.; Alvord, W.G.; Phang, J.M. Co-regulation of mitochondrial respiration by proline dehydrogenase/oxidase and succinate. Amino Acids 2016, 48, 859–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zachar, I.; Boza, G. Endosymbiosis before eukaryotes: Mitochondrial establishment in protoeukaryotes. Cell. Mol. Life Sci. 2020, 77, 3503–3523. [Google Scholar] [CrossRef] [Green Version]
- Johnston, I.G.; Williams, B.P. Evolutionary Inference across Eukaryotes Identifies Specific Pressures Favoring Mitochondrial Gene Retention. Cell Syst. 2016, 2, 101–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chrétien, D.; Bénit, P.; Ha, H.-H.; Keipert, S.; El-Khoury, R.; Chang, Y.-T.; Jastroch, M.; Jacobs, H.T.; Rustin, P.; Rak, M. Mitochondria are physiologically maintained at close to 50 °C. PLoS Biol. 2018, 16, e2003992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, J.F. Why chloroplasts and mitochondria retain their own genomes and genetic systems: Colocation for redox regulation of gene expression. Proc. Natl. Acad. Sci. USA 2015, 112, 10231–10238. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Vizarra, E.; Zeviani, M. Mitochondrial disorders of the OXPHOS system. FEBS Lett. 2020. [Google Scholar] [CrossRef]
- Iwata, S.; Lee, J.W.; Okada, K.; Lee, J.K.; Iwata, M.; Rasmussen, B.; Link, T.A.; Ramaswamy, S.; Jap, B.K. Complete Structure of the 11-Subunit Bovine Mitochondrial Cytochrome bc1 Complex. Science 1998, 281, 64–71. [Google Scholar] [CrossRef]
- Tsukihara, T.; Aoyama, H.; Yamashita, E.; Tomizaki, T.; Yamaguchi, H.; Shinzawa-Itoh, K.; Nakashima, R.; Yaono, R.; Yoshikawa, S. The Whole Structure of the 13-Subunit Oxidized Cytochrome c Oxidase at 2.8 A. Science 1996, 272, 1136–1144. [Google Scholar] [CrossRef]
- Sun, F.; Huo, X.; Zhai, Y.; Wang, A.; Xu, J.; Su, D.; Bartlam, M.; Rao, Z. Crystal Structure of Mitochondrial Respiratory Membrane Protein Complex II. Cell 2005, 121, 1043–1057. [Google Scholar] [CrossRef] [Green Version]
- Zickermann, V.; Wirth, C.; Nasiri, H.; Siegmund, K.; Schwalbe, H.; Hunte, C.; Brandt, U. Mechanistic insight from the crystal structure of mitochondrial complex I. Science 2015, 347, 44–49. [Google Scholar] [CrossRef] [Green Version]
- Zhu, J.; Vinothkumar, K.R.; Hirst, J.Z.J. Structure of mammalian respiratory complex I. Nat. Cell Biol. 2016, 536, 354–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaza, J.N.; Vinothkumar, K.R.; Hirst, J. Structure of the Deactive State of Mammalian Respiratory Complex I. Structure 2018, 26, 312–319.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agip, A.-N.A.; Blaza, J.N.; Bridges, H.R.; Viscomi, C.; Rawson, S.; Muench, S.P.; Hirst, J. Cryo-EM structures of complex I from mouse heart mitochondria in two biochemically defined states. Nat. Struct. Mol. Biol. 2018, 25, 548–556. [Google Scholar] [CrossRef]
- Fiedorczuk, K.; Letts, J.A.; Degliesposti, G.; Kaszuba, K.; Skehel, G.D.M.; Sazanov, L.A. Atomic structure of the entire mammalian mitochondrial complex I. Nat. Cell Biol. 2016, 538, 406–410. [Google Scholar] [CrossRef] [Green Version]
- Kampjut, D.; Sazanov, L.A. The coupling mechanism of mammalian respiratory complex I. Science 2020, 370, eabc4209. [Google Scholar] [CrossRef]
- Vinothkumar, K.R.; Zhu, J.; Hirst, J. Architecture of mammalian respiratory complex I. Nat. Cell Biol. 2014, 515, 80–84. [Google Scholar] [CrossRef] [Green Version]
- Yoga, E.G.; Angerer, H.; Parey, K.; Zickermann, V. Respiratory complex I—Mechanistic insights and advances in structure determination. Biochim. Biophys. Acta Bioenerg. 2020, 1861, 148153. [Google Scholar] [CrossRef]
- Cecchini, G. Function and Structure of Complex II of the Respiratory Chain. Annu. Rev. Biochem. 2003, 72, 77–109. [Google Scholar] [CrossRef] [Green Version]
- Bezawork-Geleta, A.; Rohlena, J.; Dong, L.; Pacak, K.; Neuzil, J. Mitochondrial Complex II: At the Crossroads. Trends Biochem. Sci. 2017, 42, 312–325. [Google Scholar] [CrossRef]
- Sarewicz, M.; Pintscher, S.; Pietras, R.; Borek, A.; Bujnowicz, Ł.; Hanke, G.; Cramer, W.A.; Finazzi, G.; Osyczka, A. Catalytic Reactions and Energy Conservation in the Cytochrome bc1 and b6f Complexes of Energy-Transducing Membranes. Chem. Rev. 2021, 121, 2020–2108. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Gu, J.; Wu, M.; Yang, M. Amazing structure of respirasome: Unveiling the secrets of cell respiration. Protein Cell 2016, 7, 854–865. [Google Scholar] [CrossRef] [Green Version]
- Hakvoort, T.B.; Moolenaar, K.; Lankvelt, A.H.; Sinjorgo, K.M.; Dekker, H.L.; Muijsers, A.O. Separation, stability and kinetics of monomeric and dimeric bovine heart cytochrome c oxidase. Biochim. Biophys. Acta Bioenerg. 1987, 894, 347–354. [Google Scholar] [CrossRef]
- Zong, S.; Wu, M.; Gu, J.; Liu, T.; Guo, R.; Yang, M. Structure of the intact 14-subunit human cytochrome c oxidase. Cell Res. 2018, 28, 1026–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balsa, E.; Marco, R.; Perales-Clemente, E.; Szklarczyk, R.; Calvo, E.; Landázuri, M.O.; Enríquez, J.A. NDUFA4 Is a Subunit of Complex IV of the Mammalian Electron Transport Chain. Cell Metab. 2012, 16, 378–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, B.C. The sequence of electron carriers in the reaction of cytochrome c oxidase with oxygen. J. Bioenerg. Biomembr. 1993, 25, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Kadenbach, B.; Hüttemann, M. The subunit composition and function of mammalian cytochrome c oxidase. Mitochondrion 2015, 24, 64–76. [Google Scholar] [CrossRef] [PubMed]
- Ishigami, I.; Zatsepin, N.A.; Hikita, M.; Conrad, C.E.; Nelson, G.; Coe, J.D.; Basu, S.; Grant, T.D.; Seaberg, M.H.; Sierra, R.G.; et al. Crystal structure of CO-bound cytochrome c oxidase determined by serial femtosecond X-ray crystallography at room temperature. Proc. Natl. Acad. Sci. USA 2017, 114, 8011–8016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frey, T.G.; Mannella, C.A. The internal structure of mitochondria. Trends Biochem. Sci. 2000, 25, 319–324. [Google Scholar] [CrossRef]
- Harner, M.; Körner, C.; Walther, D.; Mokranjac, D.; Kaesmacher, J.; Welsch, U.; Griffith, J.; Mann, M.; Reggiori, F.; Neupert, W. The mitochondrial contact site complex, a determinant of mitochondrial architecture. EMBO J. 2011, 30, 4356–4370. [Google Scholar] [CrossRef] [Green Version]
- Rampelt, H.; Zerbes, R.M.; van der Laan, M.; Pfanner, N. Role of the mitochondrial contact site and cristae organizing system in membrane architecture and dynamics. Biochim. Biophys. Acta Bioenerg. 2017, 1864, 737–746. [Google Scholar] [CrossRef] [PubMed]
- Stephan, T.; Brüser, C.; Deckers, M.; Steyer, A.M.; Balzarotti, F.; Barbot, M.; Behr, T.S.; Heim, G.; Hübner, W.; Ilgen, P.; et al. MICOS assembly controls mitochondrial inner membrane remodeling and crista junction redistribution to mediate cristae formation. EMBO J. 2020, 39, 104105. [Google Scholar] [CrossRef] [PubMed]
- Frezza, C.; Cipolat, S.; De Brito, O.M.; Micaroni, M.; Beznoussenko, G.V.; Rudka, T.; Bartoli, D.; Polishuck, R.S.; Danial, N.N.; De Strooper, B.; et al. OPA1 Controls Apoptotic Cristae Remodeling Independently from Mitochondrial Fusion. Cell 2006, 126, 177–189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quintana-Cabrera, R.; Quirin, C.; Glytsou, C.; Corrado, M.; Urbani, A.; Pellattiero, A.; Calvo, E.; Vázquez, J.; Enríquez, J.A.; Gerle, C.; et al. The cristae modulator Optic atrophy 1 requires mitochondrial ATP synthase oligomers to safeguard mitochondrial function. Nat. Commun. 2018, 9, 3399. [Google Scholar] [CrossRef]
- Del Dotto, V.; Fogazza, M.; Carelli, V.; Rugolo, M.; Zanna, C. Eight human OPA1 isoforms, long and short: What are they for? Biochim. Biophys. Acta Bioenerg. 2018, 1859, 263–269. [Google Scholar] [CrossRef] [PubMed]
- Paumard, P.; Vaillier, J.; Coulary, B.; Schaeffer, J.; Soubannier, V.; Mueller, D.M.; Brèthes, D.; Di Rago, J.-P.; Velours, J. The ATP synthase is involved in generating mitochondrial cristae morphology. EMBO J. 2002, 21, 221–230. [Google Scholar] [CrossRef]
- Davies, K.M.; Strauss, M.; Daum, B.; Kief, J.H.; Osiewacz, H.D.; Rycovska, A.; Zickermann, V.; Kühlbrandt, W. Macromolecular organization of ATP synthase and complex I in whole mitochondria. Proc. Natl. Acad. Sci. USA 2011, 108, 14121–14126. [Google Scholar] [CrossRef] [Green Version]
- Rampelt, H.; Van Der Laan, M. The Yin & Yang of Mitochondrial Architecture—Interplay of MICOS and F1Fo-ATP synthase in cristae formation. Microb. Cell 2017, 4, 236–239. [Google Scholar] [CrossRef]
- Kondadi, A.K.; Anand, R.; Reichert, A.S. Cristae Membrane Dynamics—A Paradigm Change. Trends Cell Biol. 2020, 30, 923–936. [Google Scholar] [CrossRef]
- Busch, K.B. Inner mitochondrial membrane compartmentalization: Dynamics across scales. Int. J. Biochem. Cell Biol. 2020, 120, 105694. [Google Scholar] [CrossRef]
- Rieger, B.; Junge, W.; Busch, K.B. Lateral pH gradient between OXPHOS complex IV and F0F1 ATP-synthase in folded mitochondrial membranes. Nat. Commun. 2014, 5, 3103. [Google Scholar] [CrossRef] [Green Version]
- Toth, A.; Meyrat, A.; Stoldt, S.; Santiago, R.; Wenzel, D.; Jakobs, S.; Von Ballmoos, C.; Ott, M. Kinetic coupling of the respiratory chain with ATP synthase, but not proton gradients, drives ATP production in cristae membranes. Proc. Natl. Acad. Sci. USA 2020, 117, 2412–2421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuart, R.A.; Cruciat, C.-M.; Brunner, S.; Baumann, F.; Neupert, W. The Cytochrome bc 1 and Cytochrome c Oxidase Complexes Associate to Form a Single Supracomplex in Yeast Mitochondria. J. Biol. Chem. 2000, 275, 18093–18098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schägger, H. Supercomplexes in the respiratory chains of yeast and mammalian mitochondria. EMBO J. 2000, 19, 1777–1783. [Google Scholar] [CrossRef] [Green Version]
- Acín-Pérez, R.; Fernández-Silva, P.; Peleato, M.L.; Pérez-Martos, A.; Enriquez, J.A. Respiratory Active Mitochondrial Supercomplexes. Mol. Cell 2008, 32, 529–539. [Google Scholar] [CrossRef]
- Milenkovic, D.; Blaza, J.N.; Larsson, N.-G.; Hirst, J. The Enigma of the Respiratory Chain Supercomplex. Cell Metab. 2017, 25, 765–776. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobo-Jarne, T.; Ugalde, C. Respiratory chain supercomplexes: Structures, function and biogenesis. Semin. Cell Dev. Biol. 2018, 76, 179–190. [Google Scholar] [CrossRef]
- Althoff, T.; Mills, D.J.; Popot, J.-L.; Kühlbrandt, W. Arrangement of electron transport chain components in bovine mitochondrial supercomplex I1III2IV1. EMBO J. 2011, 30, 4652–4664. [Google Scholar] [CrossRef] [Green Version]
- Sousa, J.S.; Mills, D.J.; Vonck, J.; Kühlbrandt, W. Functional asymmetry and electron flow in the bovine respirasome. eLife 2016, 5, e21290. [Google Scholar] [CrossRef]
- Letts, J.A.; Fiedorczuk, K.; Sazanov, J.A.L.K.F.L.A. The architecture of respiratory supercomplexes. Nat. Cell Biol. 2016, 537, 644–648. [Google Scholar] [CrossRef]
- Gu, J.; Wu, M.; Guo, R.; Yan, K.; Lei, J.; Gao, N.; Yang, M. The architecture of the mammalian respirasome. Nat. Cell Biol. 2016, 537, 639–643. [Google Scholar] [CrossRef]
- Dudkina, N.V.; Kudryashev, M.; Stahlberg, H.; Boekema, E.J. Interaction of complexes I, III, and IV within the bovine respirasome by single particle cryoelectron tomography. Proc. Natl. Acad. Sci. USA 2011, 108, 15196–15200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, K.M.; Blum, T.B.; Kühlbrandt, W. Conserved in situ arrangement of complex I and III2 in mitochondrial respiratory chain supercomplexes of mammals, yeast, and plants. Proc. Natl. Acad. Sci. USA 2018, 115, 3024–3029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, R.; Zong, S.; Wu, M.; Gu, J.; Yang, M. Architecture of Human Mitochondrial Respiratory Megacomplex I2III2IV2. Cell 2017, 170, 1247–1257.e12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letts, J.A.; Fiedorczuk, K.; Degliesposti, G.; Skehel, M.; Sazanov, L.A. Structures of Respiratory Supercomplex I+III2 Reveal Functional and Conformational Crosstalk. Mol. Cell 2019, 75, 1131–1146.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hartley, A.M.; Lukoyanova, N.; Zhang, Y.; Cabrera-Orefice, A.; Arnold, S.; Meunier, B.; Pinotsis, N.; Maréchal, A. Structure of yeast cytochrome c oxidase in a supercomplex with cytochrome bc1. Nat. Struct. Mol. Biol. 2019, 26, 78–83. [Google Scholar] [CrossRef] [Green Version]
- Azuma, K.; Ikeda, K.; Inoue, S. Functional Mechanisms of Mitochondrial Respiratory Chain Supercomplex Assembly Factors and Their Involvement in Muscle Quality. Int. J. Mol. Sci. 2020, 21, 3182. [Google Scholar] [CrossRef]
- Chen, Y.-C.; Taylor, E.B.; Dephoure, N.; Heo, J.-M.; Tonhato, A.; Papandreou, I.; Nath, N.; Denko, N.C.; Gygi, S.P.; Rutter, J. Identification of a Protein Mediating Respiratory Supercomplex Stability. Cell Metab. 2012, 15, 348–360. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, K.; Shiba, S.; Horie-Inoue, K.; Shimokata, K.; Inoue, S. A stabilizing factor for mitochondrial respiratory supercomplex assembly regulates energy metabolism in muscle. Nat. Commun. 2013, 4, 2147. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Pérez, R.; Lobo-Jarne, T.; Milenkovic, D.; Mourier, A.; Bratic, A.; García-Bartolomé, A.; Fernández-Vizarra, E.; Cadenas, S.; Delmiro, A.; García-Consuegra, I.; et al. COX7A2L Is a Mitochondrial Complex III Binding Protein that Stabilizes the III2+IV Supercomplex without Affecting Respirasome Formation. Cell Rep. 2016, 16, 2387–2398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobo-Jarne, T.; Pérez-Pérez, R.; Fontanesi, F.; Timon-Gomez, A.; Wittig, I.; Peñas, A.; Serrano-Lorenzo, P.; García-Consuegra, I.; Arenas, J.; Martín, M.A.; et al. Multiple pathways coordinate assembly of human mitochondrial complex IV and stabilization of respiratory supercomplexes. EMBO J. 2020, 39, e103912. [Google Scholar] [CrossRef]
- Lapuente-Brun, E.; Moreno-Loshuertos, R.; Acín-Pérez, R.; Latorre-Pellicer, A.; Colás, C.; Balsa, E.; Perales-Clemente, E.; Quirós, P.M.; Calvo, E.; Rodríguez-Hernández, Á.; et al. Supercomplex Assembly Determines Electron Flux in the Mitochondrial Electron Transport Chain. Science 2013, 340, 1567–1570. [Google Scholar] [CrossRef]
- Timón-Gómez, A.; Garlich, J.; Stuart, R.A.; Ugalde, C.; Barrientos, A. Distinct Roles of Mitochondrial HIGD1A and HIGD2A in Respiratory Complex and Supercomplex Biogenesis. Cell Rep. 2020, 31, 107607. [Google Scholar] [CrossRef]
- Cogliati, S.; Calvo, E.; Loureiro, M.; Guaras, A.M.; Nieto-Arellano, R.; Garcia-Poyatos, C.; Ezkurdia, I.; Mercader, C.G.-P.N.; Vázquez, J.; Enriquez, J.A. Mechanism of super-assembly of respiratory complexes III and IV. Nat. Cell Biol. 2016, 539, 579–582. [Google Scholar] [CrossRef]
- Zhang, K.; Wang, G.; Zhang, X.; Hüttemann, P.P.; Qiu, Y.; Liu, J.; Mitchell, A.; Lee, I.; Zhang, C.; Lee, J.-S.; et al. COX7AR is a Stress-inducible Mitochondrial COX Subunit that Promotes Breast Cancer Malignancy. Sci. Rep. 2016, 6, 31742. [Google Scholar] [CrossRef] [Green Version]
- Tucker, E.J.; Wanschers, B.F.J.; Szklarczyk, R.; Mountford, H.S.; Wijeyeratne, X.W.; Brand, M.A.M.V.D.; Leenders, A.M.; Rodenburg, R.J.; Reljić, B.; Compton, A.G.; et al. Mutations in the UQCC1-Interacting Protein, UQCC2, Cause Human Complex III Deficiency Associated with Perturbed Cytochrome b Protein Expression. PLoS Genet. 2013, 9, e1004034. [Google Scholar] [CrossRef] [Green Version]
- Wanschers, B.F.; Szklarczyk, R.; Brand, M.A.V.D.; Jonckheere, A.; Suijskens, J.; Smeets, R.; Rodenburg, R.J.; Stephan, K.; Helland, I.B.; Elkamil, A.; et al. A mutation in the human CBP4 ortholog UQCC3 impairs complex III assembly, activity and cytochrome b stability. Hum. Mol. Genet. 2014, 23, 6356–6365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Desmurs, M.; Foti, M.; Raemy, E.; Vaz, F.M.; Martinou, J.-C.; Bairoch, A.; Lane, L. C11orf83, a Mitochondrial Cardiolipin-Binding Protein Involved inbc1Complex Assembly and Supercomplex Stabilization. Mol. Cell. Biol. 2015, 35, 1139–1156. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Zhang, G.; Guo, F.; Li, Q.; Luo, H.; Shu, Y.; Shen, Y.; Gan, J.; Xu, L.; Yang, H. Mitochondrial UQCC3 Modulates Hypoxia Adaptation by Orchestrating OXPHOS and Glycolysis in Hepatocellular Carcinoma. Cell Rep. 2020, 33, 108340. [Google Scholar] [CrossRef] [PubMed]
- Chance, B.; Williams, G.R. A Method for the Localization of Sites for Oxidative Phosphorylation. Nat. Cell Biol. 1955, 176, 250–254. [Google Scholar] [CrossRef]
- Green, D.E.; Tzagoloff, A. The mitochondrial electron transfer chain. Arch. Biochem. Biophys. 1966, 116, 293–304. [Google Scholar] [CrossRef]
- Hackenbrock, C.R.; Chazotte, B.; Gupte, S.S. The random collision model and a critical assessment of diffusion and collision in mitochondrial electron transport. J. Bioenerg. Biomembr. 1986, 18, 331–368. [Google Scholar] [CrossRef] [PubMed]
- Hatefi, Y.; Haavik, A.; Griffiths, D. Studies on the Electron Transfer System. J. Biol. Chem. 1962, 237, 1676–1680. [Google Scholar] [CrossRef]
- Acin-Perez, R.; Enriquez, J.A. The function of the respiratory supercomplexes: The plasticity model. Biochim. Biophys. Acta Bioenerg. 2014, 1837, 444–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guarás, A.; Perales-Clemente, E.; Calvo, E.; Acín-Pérez, R.; Loureiro-Lopez, M.; Pujol, C.; Martínez-Carrascoso, I.; Nuñez, E.; García-Marqués, F.; Rodríguez-Hernández, M.A.; et al. The CoQH2/CoQ Ratio Serves as a Sensor of Respiratory Chain Efficiency. Cell Rep. 2016, 15, 197–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaza, J.N.; Serreli, R.; Jones, A.J.Y.; Mohammed, K.; Hirst, J. Kinetic evidence against partitioning of the ubiquinone pool and the catalytic relevance of respiratory-chain supercomplexes. Proc. Natl. Acad. Sci. USA 2014, 111, 15735–15740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fedor, J.G.; Hirst, J. Mitochondrial Supercomplexes Do Not Enhance Catalysis by Quinone Channeling. Cell Metab. 2018, 28, 525–531.e4. [Google Scholar] [CrossRef] [Green Version]
- Hernansanz-Agustín, P.; Enríquez, J.A. Functional segmentation of CoQ and cyt c pools by respiratory complex superassembly. Free Radic. Biol. Med. 2021, 167, 232–242. [Google Scholar] [CrossRef]
- Bianchi, C.; Genova, M.L.; Castelli, G.P.; Lenaz, G. The Mitochondrial Respiratory Chain Is Partially Organized in a Supercomplex Assembly. J. Biol. Chem. 2004, 279, 36562–36569. [Google Scholar] [CrossRef] [Green Version]
- Trouillard, M.; Meunier, B.; Rappaport, F. Questioning the functional relevance of mitochondrial supercomplexes by time-resolved analysis of the respiratory chain. Proc. Natl. Acad. Sci. USA 2011, 108, E1027–E1034. [Google Scholar] [CrossRef] [Green Version]
- Letts, J.A.; Sazanov, L.A. Clarifying the supercomplex: The higher-order organization of the mitochondrial electron transport chain. Nat. Struct. Mol. Biol. 2017, 24, 800–808. [Google Scholar] [CrossRef]
- Schägger, H.; de Coo, R.; Bauer, M.F.; Hofmann, S.; Godinot, C.; Brandt, U. Significance of Respirasomes for the Assembly/Stability of Human Respiratory Chain Complex I. J. Biol. Chem. 2004, 279, 36349–36353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moreno-Lastres, D.; Fontanesi, F.; García-Consuegra, I.; Martín, M.A.; Arenas, J.; Barrientos, A.; Ugalde, C. Mitochondrial Complex I Plays an Essential Role in Human Respirasome Assembly. Cell Metab. 2012, 15, 324–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirst, J. Open questions: Respiratory chain supercomplexes—why are they there and what do they do? BMC Biol. 2018, 16, 111. [Google Scholar] [CrossRef] [Green Version]
- Muller, F.L.; Liu, Y.; Van Remmen, H. Complex III Releases Superoxide to Both Sides of the Inner Mitochondrial Membrane. J. Biol. Chem. 2004, 279, 49064–49073. [Google Scholar] [CrossRef] [Green Version]
- Kussmaul, L.; Hirst, J. The mechanism of superoxide production by NADH: Ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc. Natl. Acad. Sci. USA 2006, 103, 7607–7612. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pryde, K.R.; Hirst, J. Superoxide Is Produced by the Reduced Flavin in Mitochondrial Complex I. J. Biol. Chem. 2011, 286, 18056–18065. [Google Scholar] [CrossRef] [Green Version]
- Maranzana, E.; Barbero, G.; Falasca, A.I.; Lenaz, G.; Genova, M.L. Mitochondrial Respiratory Supercomplex Association Limits Production of Reactive Oxygen Species from Complex I. Antioxid. Redox Signal. 2013, 19, 1469–1480. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Fabuel, I.; Le Douce, J.; Logan, A.; James, A.M.; Bonvento, G.; Murphy, M.P.; Almeida, A.; Bolaños, J.P. Complex I assembly into supercomplexes determines differential mitochondrial ROS production in neurons and astrocytes. Proc. Natl. Acad. Sci. USA 2016, 113, 13063–13068. [Google Scholar] [CrossRef] [Green Version]
- Morán, M.; Marín-Buera, L.; Gil-Borlado, M.C.; Rivera, H.; Blázquez, A.; Seneca, S.; Vázquez-López, M.; Arenas, J.; Martín, M.A.; Ugalde, C. Cellular pathophysiological consequences of BCS1L mutations in mitochondrial complex III enzyme deficiency. Hum. Mutat. 2010, 31, 930–941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz, F.; Enriquez, J.A.; Moraes, C.T. Cells Lacking Rieske Iron-Sulfur Protein Have a Reactive Oxygen Species-Associated Decrease in Respiratory Complexes I and IV. Mol. Cell. Biol. 2012, 32, 415–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernansanz-Agustín, P.; Choya-Foces, C.; Carregal-Romero, S.; Ramos, E.; Oliva, T.; Villa-Piña, T.; Moreno, L.; Izquierdo-Álvarez, A.; Cabrera-García, J.D.; Cortés, A.; et al. Na+ controls hypoxic signalling by the mitochondrial respiratory chain. Nature 2020, 586, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Tropeano, C.V.; Aleo, S.J.; Zanna, C.; Roberti, M.; Scandiffio, L.; Polosa, P.L.; Fiori, J.; Porru, E.; Roda, A.; Carelli, V.; et al. Fine-tuning of the respiratory complexes stability and supercomplexes assembly in cells defective of complex III. Biochim. Biophys. Acta Bioenerg. 2020, 1861, 148133. [Google Scholar] [CrossRef]
- Nogueira, C.; Barros, J.; Sá, M.J.; Azevedo, L.; Taipa, R.; Torraco, A.; Meschini, M.C.; Verrigni, D.; Nesti, C.; Rizza, T.; et al. Novel TTC19 mutation in a family with severe psychiatric manifestations and complex III deficiency. Neurogenetics 2013, 14, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Davoudi, M.; Kotarsky, H.; Hansson, E.; Fellman, V. Complex I Function and Supercomplex Formation Are Preserved in Liver Mitochondria Despite Progressive Complex III Deficiency. PLoS ONE 2014, 9, e86767. [Google Scholar] [CrossRef] [PubMed]
- Davoudi, M.; Kotarsky, H.; Hansson, E.; Kallijärvi, J.; Fellman, V. COX7A2L/SCAFI and Pre-Complex III Modify Respiratory Chain Supercomplex Formation in Different Mouse Strains with a Bcs1l Mutation. PLoS ONE 2016, 11, e0168774. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Castillo, S.; Baertling, F.; Kownatzki, D.; Wessels, H.J.; Arnold, S.; Brandt, U.; Nijtmans, L. The Assembly Pathway of Mitochondrial Respiratory Chain Complex I. Cell Metab. 2017, 25, 128–139. [Google Scholar] [CrossRef] [Green Version]
- Protasoni, M.; Pérez-Pérez, R.; Lobo-Jarne, T.; Harbour, M.E.; Ding, S.; Peñas, A.; Diaz, F.; Moraes, C.T.; Fearnley, I.M.; Zeviani, M.; et al. Respiratory supercomplexes act as a platform for complex III -mediated maturation of human mitochondrial complexes I and IV. EMBO J. 2020, 39, e102817. [Google Scholar] [CrossRef]
- Páleníková, P.; Harbour, M.E.; Prodi, F.; Minczuk, M.; Zeviani, M.; Ghelli, A.; Fernández-Vizarra, E. Duplexing complexome profiling with SILAC to study human respiratory chain assembly defects. Biochim. Biophys. Acta Bioenerg. 2021, 1862, 148395. [Google Scholar] [CrossRef]
- Carossa, V.; Ghelli, A.; Tropeano, C.V.; Valentino, M.L.; Iommarini, L.; Maresca, A.; Caporali, L.; La Morgia, C.; Liguori, R.; Barboni, P.; et al. A Novel in-Frame 18-bp Microdeletion in MTCYB Causes a Multisystem Disorder with Prominent Exercise Intolerance. Hum. Mutat. 2014, 35, 954–958. [Google Scholar] [CrossRef]
- Tropeano, C.V.; Fiori, J.; Carelli, V.; Caporali, L.; Daldal, F.; Ghelli, A.M.; Rugolo, M. Complex II phosphorylation is triggered by unbalanced redox homeostasis in cells lacking complex III. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 182–190. [Google Scholar] [CrossRef]
- Blakely, E.L.; Mitchell, A.L.; Fisher, N.; Meunier, B.; Nijtmans, L.G.; Schaefer, A.M.; Jackson, M.J.; Turnbull, D.M.; Taylor, R.W. A mitochondrial cytochrome b mutation causing severe respiratory chain enzyme deficiency in humans and yeast. FEBS J. 2005, 272, 3583–3592. [Google Scholar] [CrossRef]
- Lamantea, E.; Carrara, F.; Mariotti, C.; Morandi, L.; Tiranti, V.; Zeviani, M. A novel nonsense mutation (Q352X) in the mitochondrial cytochrome b gene associated with a combined deficiency of complexes I and III. Neuromuscul. Disord. 2002, 12, 49–52. [Google Scholar] [CrossRef]
- Haut, S.; Brivet, M.; Touati, G.; Rustin, P.; Lebon, S.; Garcia-Cazorla, A.; Saudubray, J.M.; Boutron, A.; Legrand, A.; Slama, A. A deletion in the human QP-C gene causes a complex III deficiency resulting in hypoglycaemia and lactic acidosis. Qual. Life Res. 2003, 113, 118–122. [Google Scholar] [CrossRef]
- Barel, O.; Shorer, Z.; Flusser, H.; Ofir, R.; Narkis, G.; Finer, G.; Shalev, H.; Nasasra, A.; Saada, A.; Birk, O.S. Mitochondrial Complex III Deficiency Associated with a Homozygous Mutation in UQCRQ. Am. J. Hum. Genet. 2008, 82, 1211–1216. [Google Scholar] [CrossRef] [Green Version]
- Gaignard, P.; Menezes, M.; Schiff, M.; Bayot, A.; Rak, M.; de Baulny, H.O.; Su, C.-H.; Gilleron, M.; Lombes, A.; Abida, H.; et al. Mutations in CYC1, Encoding Cytochrome c1 Subunit of Respiratory Chain Complex III, Cause Insulin-Responsive Hyperglycemia. Am. J. Hum. Genet. 2013, 93, 384–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyake, N.; Yano, S.; Sakai, C.; Hatakeyama, H.; Matsushima, Y.; Shiina, M.; Watanabe, Y.; Bartley, J.; Abdenur, J.E.; Wang, R.Y.; et al. Mitochondrial Complex III Deficiency Caused by a HomozygousUQCRC2Mutation Presenting with Neonatal-Onset Recurrent Metabolic Decompensation. Hum. Mutat. 2013, 34, 446–452. [Google Scholar] [CrossRef] [PubMed]
- Feichtinger, R.G.; Brunner-Krainz, M.; Alhaddad, B.; Wortmann, S.B.; Kovacs-Nagy, R.; Stojakovic, T.; Erwa, W.; Resch, B.; Windischhofer, W.; Verheyen, S.; et al. Combined Respiratory Chain Deficiency and UQCC2 Mutations in Neonatal Encephalomyopathy: Defective Supercomplex Assembly in Complex III Deficiencies. Oxidative Med. Cell. Longev. 2017, 2017, 7202589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghelli, A.; Tropeano, C.V.; Calvaruso, M.A.; Marchesini, A.; Iommarini, L.; Porcelli, A.M.; Zanna, C.; De Nardo, V.; Martinuzzi, A.; Wibrand, F.; et al. The cytochrome b p.278Y>C mutation causative of a multisystem disorder enhances superoxide production and alters supramolecular interactions of respiratory chain complexes. Hum. Mol. Genet. 2013, 22, 2141–2151. [Google Scholar] [CrossRef] [Green Version]
- Acín-Pérez, R.; Bayona-Bafaluy, M.P.; Fernández-Silva, P.; Moreno-Loshuertos, R.; Pérez-Martos, A.; Bruno, C.; Moraes, C.T.; Enríquez, J.A. Respiratory complex III is required to maintain complex I in mammalian mitochondria. Mol. Cell. 2004, 13, 805–815. [Google Scholar] [CrossRef]
- Spinazzi, M.; Radaelli, E.; Horré, K.; Arranz, A.M.; Gounko, N.V.; Agostinis, P.; Maia, T.M.; Impens, F.; Morais, V.A.; Lopez-Lluch, G.; et al. PARL deficiency in mouse causes Complex III defects, coenzyme Q depletion, and Leigh-like syndrome. Proc. Natl. Acad. Sci. USA 2019, 116, 277–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iommarini, L.; Ghelli, A.; Leone, G.; Tropeano, C.V.; Kurelac, I.; Amato, L.B.; Gasparre, G.; Porcelli, A.M. Mild phenotypes and proper supercomplex assembly in human cells carrying the homoplasmic m.15557G > A mutation in cytochrome b gene. Hum. Mutat. 2018, 39, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Bottani, E.; Lamperti, C.; Prigione, A.; Tiranti, V.; Persico, N.; Brunetti, D. Therapeutic Approaches to Treat Mitochondrial Diseases: “One-Size-Fits-All” and “Precision Medicine” Strategies. Pharmaceutics 2020, 12, 1083. [Google Scholar] [CrossRef] [PubMed]
- Ježek, P.; Hlavatá, L. Mitochondria in homeostasis of reactive oxygen species in cell, tissues, and organism. Int. J. Biochem. Cell Biol. 2005, 37, 2478–2503. [Google Scholar] [CrossRef]
- Raha, S.; Robinson, B.H. Mitochondria, oxygen free radicals, disease and ageing. Trends Biochem. Sci. 2000, 25, 502–508. [Google Scholar] [CrossRef]
- Wong, H.-S.; Dighe, P.A.; Mezera, V.; Monternier, P.-A.; Brand, M.D. Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J. Biol. Chem. 2017, 292, 16804–16809. [Google Scholar] [CrossRef] [Green Version]
- Seelert, H.; Dani, D.; Dante, S.; Hauß, T.; Krause, F.; Schafer, E.; Frenzel, M.; Poetsch, A.; Rexroth, S.; Schwaßmann, H.; et al. From protons to OXPHOS supercomplexes and Alzheimer’s disease: Structure–dynamics–function relationships of energy-transducing membranes. Biochim. Biophys. Acta Bioenerg. 2009, 1787, 657–671. [Google Scholar] [CrossRef]
- Genova, M.L.; Lenaz, G. Functional role of mitochondrial respiratory supercomplexes. Biochim. Biophys. Acta Bioenerg. 2014, 1837, 427–443. [Google Scholar] [CrossRef] [Green Version]
- Malferrari, M.; Ghelli, A.; Roggiani, F.; Valenti, G.; Paolucci, F.; Rugolo, M.; Rapino, S. Reactive Oxygen Species Produced by Mutated Mitochondrial Respiratory Chains of Entire Cells Monitored Using Modified Microelectrodes. ChemElectroChem 2019, 6, 627–633. [Google Scholar] [CrossRef] [Green Version]
- Anwar, M.R.; Saldana-Caboverde, A.; Garcia, S.; Diaz, F. The Organization of Mitochondrial Supercomplexes is Modulated by Oxidative Stress In Vivo in Mouse Models of Mitochondrial Encephalopathy. Int. J. Mol. Sci. 2018, 19, 1582. [Google Scholar] [CrossRef] [Green Version]
- Mori, M.; Goldstein, J.; Young, S.P.; Bossen, E.H.; Shoffner, J.; Koeberl, D.D. Complex III deficiency due to an in-frame MTCYB deletion presenting as ketotic hypoglycemia and lactic acidosis. Mol. Genet. Metab. Rep. 2015, 4, 39–41. [Google Scholar] [CrossRef]
- Gaignard, P.; Eyer, D.; Lebigot, E.; Oliveira, C.; Therond, P.; Boutron, A.; Slama, A. UQCRC2 mutation in a patient with mitochondrial complex III deficiency causing recurrent liver failure, lactic acidosis and hypoglycemia. J. Hum. Genet. 2017, 62, 729–731. [Google Scholar] [CrossRef] [PubMed]
- Levéen, P.; Kotarsky, H.; Mörgelin, M.; Karikoski, R.; Elmér, E.; Fellman, V. The GRACILE mutation introduced into Bcs1l causes postnatal complex III deficiency: A viable mouse model for mitochondrial hepatopathy. Hepatology 2011, 53, 437–447. [Google Scholar] [CrossRef]
- Rajendran, J.; Tomašić, N.; Kotarsky, H.; Hansson, E.; Velagapudi, V.; Kallijärvi, J.; Fellman, V. Effect of High-Carbohydrate Diet on Plasma Metabolome in Mice with Mitochondrial Respiratory Chain Complex III Deficiency. Int. J. Mol. Sci. 2016, 17, 1824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wibrand, F.; Ravn, K.; Schwartz, M.; Rosenberg, T.; Horn, N.; Vissing, J. Multisystem disorder associated with a missense mutation in the mitochondrial cytochrome b gene. Ann. Neurol. 2001, 50, 540–543. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.F.; Caudal, A.; Abell, L.; Gowda, G.A.N.; Tian, R. Targeting NAD+ Metabolism as Interventions for Mitochondrial Disease. Sci. Rep. 2019, 9, 3073. [Google Scholar] [CrossRef]
- Johnson, S.C.; Yanos, M.E.; Kayser, E.-B.; Quintana, A.; Sangesland, M.; Castanza, A.; Uhde, L.; Hui, J.; Wall, V.Z.; Gagnidze, A.; et al. mTOR Inhibition Alleviates Mitochondrial Disease in a Mouse Model of Leigh Syndrome. Science 2013, 342, 1524–1528. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Canaveras, J.C.G.; Chen, Z.; Wang, L.; Liang, L.; Jang, C.; Mayr, J.A.; Zhang, Z.; Ghergurovich, J.M.; Zhan, L.; et al. Serine Catabolism Feeds NADH when Respiration Is Impaired. Cell Metab. 2020, 31, 809–821.e6. [Google Scholar] [CrossRef]
- Jain, I.H.; Zazzeron, L.; Goli, R.; Alexa, K.; Schatzman-Bone, S.; Dhillon, H.; Goldberger, O.; Peng, J.; Shalem, O.; Sanjana, N.E.; et al. Hypoxia as a therapy for mitochondrial disease. Science 2016, 352, 54–61. [Google Scholar] [CrossRef] [Green Version]
- Seo, B.B.; Kitajima-Ihara, T.; Chan, E.K.L.; Scheffler, I.E.; Matsuno-Yagi, A.; Yagi, T. Molecular remedy of complex I defects: Rotenone-insensitive internal NADH-quinone oxidoreductase of Saccharomyces cerevisiae mitochondria restores the NADH oxidase activity of complex I-deficient mammalian cells. Proc. Natl. Acad. Sci. USA 1998, 95, 9167–9171. [Google Scholar] [CrossRef] [Green Version]
- Perales-Clemente, E.; Bayona-Bafaluy, M.P.; Pérez-Martos, A.; Barrientos, A.; Fernández-Silva, P.; Enriquez, J.A. Restoration of electron transport without proton pumping in mammalian mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 18735–18739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife 2014, 3, e02242. [Google Scholar] [CrossRef]
- Bai, Y.; Hájek, P.; Chomyn, A.; Chan, E.; Seo, B.B.; Matsuno-Yagi, A.; Yagi, T.; Attardi, G. Lack of Complex I Activity in Human Cells Carrying a Mutation in MtDNA-encoded ND4 Subunit Is Corrected by the Saccharomyces cerevisiae NADH-Quinone Oxidoreductase (NDI1) Gene. J. Biol. Chem. 2001, 276, 38808–38813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yagi, T.; Seo, B.B.; Nakamaru-Ogiso, E.; Marella, M.; Barber-Singh, J.; Yamashita, T.; Matsuno-Yagi, A. Possibility of trans-kingdom gene therapy for Complex I diseases. Biochim. Biophys. Acta Bioenerg. 2006, 1757, 708–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McElroy, G.S.; Reczek, C.R.; Reyfman, P.A.; Mithal, D.S.; Horbinski, C.M.; Chandel, N.S. NAD+ Regeneration Rescues Lifespan, but Not Ataxia, in a Mouse Model of Brain Mitochondrial Complex I Dysfunction. Cell Metab. 2020, 32, 301–308.e6. [Google Scholar] [CrossRef] [PubMed]
- El-Khoury, R.; Kemppainen, K.K.; Dufour, E.; Szibor, M.; Jacobs, H.T.; Rustin, P. Engineering the alternative oxidase gene to better understand and counteract mitochondrial defects: State of the art and perspectives. Br. J. Pharmacol. 2014, 171, 2243–2249. [Google Scholar] [CrossRef] [Green Version]
- Szibor, M.; Gainutdinov, T.; Fernandez-Vizarra, E.; Dufour, E.; Gizatullina, Z.; Debska-Vielhaber, G.; Heidler, J.; Wittig, I.; Viscomi, C.; Gellerich, F.; et al. Bioenergetic consequences from xenotopic expression of a tunicate AOX in mouse mitochondria: Switch from RET and ROS to FET. Biochim. Biophys. Acta Bioenerg. 2020, 1861, 148137. [Google Scholar] [CrossRef]
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.-H.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; DeBerardinis, R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nat. Cell Biol. 2011, 481, 385–388. [Google Scholar] [CrossRef] [Green Version]
- Kotarsky, H.; Keller, M.; Davoudi, M.; Leveen, P.; Karikoski, R.; Enot, D.P.; Fellman, V. Metabolite Profiles Reveal Energy Failure and Impaired Beta-Oxidation in Liver of Mice with Complex III Deficiency Due to a BCS1L Mutation. PLoS ONE 2012, 7, e41156. [Google Scholar] [CrossRef]
- Selak, M.A.; Armour, S.M.; MacKenzie, E.D.; Boulahbel, H.; Watson, D.G.; Mansfield, K.D.; Pan, Y.; Simon, M.; Thompson, C.B.; Gottlieb, E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-α prolyl hydroxylase. Cancer Cell 2005, 7, 77–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.; Roth, P.; Fang, H.; Wang, G. Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J. Biol. Chem. 1994, 269, 23757–23763. [Google Scholar] [CrossRef]
- Ansó, E.; Weinberg, S.E.; Diebold, L.P.; Thompson, B.J.; Malinge, S.; Schumacker, P.T.; Liu, X.; Zhang, Y.; Shao, Y.Z.Z.; Steadman, M.; et al. The mitochondrial respiratory chain is essential for haematopoietic stem cell function. Nat. Cell Biol. 2017, 19, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, R.; Yeoh, K.K.; Tian, Y.; Hillringhaus, L.; Bagg, E.A.; Rose, N.R.; Leung, I.K.H.; Li, X.S.; Woon, E.C.Y.; Yang, M.; et al. The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 2011, 12, 463–469. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roux, K.J. Marked by association: Techniques for proximity-dependent labeling of proteins in eukaryotic cells. Cell. Mol. Life Sci. 2013, 70, 3657–3664. [Google Scholar] [CrossRef]
- Varnaitė, R.; MacNeill, S.A. Meet the neighbors: Mapping local protein interactomes by proximity-dependent labeling with BioID. Proteomics 2016, 16, 2503–2518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Coo, I.F.M.; Renier, W.O.; Ruitenbeek, W.; Ter Laak, H.J.; Bakker, M.; Schägger, H.; Van Oost, B.A.; Smeets, H.J.M. A 4–base pair deletion in the mitochondrial cytochrome b gene associated with parkinsonism/MELAS overlap syndrome. Ann. Neurol. 1999, 45, 130–133. [Google Scholar] [CrossRef]
- Rana, M.; De Coo, I.; Diaz, F.; Smeets, H.; Moraes, C.T. An out-of-frame cytochrome b gene deletion from a patient with parkinsonism is associated with impaired complex III assembly and an increase in free radical production. Ann. Neurol. 2000, 48, 774–781. [Google Scholar] [CrossRef]
- Andreu, A.L.; Hanna, M.G.; Reichmann, H.; Bruno, C.; Penn, A.S.; Tanji, K.; Pallotti, F.; Iwata, S.; Bonilla, E.; Lach, B.; et al. Exercise Intolerance Due to Mutations in the Cytochrome b Gene of Mitochondrial DNA. N. Engl. J. Med. 1999, 341, 1037–1044. [Google Scholar] [CrossRef]
- Schuelke, M.; Krude, H.; Finckh, B.; Mayatepek, E.; Janssen, A.; Schmelz, M.; Trefz, F.; Trijbels, F.; Smeitink, J. Septo-optic dysplasia associated with a new mitochondrial cytochrome b mutation. Ann. Neurol. 2002, 51, 388–392. [Google Scholar] [CrossRef]
- Emmanuele, V.; Sotiriou, E.; Rios, P.G.; Ganesh, J.; Ichord, R.; Foley, A.R.; Akman, H.O.; DiMauro, S. A Novel Mutation in the Mitochondrial DNA Cytochrome b Gene (MTCYB) in a Patient with Mitochondrial Encephalomyopathy, Lactic Acidosis, and Strokelike Episodes Syndrome. J. Child Neurol. 2013, 28, 236–242. [Google Scholar] [CrossRef] [Green Version]
- Legros, F.; Chatzoglou, E.; Frachon, P.; De Baulny, H.O.; Laforêt, P.; Jardel, C.; Godinot, C.; Lombès, A. Functional characterization of novel mutations in the human cytochrome b gene. Eur. J. Hum. Genet. 2001, 9, 510–518. [Google Scholar] [CrossRef]
- Bruno, C.; Santorelli, F.M.; Assereto, S.; Tonoli, E.; Tessa, A.; Traverso, M.; Scapolan, S.; Bado, M.; Tedeschi, S.; Minetti, C. Progressive exercise intolerance associated with a new muscle-restricted nonsense mutation (G142X) in the mitochondrial cytochrome b gene. Muscle Nerve 2003, 28, 508–511. [Google Scholar] [CrossRef]
- Keightley, J.A.; Anitori, R.; Burton, M.D.; Quan, F.; Buist, N.R.; Kennaway, N.G. Mitochondrial Encephalomyopathy and Complex III Deficiency Associated with a Stop-Codon Mutation in the Cytochrome b Gene. Am. J. Hum. Genet. 2000, 67, 1400–1410. [Google Scholar] [CrossRef] [Green Version]
- Valnot, I.; Kassis, J.; Chretien, D.; De Lonlay, P.; Parfait, B.; Munnich, A.; Kachaner, J.; Rustin, P.; Rötig, A. A mitochondrial cytochrome b mutation but no mutations of nuclearly encoded subunits in ubiquinol cytochrome c reductase (complex III) deficiency. Qual. Life Res. 1999, 104, 460–466. [Google Scholar] [CrossRef]
- Dumoulin, R.; Mandon, G.; Collombet, J.M.; Blond, J.L.; Carrier, H.; Godinot, C.; Flocard, F.; Villard, J.; Guibaud, P.; Mathieu, M.; et al. Human cultured myoblasts: A model for the diagnosis of mitochondrial diseases. J. Inherit. Metab. Dis. 1993, 16, 545–547. [Google Scholar] [CrossRef]
- Bouzidi, M.F.; Carrier, H.; Godinot, C. Antimycin resistance and ubiquinol cytochrome c reductase instability associated with a human cytochrome b mutation. Biochim. Biophys. Acta Mol. Basis Dis. 1996, 1317, 199–209. [Google Scholar] [CrossRef] [Green Version]
- Fragaki, K.; Procaccio, V.; Bannwarth, S.; Serre, V.; O’Hearn, S.; Potluri, P.; Augé, G.; Casagrande, F.; Caruba, C.; Lambert, J.C.; et al. A neonatal polyvisceral failure linked to a de novo homoplasmic mutation in the mitochondrially encoded cytochrome b gene. Mitochondrion 2009, 9, 346–352. [Google Scholar] [CrossRef]
- Mancuso, M.; Filosto, M.; Stevens, J.C.; Patterson, M.; Shanske, S.; Krishna, S.; DiMauro, S. Mitochondrial myopathy and complex III deficiency in a patient with a new stop-codon mutation (G339X) in the cytochrome b gene. J. Neurol. Sci. 2003, 209, 61–63. [Google Scholar] [CrossRef]
- Andreu, A.L.; Bruno, C.; Shanske, S.; Shtilbans, A.; Hirano, M.; Krishna, S.; Hayward, L.; Systrom, D.S.; Brown, R.H.; DiMauro, S. Missense mutation in the mtDNA cytochrome b gene in a patient with myopathy. Neurology 1998, 51, 1444–1447. [Google Scholar] [CrossRef]
- Gusic, M.; Schottmann, G.; Feichtinger, R.G.; Du, C.; Scholz, C.; Wagner, M.; Mayr, J.A.; Lee, C.-Y.; Yépez, V.A.; Lorenz, N.; et al. Bi-Allelic UQCRFS1 Variants Are Associated with Mitochondrial Complex III Deficiency, Cardiomyopathy, and Alopecia Totalis. Am. J. Hum. Genet. 2020, 106, 102–111. [Google Scholar] [CrossRef]
- Hinson, J.T.; Fantin, V.R.; Schönberger, J.; Breivik, N.; Siem, G.; McDonough, B.; Sharma, P.; Keogh, I.; Godinho, R.; Santos, F.; et al. Missense Mutations in the BCS1LGene as a Cause of the Björnstad Syndrome. N. Engl. J. Med. 2007, 356, 809–819. [Google Scholar] [CrossRef]
- De Meirleir, L.; Seneca, S.; Damis, E.; Sepulchre, B.; Hoorens, A.; Gerlo, E.; Silva, M.T.G.; Hernandez, E.M.; Lissens, W.; Van Coster, R. Clinical and diagnostic characteristics of complex III deficiency due to mutations in the BCS1Lgene. Am. J. Med. Genet. Part A 2003, 121A, 126–131. [Google Scholar] [CrossRef] [PubMed]
- Blázquez, A.; Gil-Borlado, M.C.; Morán, M.; Verdú, A.; Cazorla-Calleja, M.R.; Martín, M.A.; Arenas, J.; Ugalde, C. Infantile mitochondrial encephalomyopathy with unusual phenotype caused by a novel BCS1L mutation in an isolated complex III-deficient patient. Neuromuscul. Disord. 2009, 19, 143–146. [Google Scholar] [CrossRef]
- Visapää, I.; Fellman, V.; Vesa, J.; Dasvarma, A.; Hutton, J.L.; Kumar, V.; Payne, G.S.; Makarow, M.; Van Coster, R.; Taylor, R.W.; et al. GRACILE Syndrome, a Lethal Metabolic Disorder with Iron Overload, Is Caused by a Point Mutation in BCS1L. Am. J. Hum. Genet. 2002, 71, 863–876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oláhová, M.; Berti, C.C.; Collier, J.J.; Alston, C.L.; Jameson, E.; Jones, S.A.; Edwards, N.; He, L.; Chinnery, P.F.; Horvath, R.; et al. Molecular genetic investigations identify new clinical phenotypes associated with BCS1L-related mitochondrial disease. Hum. Mol. Genet. 2019, 28, 3766–3776. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Vizarra, E.; Bugiani, M.; Goffrini, P.; Carrara, F.; Farina, L.; Procopio, E.; Donati, A.; Uziel, G.; Ferrero, I.; Zeviani, M. Impaired complex III assembly associated with BCS1L gene mutations in isolated mitochondrial encephalopathy. Hum. Mol. Genet. 2007, 16, 1241–1252. [Google Scholar] [CrossRef] [Green Version]
- De Lonlay, P.; Valnot, I.; Barrientos, A.; Gorbatyuk, M.; Tzagoloff, A.; Taanman, J.-W.; Benayoun, E.; Chrétien, D.; Kadhom, N.; Lombès, A.; et al. A mutant mitochondrial respiratory chain assembly protein causes complex III deficiency in patients with tubulopathy, encephalopathy and liver failure. Nat. Genet. 2001, 29, 57–60. [Google Scholar] [CrossRef]
- Ezgu, F.; Senaca, S.; Gunduz, M.; Tumer, L.; Hasanoglu, A.; Tiras, U.; Unsal, R.; Bakkaloglu, S.A. Severe renal tubulopathy in a newborn due to BCS1L gene mutation: Effects of different treatment modalities on the clinical course. Gene 2013, 528, 364–366. [Google Scholar] [CrossRef]
- Tuppen, H.A.; Fehmi, J.; Czermin, B.; Goffrini, P.; Meloni, F.; Ferrero, I.; He, L.; Blakely, E.L.; McFarland, R.; Horvath, R.; et al. Long-term survival of neonatal mitochondrial complex III deficiency associated with a novel BCS1L gene mutation. Mol. Genet. Metab. 2010, 100, 345–348. [Google Scholar] [CrossRef]
- Al-Owain, M.; Colak, D.; AlBakheet, A.; Al-Younes, B.; Al-Humaidi, Z.; Al-Sayed, M.; Al-Hindi, H.; Al-Sugair, A.; Al-Muhaideb, A.; Rahbeeni, Z.; et al. Clinical and biochemical features associated with BCS1L mutation. J. Inherit. Metab. Dis. 2013, 36, 813–820. [Google Scholar] [CrossRef]
- Baker, R.A.; Priestley, J.R.C.; Wilstermann, A.M.; Reese, K.J.; Mark, P.R. Clinical spectrum of BCS1L Mitopathies and their underlying structural relationships. Am. J. Med. Genet. Part A 2019, 179, 373–380. [Google Scholar] [CrossRef]
- Gil-Borlado, M.C.; González-Hoyuela, M.; Blázquez, A.; García-Silva, M.T.; Gabaldón, T.; Manzanares, J.; Vara, J.; Martín, M.A.; Seneca, S.; Arenas, J.; et al. Pathogenic mutations in the 5′ untranslated region of BCS1L mRNA in mitochondrial complex III deficiency. Mitochondrion 2009, 9, 299–305. [Google Scholar] [CrossRef]
- Kunii, M.; Doi, H.; Higashiyama, Y.; Kugimoto, C.; Ueda, N.; Hirata, J.; Tomita-Katsumoto, A.; Kashikura-Kojima, M.; Kubota, S.; Taniguchi, M.; et al. A Japanese case of cerebellar ataxia, spastic paraparesis and deep sensory impairment associated with a novel homozygous TTC19 mutation. J. Hum. Genet. 2015, 60, 187–191. [Google Scholar] [CrossRef] [PubMed]
- Melchionda, L.; Damseh, N.S.; Abu Libdeh, B.Y.; Nasca, A.; Elpeleg, O.; Zanolini, A.; Ghezzi, D. A novel mutation in TTC19 associated with isolated complex III deficiency, cerebellar hypoplasia, and bilateral basal ganglia lesions. Front. Genet. 2014, 5, 397. [Google Scholar] [CrossRef] [Green Version]
- Ghezzi, D.; Arzuffi, P.; Zordan, M.; Da Re, C.; Lamperti, C.; Benna, C.; D’Adamo, P.; Diodato, D.; Costa, R.; Mariotti, C.; et al. Mutations in TTC19 cause mitochondrial complex III deficiency and neurological impairment in humans and flies. Nat. Genet. 2011, 43, 259–263. [Google Scholar] [CrossRef]
- Koch, J.; Freisinger, P.; Feichtinger, R.G.; Zimmermann, F.A.; Rauscher, C.; Wagentristl, H.P.; Konstantopoulou, V.; Seidl, R.; Haack, T.B.; Prokisch, H.; et al. Mutations in TTC19: Expanding the molecular, clinical and biochemical phenotype. Orphanet J. Rare Dis. 2015, 10, 40. [Google Scholar] [CrossRef] [Green Version]
- Atwal, P.S.; Zschocke, J.; Gibson, K.M. Mutations in the Complex III Assembly Factor Tetratricopeptide 19 Gene TTC19 Are a Rare Cause of Leigh Syndrome. JIMD Rep. Vol. 14 2014, 14, 43–45. [Google Scholar] [CrossRef] [Green Version]
- Habibzadeh, P.; Inaloo, S.; Silawi, M.; Dastsooz, H.; Fard, M.A.F.; Sadeghipour, F.; Faghihi, Z.; Rezaeian, M.; Yavarian, M.; Böhm, J.; et al. A Novel TTC19 Mutation in a Patient with Neurological, Psychological, and Gastrointestinal Impairment. Front. Neurol. 2019, 10, 944. [Google Scholar] [CrossRef]
- Ardissone, A.; Granata, T.; Legati, A.; Diodato, D.; Melchionda, L.; Lamantea, E.; Garavaglia, B.; Ghezzi, D.; Moroni, I.; Zschocke, J. Mitochondrial Complex III Deficiency Caused by TTC19 Defects: Report of a Novel Mutation and Review of Literature. JIMD Rep. 2015, 22, 115–120. [Google Scholar] [CrossRef] [Green Version]
- Morino, H.; Miyamoto, R.; Ohnishi, S.; Maruyama, H.; Kawakami, H. Exome sequencing reveals a novel TTC19 mutation in an autosomal recessive spinocerebellar ataxia patient. BMC Neurol. 2014, 14, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mordaunt, D.A.; Jolley, A.; Balasubramaniam, S.; Thorburn, D.R.; Mountford, H.S.; Compton, A.G.; Nicholl, J.; Manton, N.; Clark, D.; Bratkovic, D.; et al. Phenotypic variation of TTC19-deficient mitochondrial complex III deficiency: A case report and literature review. Am. J. Med. Genet. Part A 2015, 167, 1330–1336. [Google Scholar] [CrossRef] [PubMed]
- Bottani, E.; Cerutti, R.; Harbour, M.E.; Ravaglia, S.; Dogan, S.A.; Giordano, C.; Fearnley, I.M.; D’Amati, G.; Viscomi, C.; Fernandez-Vizarra, E.; et al. TTC19 Plays a Husbandry Role on UQCRFS1 Turnover in the Biogenesis of Mitochondrial Respiratory Complex III. Mol. Cell 2017, 67, 96–105.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dallabona, C.; Abbink, T.E.M.; Carrozzo, R.; Torraco, A.; Legati, A.; Van Berkel, C.G.M.; Niceta, M.; Langella, T.; Verrigni, D.; Rizza, T.; et al. LYRM7 mutations cause a multifocal cavitating leukoencephalopathy with distinct MRI appearance. Brain 2016, 139, 782–794. [Google Scholar] [CrossRef] [Green Version]
- Kremer, L.S.; L’Hermitte-Stead, C.; Lesimple, P.; Gilleron, M.; Filaut, S.; Jardel, C.; Haack, T.B.; Strom, T.M.; Meitinger, T.; Azzouz, H.; et al. Severe respiratory complex III defect prevents liver adaptation to prolonged fasting. J. Hepatol. 2016, 65, 377–385. [Google Scholar] [CrossRef]
- Invernizzi, F.; Varanese, S.; Thomas, A.M.; Carrara, F.; Onofrj, M.; Zeviani, M. Two novel POLG1 mutations in a patient with progressive external ophthalmoplegia, levodopa-responsive pseudo-orthostatic tremor and parkinsonism. Neuromuscul. Disord. 2008, 18, 460–464. [Google Scholar] [CrossRef]
- Hempel, M.; Kremer, L.S.; Tsiakas, K.; Alhaddad, B.; Haack, T.B.; Löbel, U.; Feichtinger, R.G.; Sperl, W.; Prokisch, H.; Mayr, J.A.; et al. LYRM7—Associated complex III deficiency: A clinical, molecular genetic, MR tomographic, and biochemical study. Mitochondrion 2017, 37, 55–61. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Mutated Gene | Mutation | Enzymatic Activity | Isolated Complexes Assembly | Supercomplexes Assembly | Refs. | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CIII | CIII | CI | CIV | CIII | CI | CIV | CI+CIII+CIV | CI+CIII | CIII+CIV | ||
| Structural subunits | |||||||||||
| MTCYB | p.14I> * | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | [118,167,168] |
| p.34G>S | ↓ | mild ↓ | = | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | [169] | |
| p.35S>P | ↓ | n.d. | n.d | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | [170] | |
| p.40C>R | mild ↓ | mild ↓ | mild ↓ | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | [171] | |
| p.113W> * | ↓ | n.d. | n.d | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | [169] | |
| p.135W> * | ↓ | n.d. | n-d | ↓ | n.d. | n.d. | n.d. | n.d. | n.d. | [172] | |
| p.141W> * | ↓ | = | = | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | [169] | |
| p.142G> * | ↓ | ↓ | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | [173] | |
| p.151S>P | ↓ | n.d. | n.d. | ↓ | n.d. | n.d. | n.d. | n.d. | n.d. | [172] | |
| p.166G> * | ↓ | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | [174] | |
| p.166G>E | ↓ | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | [175] | |
| p.∆251-258 | ↓ | mild ↓ | = | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | [169] | |
| p.271E>K | mild ↓ | = | = | = | = | = | = | = | = | [132] | |
| p.278Y>C | ↓ | mild ↓ | n.d. | = | = | = | mild↑ | mild↑ | ↓ | [129,145] | |
| p.290G>D | ↓ | n.d. | n.d | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | [176,177] | |
| p.297S>P | ↓ | = | = | ↓ | = | = | n.d. | n.d. | n.d. | [178] | |
| p.∆300-305 | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | [113,120] | |
| p.318K>P | ↓ | ↓ | = | ↓ | ↓ | = | n.d. | n.d. | n.d. | [122] | |
| p.326W> * | ↓ | n.d. | n.d | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | [169] | |
| p.339G> * | ↓ | n.d. | n.d | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | [179] | |
| p.339G>E | ↓ | n.d. | n.d | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | [180] | |
| p.352Q> * | ↓ | ↓ | ↓ | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | [123] | |
| p.373E>K | ↓ | ↓ | = | ↓ | ↓ | = | n.d. | n.d. | n.d. | [130] | |
| UQCRB | Change at C-term | ↓ | ↓ | ↓ | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | [124] |
| UQCRQ | p.45S>F; p.45S>F | ↓ | ↓ | = | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | [125] |
| CYC1 | p.96W>C; p.215L>F | ↓ | ↓ | ↓ | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | [126] |
| UQCRC2 | p.183R>W; p.183R>W | ↓ | ↑ | ↓ | ↓ | ↓ | = | ↓ | ↓ | n.d. | [127] |
| p.183R>W; p183R>W | ↓ | ↓ | = | ↓ | ↓ | n.d. | n.d. | n.d. | n.d. | [142] | |
| UQCRFS1 | p.14V>D; p.204R> * | reduced overall respiration | ↓ | n.d. | n.d. | n.d. | n.d. | n.d. | [181] | ||
| p.72V>T81del10; p.72V>T81del10 | reduced overall respiration | ↓ | n.d. | n.d. | n.d. | n.d. | n.d. | [181] | |||
| mouse KO | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | [111] | |
| Assembly Factors | |||||||||||
| BCS1L | p.35G>R; p.184R>C | ↓ | n.d. | n.d. | ↓ | n.d. | n.d. | = | n.d. | ↓ | [182] |
| p.45R>C; p.56R> * | ↓ | ↓ | = | ↓ | ↓ | ↓ | n.d. | n.d. | n.d. | [110,183] | |
| p.50T>A; p.50T>A | mild ↓ | n.d. | n.d. | mild↓ | = | n.d. | n.d. | n.d. | n.d. | [110,184] | |
| p.R56 *; g1181A>G/g1164C>C | ↓ | = | ↓ | ↓ | = | ↓ | n.d. | n.d. | n.d. | [110,183] | |
| p.R56 *; p.327V>A | = | = | = | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | [185] | |
| p.R56 *; p.69R>C | = | = | = | ↓ | = | = | n.d. | n.d. | n.d. | [186] | |
| p.73R>C; p.368F>I | ↓ | = | = | = | = | = | = | = | = | [187] | |
| p.78S>G; p.144R>Q | = | = | = | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | [185] | |
| p.99P>L; p.99P>L | ↓ | ↓ | ↓ | ↓ | ↓ | ↓ | n.d. | n.d. | n.d. | [110,188,189] | |
| p.109R>W; p.109R>W | = | = | = | ↓ | = | ↓ | n.d. | n.d. | n.d. | [186] | |
| p.129G>R; p.129G>R | ↓ | n.d. | n.d. | ↓ | n.d. | n.d. | n.d. | n.d. | n.d. | [190,191] | |
| p.155R>P; p.353V>M | ↓ | n.d. | = | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | [188] | |
| p.183R>C; p.184R>C | ↓ | = | = | = | = | = | = | = | = | [187] | |
| p.184R>C; g1892A>G | ↓ | = | = | = | mild ↓ | = | = | n.d. | ↓ | [110] | |
| p.184R>C; p.280L>F | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | [192] | |
| p.277S>N; p.277S>N | ↓ | n.d. | = | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | [188] | |
| decreased levels BCS1L | ↓ | n.d. | ↓ | ↓ | = | = | n.d. | n.d. | n.d. | [193] | |
| mouse p.78S>G: p.78S>G | ↓ | = | = | ↓ | = | = | mild ↓ | mild ↓ | mild ↓ | [115] | |
| mouse KO | [116] | ||||||||||
| TTC19 | p.54P>A * | ↓ | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | [194] |
| p.77Q>R *; p.77Q>R * | ↓ | n.d. | n.d. | = | n.d. | n.d. | n.d. | n.d. | n.d. | [195] | |
| p.173Q> * | ↓ | = | = | ↓ | n.d. | n.d. | n.d. | n.d. | n.d. | [196] | |
| p.185L>P | n.d. | n.d. | n.d. | = | n.d. | n.d. | n.d. | n.d. | n.d. | [197] | |
| p.186W> *; p.322G>M * | ↓ | n.d. | ↓ | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | [198] | |
| p.194R>N * | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | [199] | |
| p.219L> * | ↓ | = | = | ↓ | n.d. | n.d. | n.d. | n.d. | n.d. | [196] | |
| p.261E>G *; p.261A>G * | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | [200] | |
| p.277Q> *; p.277Q> * | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | [201] | |
| p.313Q> * | ↓ | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | [202] | |
| p.321A> *; p.321A> * | ↓ | n.d. | n.d. | ↓ | n.d. | n.d. | n.d. | n.d. | ↓ | [114] | |
| p.324L>P | n.d. | n.d. | n.d. | = | n.d. | n.d. | n.d. | n.d. | n.d. | [197] | |
| mouse and human KO | ↓ | = | = | mild ↓ | = | = | = | = | = | [203] | |
| UQCC2 | Protein absent | ↓ | ↓ | ↓ | ↓ | mild ↓ | mild↑ | ↓ | ↓ | ↓ | [85] |
| p.[8R>P;10L>F];[8R>P;10L>F] | ↓ | ↓ | = | ↓ | ↓ | = | ↓ | ↓ | ↓ | [128] | |
| UQCC3 | p.20V>E; p.20V>E | ↓ | mild ↓ | = | ↓ | ↓ | = | n.d. | n.d. | n.d. | [86] |
| LYRM7 | p.13T>H *; p.13T>H * | ↓ | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | [204] |
| LYRM7 | p.18R>D *; p.18R>A * | ↓ | mild ↓ | mild ↓ | ↓ | mild ↓ | mild ↓ | n.d. | n.d. | n.d. | [205] |
| p.25D>N; p25D>N | ↓ | n.d. | n.d. | ↓ | n.d. | = | n.d. | n.d. | n.d. | [206] | |
| p.25D>N; p25D>N | ↓ | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | [204] | |
| p.72Q> *; p72Q> * | ↓ | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | [204] | |
| p.82K>N *; p.82K>N * | ↓ | n.d. | n.d. | ↓ | n.d. | n.d. | n.d. | n.d. | n.d. | [204] | |
| Protein absent | ↓ | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. | [207] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rugolo, M.; Zanna, C.; Ghelli, A.M. Organization of the Respiratory Supercomplexes in Cells with Defective Complex III: Structural Features and Metabolic Consequences. Life 2021, 11, 351. https://doi.org/10.3390/life11040351
Rugolo M, Zanna C, Ghelli AM. Organization of the Respiratory Supercomplexes in Cells with Defective Complex III: Structural Features and Metabolic Consequences. Life. 2021; 11(4):351. https://doi.org/10.3390/life11040351
Chicago/Turabian StyleRugolo, Michela, Claudia Zanna, and Anna Maria Ghelli. 2021. "Organization of the Respiratory Supercomplexes in Cells with Defective Complex III: Structural Features and Metabolic Consequences" Life 11, no. 4: 351. https://doi.org/10.3390/life11040351
APA StyleRugolo, M., Zanna, C., & Ghelli, A. M. (2021). Organization of the Respiratory Supercomplexes in Cells with Defective Complex III: Structural Features and Metabolic Consequences. Life, 11(4), 351. https://doi.org/10.3390/life11040351