
An Unsupervised Algorithm for Host Identification in Flaviviruses



Abstract
1. Introduction
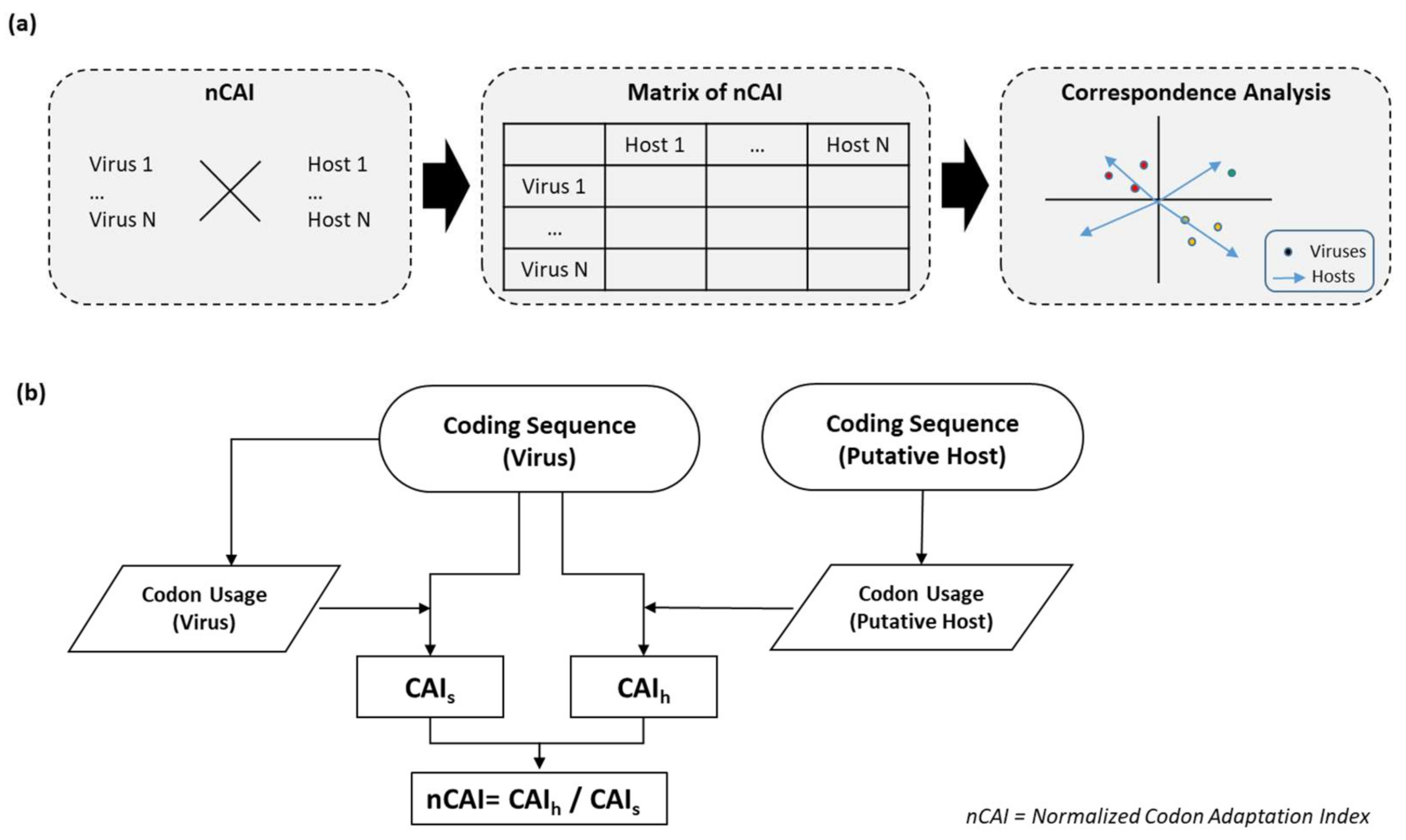
2. Materials and Methods
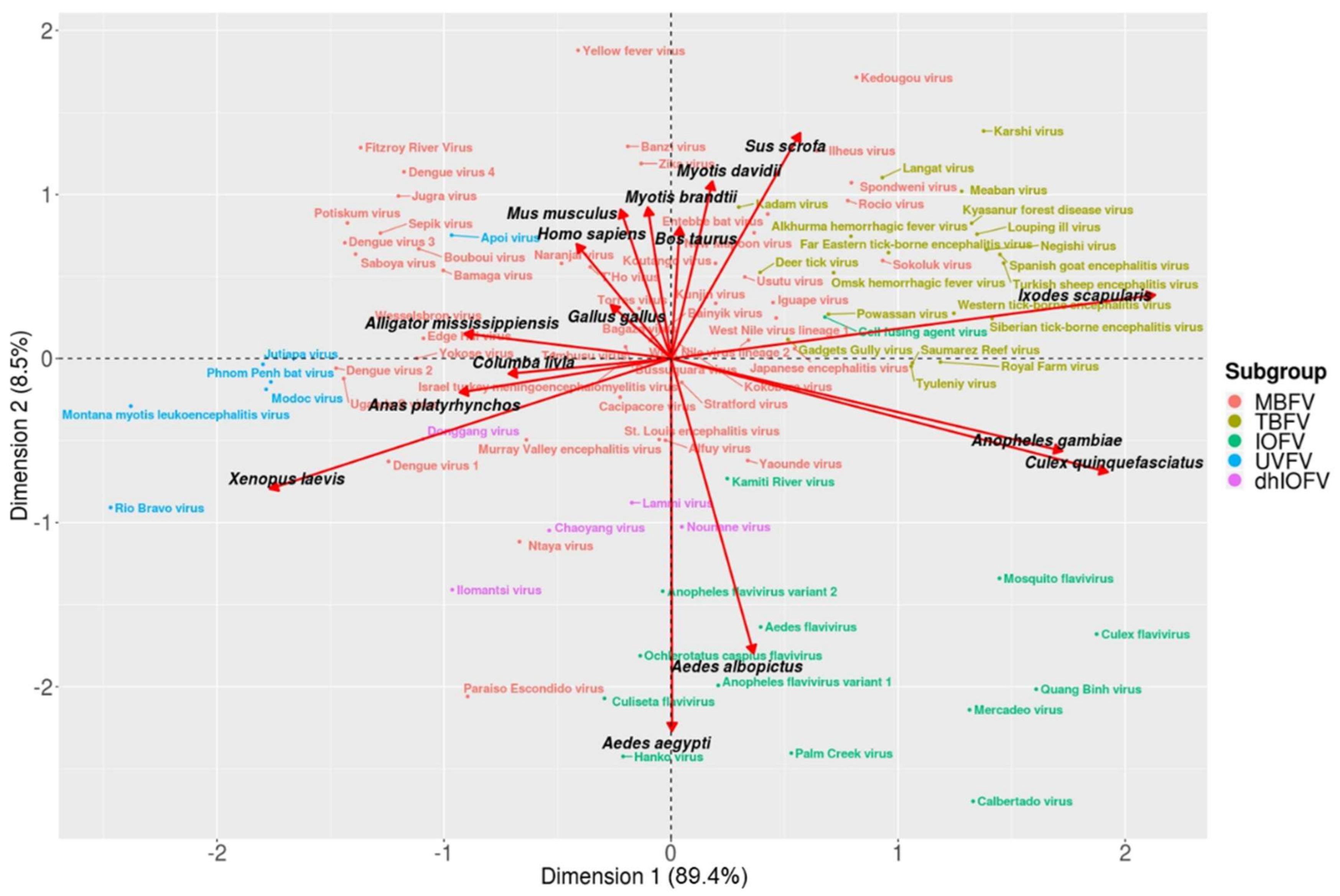
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kindhauser, M.K.; Allen, T.; Frank, V.; Santhana, R.S.; Dye, C. Zika: The origin and spread of a mosquito-borne virus. Bull. World Health Organ. 2016, 94, 675C–686C. [Google Scholar] [CrossRef] [PubMed]
- Omilabu, S.A.; Salu, O.B.; Oke, B.O.; James, A.B. The West African ebola virus disease epidemic 2014–2015: A commissioned review. Niger. Postgrad. Med. J. 2016, 23, 49–56. [Google Scholar] [CrossRef]
- Wang, C.; Horby, P.W.; Hayden, F.G.; Gao, G.F. A novel coronavirus outbreak of global health concern. Lancet 2020, 395, 470–473. [Google Scholar] [CrossRef]
- Girard, M.P.; Tam, J.S.; Assossou, O.M.; Kieny, M.P. The 2009 A (H1N1) influenza virus pandemic: A review. Vaccine 2010, 28, 4895–4902. [Google Scholar] [CrossRef] [PubMed]
- Gates, B. Responding to Covid-19-A Once-in-a-Century Pandemic? N. Engl. J. Med. 2020, 382, 1677–1679. [Google Scholar] [CrossRef]
- Smith, D. Applications of bioinformatics and computational biology to influenza surveillance and vaccine strain selection. Vaccine 2003, 21, 1758–1761. [Google Scholar] [CrossRef][Green Version]
- Sanjuán, R.; Nebot, M.R.; Chirico, N.; Mansky, L.M.; Belshaw, R. Viral mutation rates. J. Virol. 2010, 84, 9733–9748. [Google Scholar] [CrossRef]
- Kuno, G. Host range specificity of flaviviruses: Correlation with in vitro replication. J. Med. Entomol. 2007, 44, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Di Giallonardo, F.; Schlub, T.E.; Shi, M.; Holmes, E.C. Dinucleotide Composition in Animal RNA Viruses Is Shaped More by Virus Family than by Host Species. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Parry, R.; Asgari, S. Discovery of Novel Crustacean and Cephalopod Flaviviruses: Insights into the Evolution and Circulation of Flaviviruses between Marine Invertebrate and Vertebrate Hosts. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Jenkins, G.M.; Holmes, E.C. The extent of codon usage bias in human RNA viruses and its evolutionary origin. Virus Res. 2003, 92, 1–7. [Google Scholar] [CrossRef]
- Chambers, T.J.; Hahn, C.S.; Galler, R.; Rice, C.M. Flavivirus genome organization, expression, and replication. Annu. Rev. Microbiol. 1990, 44, 649–688. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Becher, P.; Bukh, J.; Gould, E.A.; Meyers, G.; Monath, T.; Muerhoff, S.; Pletnev, A.; Rico-Hesse, R.; Smith, D.B.; et al. Ictv Report Consortium ICTV virus taxonomy profile: Flaviviridae. J. Gen. Virol. 2017, 98, 2–3. [Google Scholar] [CrossRef] [PubMed]
- Huhtamo, E.; Cook, S.; Moureau, G.; Uzcátegui, N.Y.; Sironen, T.; Kuivanen, S.; Putkuri, N.; Kurkela, S.; Harbach, R.E.; Firth, A.E.; et al. Novel flaviviruses from mosquitoes: Mosquito-specific evolutionary lineages within the phylogenetic group of mosquito-borne flaviviruses. Virology 2014, 464–465, 320–329. [Google Scholar] [CrossRef]
- Alkan, C.; Zapata, S.; Bichaud, L.; Moureau, G.; Lemey, P.; Firth, A.E.; Gritsun, T.S.; Gould, E.A.; de Lamballerie, X.; Depaquit, J.; et al. Ecuador Paraiso Escondido Virus, a New Flavivirus Isolated from New World Sand Flies in Ecuador, Is the First Representative of a Novel Clade in the Genus Flavivirus. J. Virol. 2015, 89, 11773–11785. [Google Scholar] [CrossRef]
- Lobo, F.P.; Mota, B.E.F.; Pena, S.D.J.; Azevedo, V.; Macedo, A.M.; Tauch, A.; Machado, C.R.; Franco, G.R. Virus-host coevolution: Common patterns of nucleotide motif usage in Flaviviridae and their hosts. PLoS ONE 2009, 4, e6282. [Google Scholar] [CrossRef] [PubMed]
- Guarner, J.; Hale, G.L. Four human diseases with significant public health impact caused by mosquito-borne flaviviruses: West Nile, Zika, dengue and yellow fever. Semin. Diagn. Pathol. 2019, 36, 170–176. [Google Scholar] [CrossRef]
- Bahir, I.; Fromer, M.; Prat, Y.; Linial, M. Viral adaptation to host: A proteome-based analysis of codon usage and amino acid preferences. Mol. Syst. Biol. 2009, 5, 311. [Google Scholar] [CrossRef] [PubMed]
- Sharp, P.M.; Li, W.H. The codon Adaptation Index--a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15, 1281–1295. [Google Scholar] [CrossRef]
- Puigbò, P.; Bravo, I.G.; Garcia-Vallve, S. CAIcal: A combined set of tools to assess codon usage adaptation. Biol. Direct 2008, 3, 38. [Google Scholar] [CrossRef]
- Nakamura, Y.; Gojobori, T.; Ikemura, T. Codon usage tabulated from international DNA sequence databases: Status for the year 2000. Nucleic Acids Res. 2000, 28, 292. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2013, 41, D36–D42. [Google Scholar] [CrossRef] [PubMed]
- Gaunt, M.W.; Sall, A.A.; de Lamballerie, X.; Falconar, A.K.; Dzhivanian, T.I.; Gould, E.A. Phylogenetic relationships of flaviviruses correlate with their epidemiology, disease association and biogeography. J. Gen. Virol. 2001, 82, 1867–1876. [Google Scholar] [CrossRef] [PubMed]
- Kuno, G.; Chang, G.J.; Tsuchiya, K.R.; Karabatsos, N.; Cropp, C.B. Phylogeny of the genus Flavivirus. J. Virol. 1998, 72, 73–83. [Google Scholar] [CrossRef] [PubMed]
- Grard, G.; Moureau, G.; Charrel, R.N.; Holmes, E.C.; Gould, E.A.; de Lamballerie, X. Genomics and evolution of Aedes-borne flaviviruses. J. Gen. Virol. 2010, 91, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Mihara, T.; Nishimura, Y.; Shimizu, Y.; Nishiyama, H.; Yoshikawa, G.; Uehara, H.; Hingamp, P.; Goto, S.; Ogata, H. Linking Virus Genomes with Host Taxonomy. Viruses 2016, 8, 66. [Google Scholar] [CrossRef]
- Sharp, P.M.; Li, W.H. An evolutionary perspective on synonymous codon usage in unicellular organisms. J. Mol. Evol. 1986, 24, 28–38. [Google Scholar] [CrossRef]
- Sharp, P.M.; Tuohy, T.M.; Mosurski, K.R. Codon usage in yeast: Cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res. 1986, 14, 5125–5143. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef]
- Federhen, S. The NCBI Taxonomy database. Nucleic Acids Res. 2012, 40, D136–D143. [Google Scholar] [CrossRef]
- Lawrie, C.H.; Uzcátegui, N.Y.; Armesto, M.; Bell-Sakyi, L.; Gould, E.A. Susceptibility of mosquito and tick cell lines to infection with various flaviviruses. Med. Vet. Entomol. 2004, 18, 268–274. [Google Scholar] [CrossRef]
- Sotomayor-Bonilla, J.; Tolsá-García, M.J.; García-Peña, G.E.; Santiago-Alarcon, D.; Mendoza, H.; Alvarez-Mendizabal, P.; Rico-Chávez, O.; Sarmiento-Silva, R.E.; Suzán, G. Insights into the Host Specificity of Mosquito-Borne Flaviviruses Infecting Wild Mammals. Ecohealth 2019, 16, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Wagner, S.; Mathis, A.; Schönenberger, A.C.; Becker, S.; Schmidt-Chanasit, J.; Silaghi, C.; Veronesi, E. Vector competence of field populations of the mosquito species Aedes japonicus japonicus and Culex pipiens from Switzerland for two West Nile virus strains. Med. Vet. Entomol. 2018, 32, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhou, T.; Lai, Z.; Zhang, Z.; Jia, Z.; Zhou, G.; Williams, T.; Xu, J.; Gu, J.; Zhou, X.; et al. Competence of Aedes aegypti, Ae. albopictus, and Culex quinquefasciatus Mosquitoes as Zika Virus Vectors, China. Emerg. Infect. Dis. 2017, 23, 1085–1091. [Google Scholar] [CrossRef] [PubMed]
- Vaidyanathan, R.; Scott, T.W. Geographic variation in vector competence for West Nile virus in the Culex pipiens (Diptera: Culicidae) complex in California. Vector Borne Zoonotic Dis. 2007, 7, 193–198. [Google Scholar] [CrossRef]
- Ndiaye, E.H.; Fall, G.; Gaye, A.; Bob, N.S.; Talla, C.; Diagne, C.T.; Diallo, D.; Yamar, B.A.; Dia, I.; Kohl, A.; et al. Vector competence of Aedes vexans (Meigen), Culex poicilipes (Theobald) and Cx. quinquefasciatus Say from Senegal for West and East African lineages of Rift Valley fever virus. Parasit. Vectors 2016, 9, 94. [Google Scholar] [CrossRef]
- Nanfack Minkeu, F.; Vernick, K.D. A systematic review of the natural virome of anopheles mosquitoes. Viruses 2018, 10, 222. [Google Scholar] [CrossRef]
- Lequime, S.; Lambrechts, L. Discovery of flavivirus-derived endogenous viral elements in Anopheles mosquito genomes supports the existence of Anopheles-associated insect-specific flaviviruses. Virus Evol. 2017, 3, vew035. [Google Scholar] [CrossRef]
- Colmant, A.M.G.; Hobson-Peters, J.; Bielefeldt-Ohmann, H.; van den Hurk, A.F.; Hall-Mendelin, S.; Chow, W.K.; Johansen, C.A.; Fros, J.; Simmonds, P.; Watterson, D.; et al. A New Clade of Insect-Specific Flaviviruses from Australian Anopheles Mosquitoes Displays Species-Specific Host Restriction. mSphere 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Gao, Z.; Zhang, J.; Chen, H.; Pejsak, Z.; Ma, L.; Ding, Y.; Liu, Y. Comparative [corrected] codon usage between the three main viruses in pestivirus genus and their natural susceptible livestock. Virus Genes 2012, 44, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Rocha, E.P.C. Codon usage bias from tRNA’s point of view: Redundancy, specialization, and efficient decoding for translation optimization. Genome Res. 2004, 14, 2279–2286. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Tick | Aedes | Anopheles | Culex | Mammals | Other Vertebrates | |||
|---|---|---|---|---|---|---|---|---|
| Flavivirus Groups | %GC3 | 72.0% | 58.1% | 69.6% | 69.3% | 60.9% | 52.9% | |
| - | ±1.8% | - | - | ±2.5% | ±4.5% | |||
| dhIOFV (n = 5) | 50.0% | nCAI | 0.738 | 0.949 | 0.729 | 0.764 | 0.889 | 1.027 |
| ±2.3% | ±0.048 | ±0.053 | ±0.051 | ±0.049 | ±0.062 | ±0.049 | ||
| IOFV (n = 14) | 54.3% | nCAI | 0.748 | 0.938 | 0.734 | 0.780 | 0.866 | 0.982 |
| ±3.8% | ±0.046 | ±0.044 | ±0.042 | ±0.047 | 0.051 | ±0.037 | ||
| MBFV (n = 49) | 52.1% | nCAI | 0.765 | 0.967 | 0.752 | 0.792 | 0.925 | 1.053 |
| ±3.7% | ±0.033 | ±0.033 | ±0.029 | ±0.032 | ±0.051 | ±0.035 | ||
| TBFV (n = 20) | 58.9% | nCAI | 0.812 | 0.979 | 0.795 | 0.842 | 0.938 | 1.045 |
| ±1.8% | ±0.021 | ±0.024 | ±0.020 | ±0.020 | ±0.043 | ±0.028 | ||
| UVFV (n = 6) | 44.4% | nCAI | 0.705 | 0.948 | 0.706 | 0.746 | 0.907 | 1.057 |
| ±3.3% | ±0.012 | ±0.028 | ±0.010 | ±0.010 | ±0.053 | ±0.038 | ||
| Mosquito (n = 14) | 54.3% | nCAI | 0.748 | 0.938 | 0.734 | 0.780 | 0.866 | 0.982 |
| ±3.8% | ±0.046 | ±0.044 | ±0.042 | ±0.047 | ±0.051 | ±0.037 | ||
| Tick (n = 20) | 58.9% | nCAI | 0.812 | 0.979 | 0.795 | 0.842 | 0.938 | 1.045 |
| ±1.8% | ±0.021 | ±0.024 | ±0.020 | ±0.020 | ±0.043 | ±0.028 | ||
| Vertebrate (n = 60) | 51.2% | nCAI | 0.757 | 0.963 | 0.746 | 0.785 | 0.920 | 1.052 |
| ±4.3% | ±0.038 | ±0.035 | ±0.033 | ±0.035 | ±0.053 | ±0.037 | ||
| Virus Group | dhIOFV | IOFV | MBFV | TBFV | UVFV | Host Type | Mosquito | Tick | Vertebrate | |
|---|---|---|---|---|---|---|---|---|---|---|
| Accuracy 1 | 91.9% | 94.7% | 96.8% | 81.9% | 95.7% | 90.4% | 86.1% | 78.8% | 86.7% | 92.9% |
| Specificity 2 | 94.9% | 94.4% | 100.0% | 95.6% | 94.6% | 90.9% | 79.6% | 75.3% | 84.0% | 79.5% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Truong Nguyen, P.; Garcia-Vallvé, S.; Puigbò, P. An Unsupervised Algorithm for Host Identification in Flaviviruses. Life 2021, 11, 442. https://doi.org/10.3390/life11050442
Truong Nguyen P, Garcia-Vallvé S, Puigbò P. An Unsupervised Algorithm for Host Identification in Flaviviruses. Life. 2021; 11(5):442. https://doi.org/10.3390/life11050442
Chicago/Turabian StyleTruong Nguyen, Phuoc, Santiago Garcia-Vallvé, and Pere Puigbò. 2021. "An Unsupervised Algorithm for Host Identification in Flaviviruses" Life 11, no. 5: 442. https://doi.org/10.3390/life11050442
APA StyleTruong Nguyen, P., Garcia-Vallvé, S., & Puigbò, P. (2021). An Unsupervised Algorithm for Host Identification in Flaviviruses. Life, 11(5), 442. https://doi.org/10.3390/life11050442