
Microsatellite Loci Reveal Genetic Diversity of Asian Callery Pear (Pyrus calleryana) in the Species Native Range and in the North American Cultivars

 , , , , , and
, , , , , and
Abstract
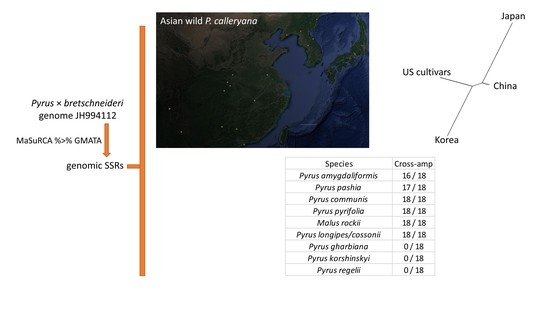
:
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. gDNA Extraction
2.3. Microsatellite Primers and Genotyping Conditions
2.4. Data Analysis
2.5. Population Genetics of P. calleryana
2.5.1. Genetic Diversity
2.5.2. Population Structure
Mantel Test for Isolation by Distance
Bayesian Clustering Using Structure and DAPC
3. Results
3.1. gSSR Development and Selection
3.2. Cross-Amplification
3.3. Population Genetics of P. calleryana
3.3.1. Genetic Diversity
3.3.2. Population Structure
Mantel Test for Isolation by Distance
Bayesian Clustering Using Structure and DAPC
4. Discussion
5. Conclusions and Outlook
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Taberlet, P.; Fumagalli, L.; Wust-Saucy, A.G.; Cosson, J.F. Comparative phylogeography and postglacial colonization routes in Europe. Mol. Ecol. 1998, 7, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Bossdorf, O.; Auge, H.; Lafuma, L.; Rogers, W.E.; Siemann, E.; Prati, D. Phenotypic and genetic differentiation between native and introduced plant populations. Oecologia 2005, 144, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Goolsby, J.A.; de Barro, P.J.; Makinson, J.R.; Pemberton, R.W.; Hartley, D.M.; Frohlich, D.R. Matching the origin of an invasive weed for selection of a herbivore haplotype for a biological control programme. Mol. Ecol. 2006, 15, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Schierenbeck, K.A.; Ainouche, M.L. Conceptual Ecology and Invasion Biology: Reciprocal Approaches to Nature; Springer: Amsterdam, The Netherlands, 2006; pp. 193–221. [Google Scholar]
- Bell, R.; Zimmerman, R. Combining ability analysis of juvenile period in pear. Hort. Sci. 1990, 25, 1425–1427. [Google Scholar] [CrossRef] [Green Version]
- Cuizhi, G.; Spongberg, S. Pyrus. Flora China 2003, 9, 173–179. [Google Scholar]
- Culley, T.M.; Hardiman, N.A. The role of intraspecific hybridization in the evolution of invasiveness: A case study of the ornamental pear tree Pyrus calleryana. Biol. Invasions 2009, 11, 1107–1119. [Google Scholar] [CrossRef]
- Culley, T.M. The rise and fall of the ornamental Callery pear tree. Arnoldia 2017, 74, 2–11. [Google Scholar]
- Swearingen, J.; Reshetiloff, K.; Slattery, B.; Zwicker, S. Plant Invaders of Mid-Atlantic Natural Areas. 2002021-06-05. Available online: http://www.maipc.org/PlantInvadersMidAtlanticNaturalAreas5thEdition.pdf (accessed on 4 June 2021).
- Dickson, E.; Arumuganathan, K.; Kresovich, S.; Doyle, J. Nuclear DNA content variation within the Rosaceae. Am. J. Bot. 1992, 79, 1081–1086. [Google Scholar] [CrossRef] [Green Version]
- Serota, T.; Culley, T. Seed germination and seedling survival of invasive Callery Pear (Pyrus calleryana Decene.) 11 years after fruit collection. Castanea 2019, 84, 47–52. [Google Scholar] [CrossRef]
- Culley, T.M.; Hardiman, N.A. The beginning of a new invasive plant: A history of the ornamental Callery pear in the United States. BioScience 2007, 57, 956–964. [Google Scholar] [CrossRef] [Green Version]
- Vincent, M.A. On the spread and current distribution of Pyrus calleryana in the United States. Castanea 2005, 70, 20–32. [Google Scholar] [CrossRef] [Green Version]
- Makimura, F.; Tsuruta, M.; Yamasaki, K.; Mukai, Y. Visitors to flowers of the endangered Callery pear, Pyrus calleryana Decne. var. dimorphopheylla (Makino) Koidz.; in the Tado population, Mie Prefecture. Jpn. J. Conserv. Ecol. 2015, 20, 197–202. [Google Scholar]
- Reichard, S.H.; Hamilton, C.W. Predicting invasions of woody plants introduced into North America: Predicción de invasiones de plantas leñosas introducidas a Norteamérica. Conserv. Biol. 1997, 11, 193–203. [Google Scholar] [CrossRef]
- EDDMapS. Early Detection; Distribution Mapping System; The University of Georgia–Center for Invasive Species and Ecosystem Health: Tifton, GA, USA, 2019. [Google Scholar]
- GBIF. GBIF Occurrence of Pyrus Calleryana. Available online: https://www.gbif.org/species/5363119 (accessed on 29 May 2021).
- Nesom, G.L. Callery pear (Pyrus calleryana—Rosaceae) naturalized in North Carolina. Rhodora 2000, 102, 361–364. [Google Scholar]
- White, J.; McClain, W.E.; Ebinger, J.E. Naturalized callery pear (Pyrus calleryana Decne.) in Illinois. Trans. Ill. State Acad. Sci. 2005, 98, 123–130. [Google Scholar]
- Randall, R.P. A Global Compendium of Weeds; R.G. and F.J. Richardson: Victoria, Australia, 2002. [Google Scholar]
- Warrix, A.R.; Marshall, J.M. Callery pear (Pyrus calleryana) response to fire in a managed prairie ecosystem. Invasive Plant Sci. Manag. 2018, 11, 27–32. [Google Scholar] [CrossRef]
- Purugganan, M.D. The molecular population genetics of regulatory genes. Mol. Ecol. 2000, 9, 1451–1461. [Google Scholar] [CrossRef] [Green Version]
- Hadziabdic, D.; Wadl, P.A.; Vito, L.M.; Boggess, S.L.; Scheffler, B.E.; Windham, M.T.; Trigiano, R.N. Development and characterization of sixteen microsatellite loci for Geosmithia morbida, the causal agent of thousand canker disease in black walnut (Juglans nigra). Conserv. Genet. Resour. 2012, 4, 287–289. [Google Scholar] [CrossRef]
- Kamvar, Z.N.; Tabima, J.F.; Grünwald, N.J. Poppr: An R package for genetic analysis of populations with clonal, partially clonal, and/or sexual reproduction. PeerJ 2014, 2, e281. [Google Scholar] [CrossRef] [Green Version]
- Nowicki, M.; Zhao, Y.; Boggess, S.L.; Fluess, H.; Paya-Milans, M.; Staton, M.E.; Houston, L.C.; Hadziabdic, D.; Trigiano, R.N. Taraxacum kok-saghyz (rubber dandelion) genomic microsatellite loci reveal modest genetic diversity and cross-amplify broadly to related species. Sci. Rep. 2019, 9, 1–17. [Google Scholar] [CrossRef]
- Vieira, M.L.C.; Santini, L.; Diniz, A.L.; Munhoz, C.d.F. Microsatellite markers: What they mean and why they are so useful. Genet. Mol. Biol. 2016, 39, 312–328. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Imai, A.; Rie, N.; Mukai, Y. Population genetic structure in a threatened tree, Pyrus calleryana var. dimorphophylla revealed by chloroplast DNA and nuclear SSR locus polymorphisms. Conserv. Genet. Resour. 2013, 14, 983–996. [Google Scholar] [CrossRef]
- Bao, L.; Chen, K.; Zhang, D.; Cao, Y.T.; Yamamoto, T.; Teng, Y. Genetic diversity and similarity of pear (Pyrus, L.) cultivars native to East Asia revealed by SSR (simple sequence repeat) markers. Genet. Resour. Crop Evol. 2007, 54, 959–971. [Google Scholar] [CrossRef]
- Yamamoto, T.; Kimura, T.; Sawamura, Y.; Kotobuki, K.; Ban, Y.; Hayashi, T.; Matsuta, N. SSRs isolated from apple can identify polymorphism and genetic diversity in pear. Theor. Appl. Genet. 2001, 102, 865–870. [Google Scholar] [CrossRef]
- Yamamoto, T.; Kimura, T.; Sawamura, Y.; Manabe, T.; Kotobuki, K.; Hayashi, T.; Ban, Y.; Matsuta, N. Simple sequence repeats for genetic analysis in pear. Euphytica 2002, 124, 129. [Google Scholar] [CrossRef]
- Yamamoto, T.; Kimura, T.; Shoda, M.; Ban, Y.; Hayashi, T.; Matsuta, N. Development of microsatellite markers in the Japanese pear (Pyrus pyrifolia Nakai). Mol. Ecol. Resour. 2002, 2, 14–16. [Google Scholar] [CrossRef]
- Bao, L.; Chen, K.; Zhang, D.; Li, X.; Teng, Y. An assessment of genetic variability and relationships within Asian pears based on AFLP (amplified fragment length polymorphism) markers. Sci. Hortic. 2008, 116, 374–380. [Google Scholar] [CrossRef]
- Yuanwen Teng, Y.; Tanabe, K.; Tamura, F.; Itai, A. Genetic relationships of pear cultivars in Xinjiang, China, as measured by RAPD markers. J. Hortic. Sci. Biotechnol. 2001, 76, 771–779. [Google Scholar] [CrossRef]
- Iketani, H.; Manabe, T.; Matsuta, N.; Akihama, T.; Hayashi, T. Incongruence between RFLPs of chloroplast DNA and morphological classification in east Asian pear (Pyrus spp.). Genet. Resour. Crop Evol. 1998, 45, 533–539. [Google Scholar] [CrossRef]
- Kimura, T.; Iketani, H.; Kotobuki, K.; Matsuta, N.; Ban, Y.; Hayashi, T.; Yamamoto, T. Genetic characterization of pear varieties revealed by chloroplast DNA sequences. J. Hortic. Sci. Biotechnol. 2003, 78, 241–247. [Google Scholar] [CrossRef]
- Liu, J.; Zheng, X.; Potter, D.; Hu, C.; Teng, Y. Genetic diversity and population structure of Pyrus calleryana (Rosaceae) in Zhejiang province, China. Biochem. Syst. Ecol. 2012, 45, 69–78. [Google Scholar] [CrossRef]
- Lalk, S.; Hartshorn, J.; Coyle, D.R. Invasive woody plants and their effects on arthropods in the United States: Challenges and opportunities. Ann. Entomol. Soc. Am. 2021, 114, 192–205. [Google Scholar] [CrossRef]
- Kim, H.; Chang, K.S.; Chang, C.-S. EH Wilson’s expedition to Korea from 1917 to 1919: Resolving place names of his collections. J. Jpn. Bot. 2010, 85, 99–117. [Google Scholar]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 1987, 19, 11–15. [Google Scholar]
- Wu, J.; Wang, Z.; Shi, Z.; Zhang, S.; Ming, R.; Zhu, S.; Khan, M.A.; Tao, S.; Korban, S.S.; Wang, H.; et al. The genome of the pear (Pyrus bretschneideri Rehd.). Genome Res. 2013, 23, 396–408. [Google Scholar] [CrossRef] [Green Version]
- Zimin, A.V.; Marcais, G.; Puiu, D.; Roberts, M.; Salzberg, S.L.; Yorke, J.A. The MaSuRCA genome assembler. Bioinformatics 2013, 29, 2669–2677. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Wang, L. GMATA: An integrated software package for genome-scale SSR mining, marker development and viewing. Front. Plant Sci. 2016, 7, 1350. [Google Scholar] [CrossRef] [Green Version]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR Protoc. Guide Methods Appl. 1990, 18, 315–322. [Google Scholar]
- Nowicki, M.; Boggess, S.L.; Saxton, A.M.; Hadziabdic, D.; Xiang, Q.-Y.J.; Molnar, T.; Huff, M.L.; Staton, M.E.; Zhao, Y.; Trigiano, R.N. Haplotyping of Cornus florida and C. kousa chloroplasts: Insights into species-level differences and patterns of plastic DNA variation in cultivars. PLoS ONE 2018, 13, e0205407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korbie, D.J.; Mattick, J.S. Touchdown PCR for increased specificity and sensitivity in PCR amplification. Nat. Protoc. 2008, 3, 1452. [Google Scholar] [CrossRef] [PubMed]
- Amos, W.; Hoffman, I.; Frodsham, A.; Zhang, L.; Best, S.; Hill, A.V.S. Automated binning of microsatellite alleles: Problems and solutions. Mol. Ecol. Notes 2007, 7, 10–14. [Google Scholar] [CrossRef]
- Lischer, H.E.; Excoffier, L. PGDSpider: An automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics 2012, 28, 298–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Goudet, J. Hierfstat, a package for R to compute and test hierarchical F-statistics. Mol. Ecol. Notes 2005, 5, 184–186. [Google Scholar] [CrossRef] [Green Version]
- Hardy, O.; Vekemans, X. SPAGeDi 1.5. A Program for Spatial Pattern Analysis of Genetic Diversity; Program Manual; Universite Libre de Bruxelles: Brussels, Belgium, 2015. [Google Scholar]
- Hardy, O.J.; Vekemans, X. SPAGeDi: A versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol. Ecol. Notes 2002, 2, 618–620. [Google Scholar] [CrossRef] [Green Version]
- Slatkin, M. A measure of population subdivision based on microsatellite allele frequencies. Genetics 1995, 139, 457–462. [Google Scholar] [CrossRef]
- Peakall, R.; Smouse, P.E. GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Pons, O.; Petit, R. Measwring and testing genetic differentiation with ordered versus unordered alleles. Genetics 1996, 144, 1237–1245. [Google Scholar] [CrossRef]
- Agapow, P.M.; Burt, A. Indices of multilocus linkage disequilibrium. Mol. Ecol. Notes 2001, 1, 101–102. [Google Scholar] [CrossRef]
- Ripley, B.; Venables, B.; Bates, D.M.; Hornik, K.; Gebhardt, A. Package ‘mass’. Cran R 2013, 538, 113–120. [Google Scholar]
- Dray, S.; Dufour, A. The ade4 package: Implementing the duality diagram for ecologists. J. Stat. Softw. 2007, 22, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P. Package ‘vegan’: Community Ecology Package, version 2.0-10. Cran R 2013, 2, 1–295. [Google Scholar]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Francis, R.M. Pophelper: An R package and web app to analyse and visualize population structure. Mol. Ecol. Resour. 2017, 17, 27–32. [Google Scholar] [CrossRef] [Green Version]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [Green Version]
- Gayevskiy, V.; Klaere, S.; Knight, S.; Goddard, M.R. ObStruct: A method to objectively analyse factors driving population structure using Bayesian ancestry profiles. PLoS ONE 2014, 9, e85196. [Google Scholar]
- Jombart, T. adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef] [Green Version]
- Nei, M. Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 1978, 89, 583–590. [Google Scholar] [CrossRef]
- Alioto, T. Transposons played a major role in the diversification between the closely related almond and peach genomes: Results from the almond genome sequence. Plant J. 2020, 101, 455–472. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, R.; Tarpy, D.R.; Reeve, H.K. Estimating effective paternity number in social insects and the effective number of alleles in a population. Mol. Ecol. 2003, 12, 3157–3164. [Google Scholar] [CrossRef]
- Shannon, C.E. A mathematical theory of communication. ACM SIGMOBILE Mob. Comput. Commun. Rev. 2001, 5, 3–55. [Google Scholar] [CrossRef]
- Stoddart, J.A.; Taylor, J.F. Genotypic diversity: Estimation and prediction in samples. Genetics 1988, 118, 705–711. [Google Scholar] [CrossRef]
- Simpson, E.H. Measurement of diversity. Nature 1949, 163, 688. [Google Scholar] [CrossRef]
- Jost, L. GST and its relatives do not measure differentiation. Mol. Ecol. 2008, 17, 4015–4026. [Google Scholar] [CrossRef]
- Hamrick, J.L.; Godt, M.W. Effects of life history traits on genetic diversity in plant species. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 1996, 351, 1291–1298. [Google Scholar]
- Ony, M.A.; Nowicki, M.; Boggess, S.l.; Klingeman, W.E.; Zobel, J.M.; Trigiano, R.N.; Hadziabdic, D. Habitat fragmentation influences genetic diversity and differentiation: Fine-scale population structure of Cercis canadensis (eastern redbud). Ecol. Evol. 2020, 10, 3655–3670. [Google Scholar] [CrossRef] [Green Version]
- Nowicki, M.; Houston, L.C.; Boggess, S.L.; Aiello, A.S.; Paya-MIlans, M.; Staton, M.E.; Hayashida, M.; Yamanaka, M.; Eda, S.; Trigiano, R.N. Species diversity and phylogeography of Cornus kousa (Asian dogwood) captured by genomic and genic microsatellites. Ecol. Evol. 2020, 10, 8299–8312. [Google Scholar] [CrossRef]
- Wu, J.; Wang, Y.; Xu, J.; Korban, S.S.; Fei, Z.; Tao, S.; Ming, R.; Tai, S.; Khan, A.M.; Postman, J.D.; et al. Diversification and independent domestication of Asian and European pears. Genome Biol. 2018, 19, 1–16. [Google Scholar] [CrossRef]
- Caballero, A.; Rodríguez-Ramilo, S.T.; Avila, V.; Fernández, J. Management of genetic diversity of subdivided populations in conservation programmes. Conserv. Genet. 2010, 11, 409–419. [Google Scholar] [CrossRef]
- Leimu, R.; Mutikainen, P.; Koricheva, J.; Fischer, M. How general are positive relationships between plant population size, fitness and genetic variation? J. Ecol. 2006, 94, 942–952. [Google Scholar] [CrossRef]
- Sakai, A.K.; Allendorf, F.W.; Holt, J.S.; Lodge, D.M.; Molofsky, J.; With, K.A.; Baughman, S.; Cabin, R.J.; Cohen, J.E.; Ellstrand, N.C.; et al. The population biology of invasive species. Annu. Rev. Ecol. Syst. 2001, 32, 305–332. [Google Scholar] [CrossRef] [Green Version]
- Hamrick, J.L.; Godt, M.J.W.; Sherman-Broyles, S.L. Factors Influencing Levels of Genetic Diversity in Woody Plant Species. In Population Genetics of Forest Trees; Springer: Berlin/Heidelberg, Germany, 1992; pp. 95–124. [Google Scholar]
- Meyer, L.; Causse, R.; Pernin, F.; Scalone, R.; Bailly, G.; Chauvel, B.; Delye, C.; Le Corre, V. New gSSR and EST-SSR markers reveal high genetic diversity in the invasive plant Ambrosia artemisiifolia L. and can be transferred to other invasive Ambrosia species. PLoS ONE 2017, 12, e0176197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunphy, B.; Hamrick, J. Gene flow among established Puerto Rican populations of the exotic tree species, Albizia lebbeck. Heredity 2005, 94, 418–425. [Google Scholar] [CrossRef]
- Pappert, R.A.; Hamrick, J.; Donovan, L.A. Genetic variation in Pueraria lobata (Fabaceae), an introduced, clonal, invasive plant of the southeastern United States. Am. J. Bot. 2000, 87, 1240–1245. [Google Scholar] [CrossRef]
- Slatkin, M. Gene flow and the geographic structure of natural populations. Science 1987, 236, 787–792. [Google Scholar] [CrossRef]
- Gaskin, J.F.; Schwarzlander, M.; Grevstad, F.S.; Haverhals, M.A.; Bourchier, R.S.; Miller, T.W. Extreme differences in population structure and genetic diversity for three invasive congeners: Knotweeds in western North America. Biol. Invasions 2014, 16, 2127–2136. [Google Scholar] [CrossRef]
- Cullingham, C.I.; Miller, J.M.; Peery, R.M.; Dupuis, J.R.; Malenfant, R.M.; Gorrell, J.C.; Janes, J.K. Confidently identifying the correct K value using the ΔK method: When does K = 2? Mol. Ecol. 2020, 29, 862–869. [Google Scholar] [CrossRef]
- Lombaert, E.; Guillemaud, T.; Deleury, E. Biases of STRUCTURE software when exploring introduction routes of invasive species. Heredity 2018, 120, 485–499. [Google Scholar] [CrossRef] [Green Version]
- Santamour, F.S., Jr.; Demuth, P. Identification of Callery pear cultivars by peroxidase isozyme patterns. J. Hered. 1980, 71, 447–449. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | N | % Missing | # Alleles | NAe | H | G | FIS | Ar | λ | He | Ho | d | Pa |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| China | 20 | 15.28 | 8.06 | 6.40 | 3.00 | 20 | 0.636 *** | 4.61 | 0.95 | 0.80 | 0.30 | 0.10 ns | 37 |
| Japan | 11 | 5.56 | 5.72 | 4.74 | 2.40 | 11 | 0.511 *** | 4.02 | 0.91 | 0.74 | 0.37 | 0.16 *** | 19 |
| Korea | 5 | 7.78 | 2.56 | 2.41 | 1.61 | 5 | 0.569 *** | 2.48 | 0.80 | 0.51 | 0.24 | 0.03 ns | 10 |
| USA | 21 | 7.94 | 7.22 | 4.62 | 3.04 | 21 | 0.397 *** | 4.06 | 0.95 | 0.72 | 0.44 | 0.11 ns | 22 |
| Total | 57 | 10.04 | 11.78 | 4.54 | 4.04 | 57 | 0.552 *** | 3.79 | 0.98 | 0.80 | 0.36 | 0.12 ns | 88 |
| SSR Locus | No. | % Missing | Ar | Ho | He | Dest | G | FST | FIS | RST | RIS | Nm |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PyC006 | 10 | 12.28 | 3.89 | 0.13 | 0.72 | 0.20 | 3.53 | 0.08 ns | 0.74 **** | 0.18 * | −0.08 ns | 5.58 |
| PyC008 | 16 | 3.51 | 4.86 | 0.36 | 0.87 | 0.82 | 7.36 | 0.16 **** | 0.38 **** | 0.47 **** | 0.69 *** | 0.85 |
| PyC009 | 15 | 12.28 | 5.39 | 0.25 | 0.85 | −0.14 | 6.13 | −0.02 ns | 0.72 **** | 0.18 * | 0.70 **** | NA |
| PyC012 | 13 | 26.32 | 4.73 | 0.08 | 0.89 | 0.78 | 8.34 | 0.14 *** | 0.85 **** | 0.17 ns | 0.85 **** | 1.07 |
| PyC013 | 14 | 0.00 | 4.34 | 0.43 | 0.84 | 0.50 | 5.86 | 0.11 *** | 0.46 **** | 0.11 * | 0.36 * | 1.23 |
| PyC014 | 14 | 7.02 | 5.45 | 0.21 | 0.86 | 0.72 | 6.84 | 0.16 **** | 0.70 **** | 0.50 **** | 0.67 **** | 0.94 |
| PyC015 | 18 | 5.26 | 5.40 | 0.12 | 0.87 | 0.85 | 7.03 | 0.20 **** | 0.85 **** | 0.74 **** | 0.87 **** | 0.67 |
| PyC017 | 20 | 21.05 | 6.48 | 0.60 | 0.92 | 0.46 | 10.80 | 0.06 ** | 0.19 ** | 0.18 * | 0.37 * | 3.34 |
| PyC018 | 16 | 8.77 | 5.89 | 0.65 | 0.91 | 0.51 | 10.18 | 0.05 ** | 0.17 ** | −0.04 ns | 0.09 ns | 3.20 |
| PyC020 | 13 | 0.00 | 4.89 | 0.39 | 0.80 | 0.20 | 4.82 | 0.03 ns | 0.49 **** | 0.07 ns | 0.34 * | 5.50 |
| PyC031 | 20 | 8.77 | 6.22 | 0.84 | 0.92 | 0.57 | 11.76 | 0.04 * | 0.13 * | 0.03 ns | −0.38 ** | 6.86 |
| PyC032 | 6 | 19.30 | 4.24 | 0.09 | 0.74 | 0.58 | 3.77 | 0.22 **** | 0.86 **** | 0.37 *** | 0.73 **** | 0.93 |
| PyC035 | 13 | 1.75 | 4.77 | 0.41 | 0.85 | 0.31 | 6.54 | 0.03 ns | 0.49 **** | 0.14 * | 0.17 ns | 7.91 |
| PyC038 | 6 | 0.00 | 4.06 | 0.93 | 0.76 | −0.04 | 4.12 | −0.01 ns | 0.23 *** | 0 ns | −0.75 **** | NA |
| PyC041 | 8 | 14.04 | 4.08 | 0.29 | 0.81 | 0.04 | 5.04 | 0.01 ns | 0.65 **** | 0.08 ns | 0.22 **** | 7.04 |
| PyC042 | 7 | 15.79 | 3.57 | 0.06 | 0.81 | 0.70 | 5.08 | 0.23 **** | 0.89 **** | 0.62 **** | 0.82 **** | 0.61 |
| PyC047 | 6 | 12.28 | 3.64 | 0.18 | 0.76 | 0.36 | 3.97 | 0.10 * | 0.61 **** | 0.19 * | 0.75 **** | 2.36 |
| PyC050 | 5 | 12.28 | 2.74 | 0.03 | 0.41 | 0.07 | 1.68 | 0.10 ns | 0.90 **** | 0.09 ns | 0.92 **** | 2.74 |
| Average | 12.2 | 10.04 | 4.70 | 0.34 | 0.81 | 0.42 | 6.27 | 0.09 **** | 0.52 **** | 0.33 **** | 0.31 **** | 1.79 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sapkota, S.; Boggess, S.L.; Trigiano, R.N.; Klingeman, W.E.; Hadziabdic, D.; Coyle, D.R.; Olukolu, B.A.; Kuster, R.D.; Nowicki, M. Microsatellite Loci Reveal Genetic Diversity of Asian Callery Pear (Pyrus calleryana) in the Species Native Range and in the North American Cultivars. Life 2021, 11, 531. https://doi.org/10.3390/life11060531
Sapkota S, Boggess SL, Trigiano RN, Klingeman WE, Hadziabdic D, Coyle DR, Olukolu BA, Kuster RD, Nowicki M. Microsatellite Loci Reveal Genetic Diversity of Asian Callery Pear (Pyrus calleryana) in the Species Native Range and in the North American Cultivars. Life. 2021; 11(6):531. https://doi.org/10.3390/life11060531
Chicago/Turabian StyleSapkota, Shiwani, Sarah L. Boggess, Robert N. Trigiano, William E. Klingeman, Denita Hadziabdic, David R. Coyle, Bode A. Olukolu, Ryan D. Kuster, and Marcin Nowicki. 2021. "Microsatellite Loci Reveal Genetic Diversity of Asian Callery Pear (Pyrus calleryana) in the Species Native Range and in the North American Cultivars" Life 11, no. 6: 531. https://doi.org/10.3390/life11060531