
Strong Shift to ATR-Dependent Regulation of the G2-Checkpoint after Exposure to High-LET Radiation



Abstract
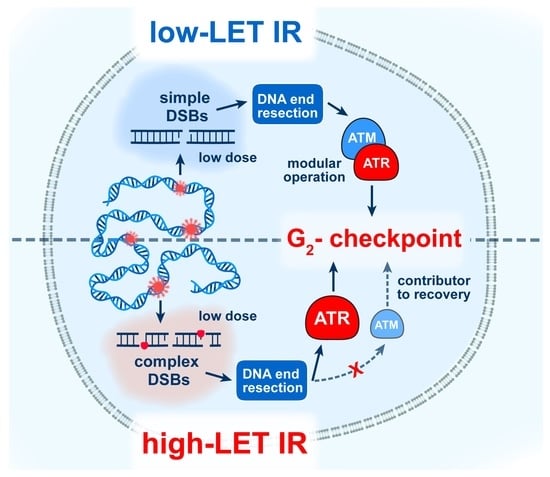
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Inhibitors
2.2. Radiation Exposure
2.3. Colony Formation Assay
2.4. Multicolor Fluorescence In Situ Hybridization (mFISH)
2.5. Flow Cytometry Analysis of Mitotic Index Using H3-pS10 Staining
2.6. Indirect Immunofluorescence for Detection of pATM-S1981 Foci
3. Results
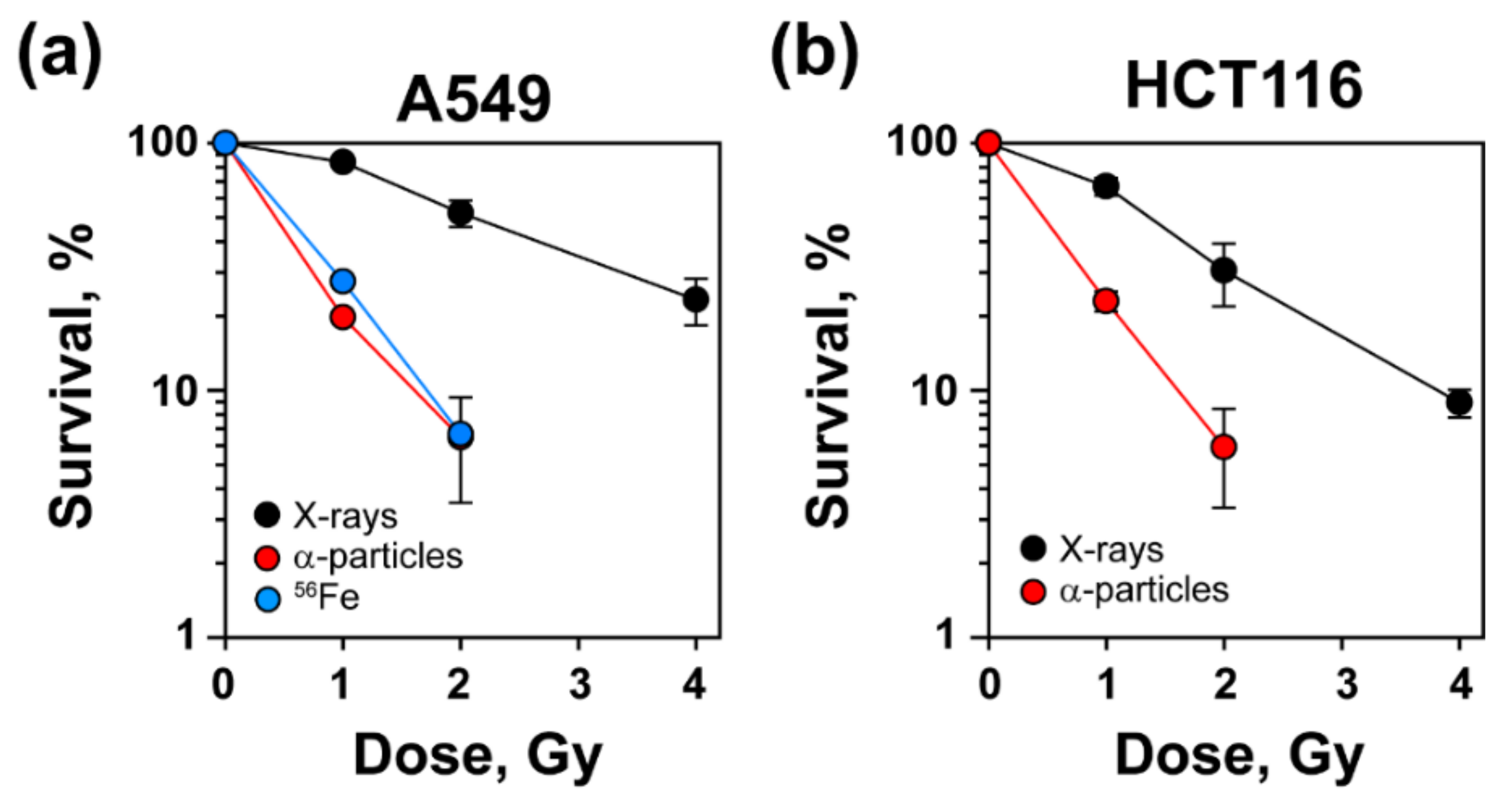
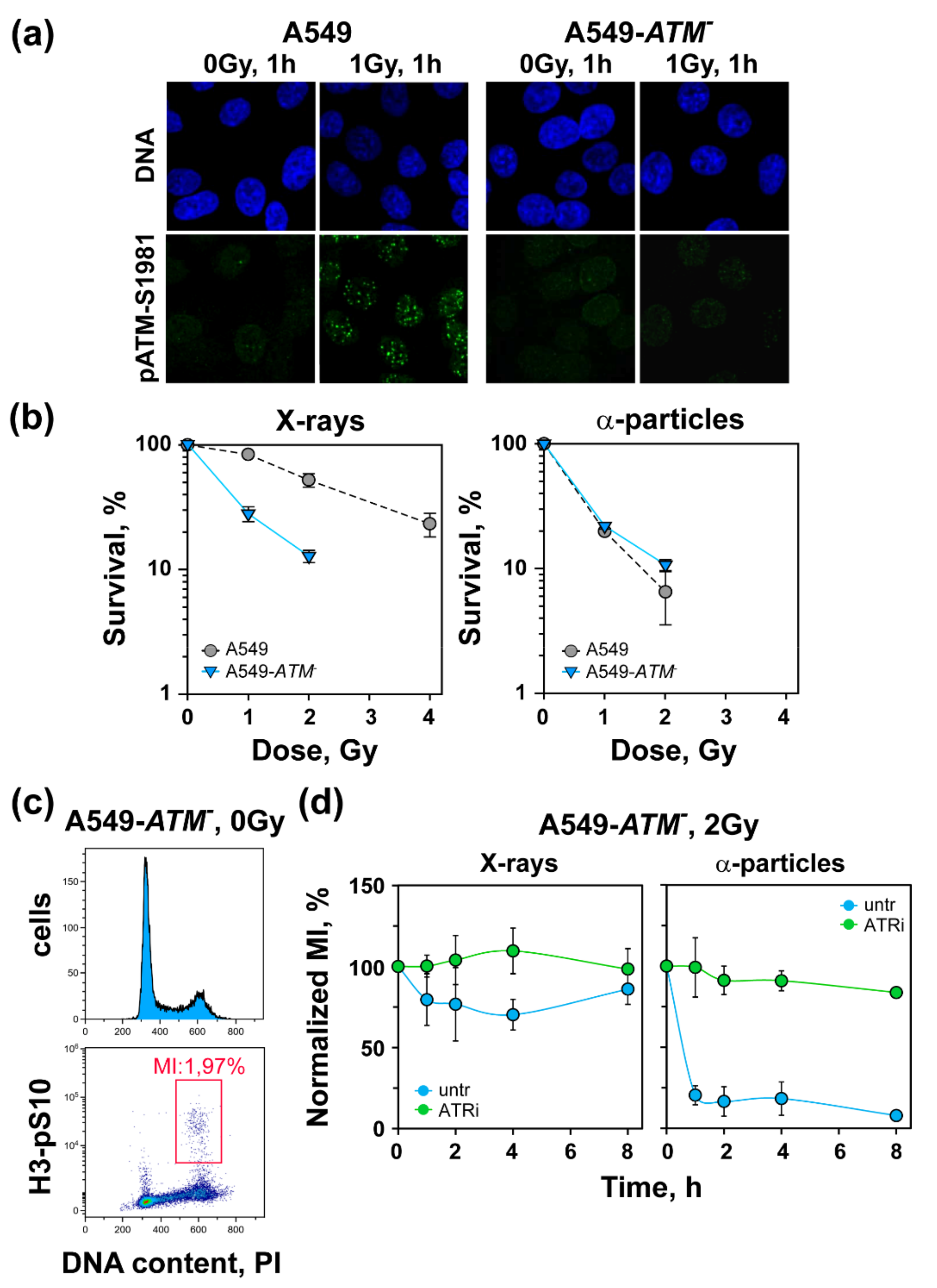
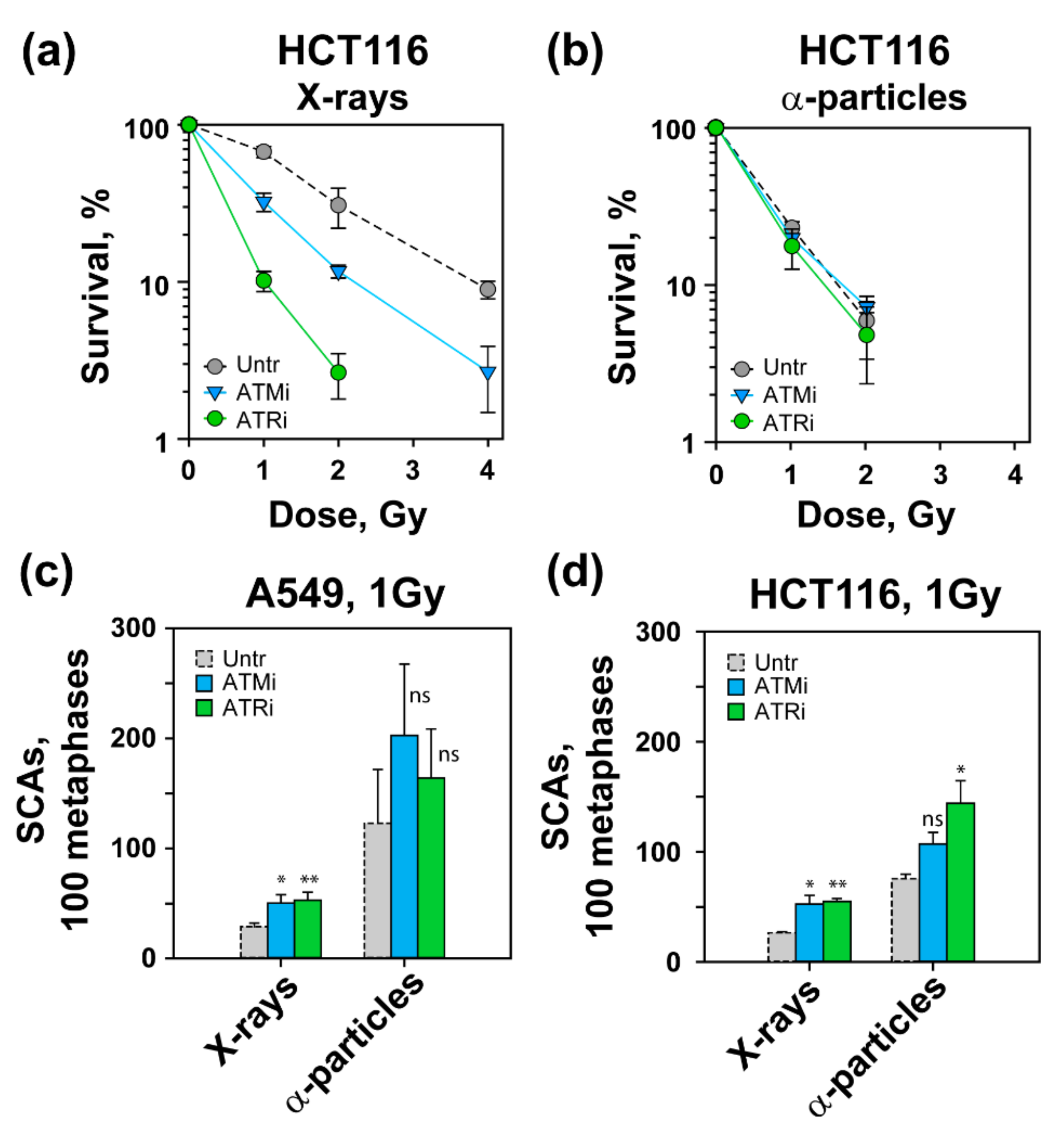
3.1. Increased Radiosensitivity after Exposure to High-LET IR
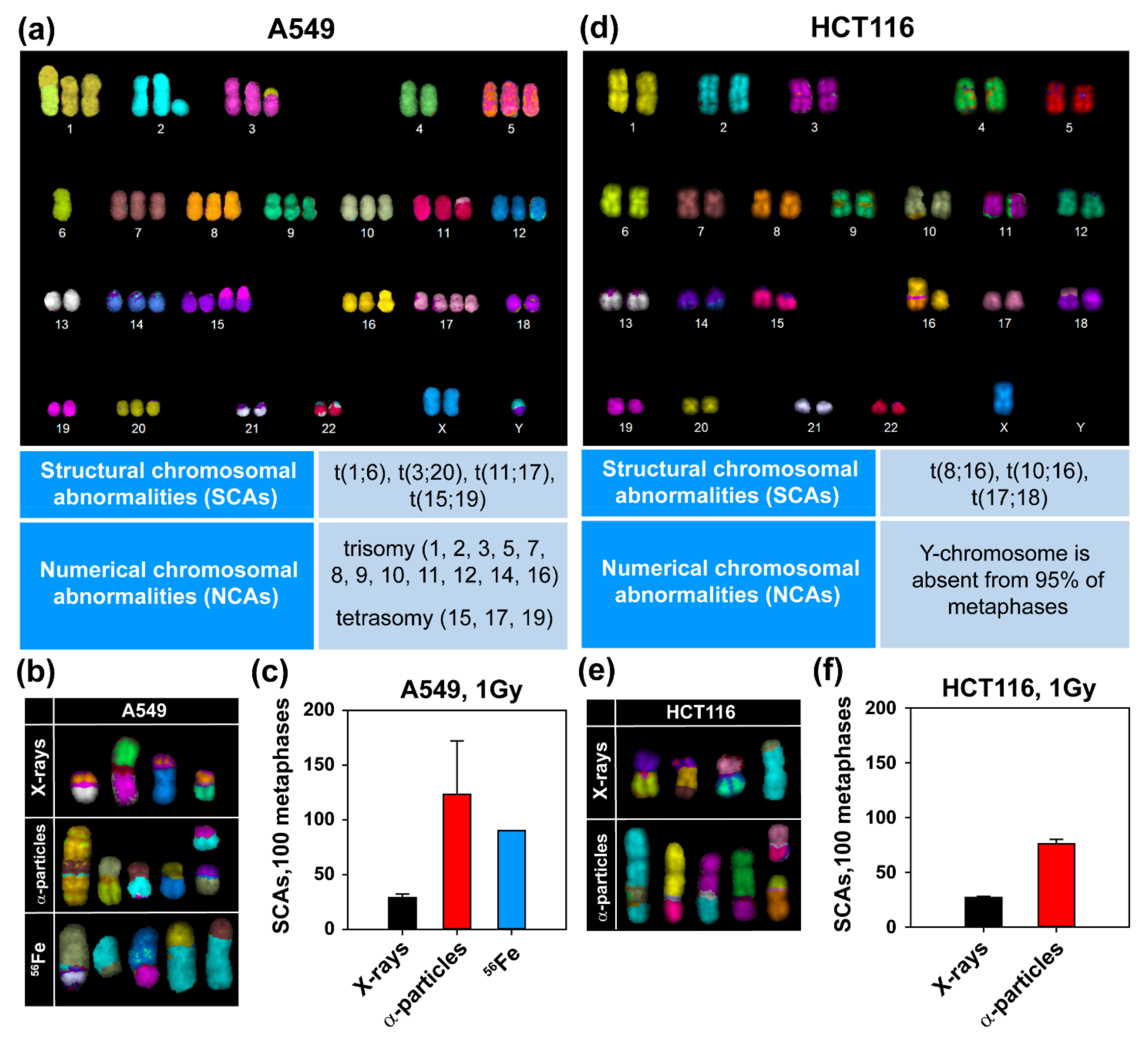
3.2. High-LET IR Increases the Incidence of SCAs
3.3. High-LET IR Alters the ATM/ATR Dependent Regulation of the G2-Checkpoint
3.4. Effects of ATM and ATR Inhibition on Cell Survival and SCA Formation in A549 Cells
4. Discussion
4.1. High Incidence of SCAs and Increased Cell Killing after Exposure of Cells to High-LET-IR
4.2. Altered Regulation of the G2-Checkpoint after Exposure to High-LET IR
4.3. LET-Dependent Radiosensitization and SCA Induction by ATMi or ATRi
4.4. Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Durante, M.; Cucinotta, F.A. Physical basis of radiation protection in space travel. Rev. Mod. Phys. 2011, 83, 1245–1281. [Google Scholar] [CrossRef]
- Durante, M.; Cucinotta, F.A. Heavy ion carcinogenesis and human space exploration. Nat. Rev. Cancer 2008, 8, 465–472. [Google Scholar] [CrossRef] [PubMed]
- Cucinotta, F.; Durante, M. Cancer risk from exposure to galactiv cosmic rays: Implications for space exploration by humans beings. Lancet Oncol. 2006, 7, 431–435. [Google Scholar] [CrossRef] [Green Version]
- Furukawa, S.; Nagamatsu, A.; Nenoi, M.; Fujimori, A.; Kakinuma, S.; Katsube, T.; Wang, B.; Tsuruoka, C.; Shirai, T.; Nakamura, A.J.; et al. Space Radiation Biology for “Living in Space”. Biomed. Res. Int. 2020, 2020, 4703286. [Google Scholar] [CrossRef] [Green Version]
- Garrett-Bakelman, F.E.; Darshi, M.; Green, S.J.; Gur, R.C.; Lin, L.; Macias, B.R.; McKenna, M.J.; Meydan, C.; Mishra, T.; Nasrini, J.; et al. The NASA Twins Study: A multidimensional analysis of a year-long human spaceflight. Science 2019, 364, eaau8650. [Google Scholar] [CrossRef] [PubMed]
- Cortese, F.; Klokov, D.; Osipov, A.; Stefaniak, J.; Moskalev, A.; Schastnaya, J.; Cantor, C.; Aliper, A.; Mamoshina, P.; Ushakov, I.; et al. Vive la radioresistance! Converging research in radiobiology and biogerontology to enhance human radioresistance for deep space exploration and colonization. Oncotarget 2018, 9, 14692–14722. [Google Scholar] [CrossRef]
- Mladenova, V.; Mladenov, E.; Iliakis, G. Novel Biological Approaches for Testing the Contributions of Single DSBs and DSB Clusters to the Biological Effects of High LET Radiation. Front. Oncol. 2016, 6, 163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hada, M.; Georgakilas, A.G. Formation of Clustered DNA Damage after High-LET Irradiation: A Review. J. Radiat. Res. 2008, 49, 203–210. [Google Scholar] [CrossRef]
- Schipler, A.; Mladenova, V.; Soni, A.; Nikolov, V.; Saha, J.; Mladenov, E.; Iliakis, G. Chromosome thripsis by DNA double strand break clusters causes enhanced cell lethality, chromosomal translocations and 53BP1-recruitment. Nucleic Acids Res. 2016, 44, 7673–7690. [Google Scholar] [CrossRef] [Green Version]
- Schipler, A.; Iliakis, G. DNA double-strand-break complexity levels and their possible contributions to the probability for error-prone processing and repair pathway choice. Nucleic Acids Res. 2013, 41, 7589–7605. [Google Scholar] [CrossRef] [Green Version]
- Hada, M.; Wu, H.; Cucinotta, F.A. mBAND analysis for high- and low-LET radiation-induced chromosome aberrations: A review. Mutat. Res. Fundam. Mol. Mech. Mutagenes. 2011, 711, 187–192. [Google Scholar] [CrossRef]
- Lee, R.; Sommer, S.; Hartel, C.; Nasonova, E.; Durante, M.; Ritter, S. Complex exchanges are responsible for the increased effectiveness of C-ions compared to X-rays at the first post-irradiation mitosis. Mutat. Res. Genet. Toxicol. Environ. Mutagenes. 2010, 701, 52–59. [Google Scholar] [CrossRef]
- Durante, M.; Formenti, S.C. Radiation-Induced Chromosomal Aberrations and Immunotherapy: Micronuclei, Cytosolic DNA, and Interferon-Production Pathway. Front. Oncol. 2018, 8, 192. [Google Scholar] [CrossRef] [PubMed]
- De Coster, W.; De Rijk, P.; De Roeck, A.; De Pooter, T.; D’Hert, S.; Strazisar, M.; Sleegers, K.; Van Broeckhoven, C. Structural variants identified by Oxford Nanopore PromethION sequencing of the human genome. Genome Res. 2019, 29, 1178–1187. [Google Scholar] [CrossRef] [Green Version]
- Soni, A.; Murmann-Konda, T.; Siemann-Loekes, M.; Pantelias, G.E.; Iliakis, G. Chromosome breaks generated by low doses of ionizing radiation in G2-phase are processed exclusively by gene conversion. DNA Repair 2020, 89, 102828. [Google Scholar] [CrossRef] [PubMed]
- Mladenov, E.; Staudt, C.; Soni, A.; Murmann-Konda, T.; Siemann-Loekes, M.; Iliakis, G. Strong suppression of gene conversion with increasing DNA double-strand break load delimited by 53BP1 and RAD52. Nucleic Acids Res. 2020, 48, 1905–1924. [Google Scholar] [CrossRef] [Green Version]
- Iliakis, G.; Mladenov, E.; Mladenova, V. Necessities in the Processing of DNA Double Strand Breaks and Their Effects on Genomic Instability and Cancer. Cancers 2019, 11, 1671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iliakis, G.; Murmann, T.; Soni, A. Alternative end-joining repair pathways are the ultimate backup for abrogated classical non-homologous end-joining and homologous recombination repair: Implications for the formation of chromosome translocations. Mutat. Res. Genet. Toxicol. Environ. Mutagenes. 2015, 793, 166–175. [Google Scholar] [CrossRef]
- Dueva, R.; Iliakis, G. Alternative pathways of non-homologous end joining (NHEJ) in genomic instability and cancer. Transl. Cancer Res. 2013, 2, 163–177. [Google Scholar]
- Lee, R.; Nasonova, E.; Hartel, C.; Durante, M.; Ritter, S. Chromosome aberration measurements in mitotic and G2-PCC lymphocytes at the standard sampling time of 48 h underestimate the effectiveness of high-LET particles. Radiat. Environ. Biophys. 2011, 50, 371–381. [Google Scholar] [CrossRef]
- Wang, M.; Wu, W.; Wu, W.; Rosidi, B.; Zhang, L.; Wang, H.; Iliakis, G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006, 34, 6170–6182. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Wang, M.; Wu, W.; Singh, S.K.; Mussfeldt, T.; Iliakis, G. Repair of radiation induced DNA double strand breaks by backup NHEJ is enhanced in G2. DNA Repair 2008, 7, 329–338. [Google Scholar] [CrossRef]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanz, M.C.; Dibitetto, D.; Smolka, M.B. DNA damage kinase signaling: Checkpoint and repair at 30 years. EMBO J. 2019, 38, e101801. [Google Scholar] [CrossRef] [PubMed]
- Waterman, D.P.; Haber, J.E.; Smolka, M.B. Checkpoint Responses to DNA Double-Strand Breaks. Annu. Rev. Biochem. 2020, 89, 103–133. [Google Scholar] [CrossRef] [Green Version]
- Mladenov, E.; Iliakis, G. Induction and repair of DNA double strand breaks: The increasing spectrum of non-homologous end joining pathways. Mutat. Res. 2011, 711, 61–72. [Google Scholar] [CrossRef]
- Mladenov, E.; Magin, S.; Soni, A.; Iliakis, G. DNA double-strand-break repair in higher eukaryotes and its role in genomic instability and cancer: Cell cycle and proliferation-dependent regulation. Semin. Cancer Biol. 2016, 37–38, 51–64. [Google Scholar] [CrossRef]
- Mladenov, E.; Fan, X.; Dueva, R.; Soni, A.; Iliakis, G. Radiation-dose-dependent functional synergisms between ATM, ATR and DNA-PKcs in checkpoint control and resection in G2-phase. Sci. Rep. 2019, 9, 8255. [Google Scholar] [CrossRef] [Green Version]
- Mladenov, E.; Fan, X.; Paul-Konietzko, K.; Soni, A.; Iliakis, G. DNA-PKcs and ATM epistatically suppress DNA end resection and hyperactivation of ATR-dependent G2-checkpoint in S-phase irradiated cells. Sci. Rep. 2019, 9, 14597. [Google Scholar] [CrossRef]
- Iliakis, G. The mutagenicity of alpha particles in Ehrlich Ascites tumor cells. Radiat. Res. 1984, 99, 52–58. [Google Scholar] [CrossRef]
- Singh, S.K.; Bencsik-Theilen, A.; Mladenov, E.; Jakob, B.; Taucher-Scholz, G.; Iliakis, G. Reduced contribution of thermally labile sugar lesions to DNA double strand break formation after exposure to heavy ions. Radiat. Oncol. 2013, 8, 77. [Google Scholar] [CrossRef] [Green Version]
- Xue, L.; Furusawa, Y.; Okayasu, R.; Miura, M.; Cui, X.; Liu, C.; Hirayama, R.; Matsumoto, Y.; Yajima, H.; Yu, D. The complexity of DNA double strand break is a crucial factor for activating ATR signaling pathway for G2/M checkpoint regulation regardless of ATM function. DNA Repair 2015, 25, 72–83. [Google Scholar] [CrossRef]
- Frankenberg, D.; Brede, H.J.; Schrewe, U.J.; Steinmetz, C.; Frankenberg-Schwager, M.; Kasten, G.; Pralle, E. Induction of DNA Double-Strand Breaks by 1 H and 4 He Ions in Primary Human Skin Fibroblasts in the LET Range of 8 to 124 keV/μm. Radiat. Res. 1999, 151, 540–549. [Google Scholar] [CrossRef]
- Mladenov, E.; Saha, J.; Iliakis, G. Processing-Challenges Generated by Clusters of DNA Double-Strand Breaks Underpin Increased Effectiveness of High-LET Radiation and Chromothripsis. Adv. Exp. Med. Biol. 2018, 1044, 149–168. [Google Scholar] [CrossRef] [PubMed]
- Gerelchuluun, A.; Zhu, J.; Su, F.; Asaithamby, A.; Chen, D.J.; Tsuboi, K. Homologous recombination pathway may play a major role in high-LET radiation-induced DNA double-strand break repair. J. Radiat. Res. 2014, 55, i83–i84. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, A.; Kubo, M.; Ma, H.; Nakagawa, A.; Yoshida, Y.; Isono, M.; Kanai, T.; Ohno, T.; Furusawa, Y.; Funayama, T.; et al. Nonhomologous End-Joining Repair Plays a More Important Role than Homologous Recombination Repair in Defining Radiosensitivity after Exposure to High-LET Radiation. Radiat. Res. 2014, 182, 338–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iliakis, G.; Mladenova, V.; Sharif, M.; Chaudhary, S.; Mavragani, I.V.; Soni, A.; Saha, J.; Schipler, A.; Mladenov, E. Defined Biological Models of High-Let Radiation Lesions. Radiat. Prot. Dosim. 2019, 183, 60–68. [Google Scholar] [CrossRef]
- Fujisawa, H.; Nakajima, N.I.; Sunada, S.; Lee, Y.; Hirakawa, H.; Yajima, H.; Fujimori, A.; Uesaka, M.; Okayasu, R. VE-821, an ATR inhibitor, causes radiosensitization in human tumor cells irradiated with high LET radiation. Radiat. Oncol. 2015, 10, 175. [Google Scholar] [CrossRef] [Green Version]
- Zhao, B.; Rothenberg, E.; Ramsden, D.A.; Lieber, M.R. The molecular basis and disease relevance of non-homologous DNA end joining. Nat. Rev. Mol. Cell Biol. 2020, 21, 765–781. [Google Scholar] [CrossRef]
- Bhargava, R.; Onyango, D.O.; Stark, J.M. Regulation of Single-Strand Annealing and its Role in Genome Maintenance. Trends Genet. TIG 2016, 32, 566–575. [Google Scholar] [CrossRef] [Green Version]
- Taverna, P.; Liu, L.; Hanson, A.J.; Monks, A.; Gerson, S.L. Characterization of MLH1 and MSH2 DNA mismatch repair proteins in cell lines of the NCI anticancer drug screen. Cancer Chemother. Pharm. 2000, 46, 507–516. [Google Scholar] [CrossRef]
- Garner, K.M.; Eastman, A. Variations in Mre11/Rad50/Nbs1 status and DNA damage-induced S-phase arrest in the cell lines of the NCI60 panel. BMC Cancer 2011, 11, 206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannini, G.; Ristori, E.; Cerignoli, F.; Rinaldi, C.; Zani, M.; Viel, A.; Ottini, L.; Crescenzi, M.; Martinotti, S.; Bignami, M.; et al. Human MRE11 is inactivated in mismatch repair-deficient cancers. EMBO Rep. 2002, 3, 248–254. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Shi, L.; Kinomura, A.; Fukuto, A.; Horikoshi, Y.; Oma, Y.; Harata, M.; Ikura, M.; Ikura, T.; Kanaar, R.; et al. Distinct roles of ATM and ATR in the regulation of ARP8 phosphorylation to prevent chromosome translocations. Elife 2018, 7. [Google Scholar] [CrossRef]
- White, J.S.; Choi, S.; Bakkenist, C.J. Irreversible chromosome damage accumulates rapidly in the absence of ATM kinase acitvity. Cell Cycle 2008, 7, 1277–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, J.S.; Choi, S.; Bakkenist, C.J. Transient ATM kinase inhibition disrupts DNA damage-induced sister chromatid exchange. Sci. Signal. 2010, 3, ra44. [Google Scholar] [CrossRef] [Green Version]
- Walls, G.M.; Oughton, J.B.; Chalmers, A.J.; Brown, S.; Collinson, F.; Forster, M.D.; Franks, K.N.; Gilbert, A.; Hanna, G.G.; Hannaway, N.; et al. CONCORDE: A phase I platform study of novel agents in combination with conventional radiotherapy in non-small-cell lung cancer. Clin. Transl. Radiat. Oncol. 2020, 25, 61–66. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mladenova, V.; Mladenov, E.; Scholz, M.; Stuschke, M.; Iliakis, G. Strong Shift to ATR-Dependent Regulation of the G2-Checkpoint after Exposure to High-LET Radiation. Life 2021, 11, 560. https://doi.org/10.3390/life11060560
Mladenova V, Mladenov E, Scholz M, Stuschke M, Iliakis G. Strong Shift to ATR-Dependent Regulation of the G2-Checkpoint after Exposure to High-LET Radiation. Life. 2021; 11(6):560. https://doi.org/10.3390/life11060560
Chicago/Turabian StyleMladenova, Veronika, Emil Mladenov, Michael Scholz, Martin Stuschke, and George Iliakis. 2021. "Strong Shift to ATR-Dependent Regulation of the G2-Checkpoint after Exposure to High-LET Radiation" Life 11, no. 6: 560. https://doi.org/10.3390/life11060560
APA StyleMladenova, V., Mladenov, E., Scholz, M., Stuschke, M., & Iliakis, G. (2021). Strong Shift to ATR-Dependent Regulation of the G2-Checkpoint after Exposure to High-LET Radiation. Life, 11(6), 560. https://doi.org/10.3390/life11060560