
Exploring the Ability of LARS2 Carboxy-Terminal Domain in Rescuing the MELAS Phenotype

 , , , ,
, , , , 

Abstract
1. Introduction
2. Materials and Methods
2.1. Tissue Culture, Transfections and Viability Assay
2.2. Heteroplasmy Determination by PCR/RFLP Analysis
2.3. RNA Extraction and RT-qPCR
2.4. Whole Cell Extracts Preparation and Mitochondria Isolation
2.5. RNA Immunoprecipitation
2.6. SDS-PAGE, BN-PAGE and Immunoblotting
2.7. [35S]-Labelling of Mitochondrial Translation Products
2.8. High Resolution Northern Blot Analysis
2.9. Complex IV Activity Determination
2.10. Mitochondrial Oxygen Consumption Measurement
2.11. Lactate Assay
3. Results
3.1. Cterm Domain Interacts with the Mutated mt-tRNALeu(UUR) in MELAS Cybrids
3.2. Cterm Rescues Viability and Mitochondrial Translation in MELAS Cybrids
3.3. Cterm Has No Effect on mt-tRNALeu(UUR) Steady-State Level and Aminoacylation
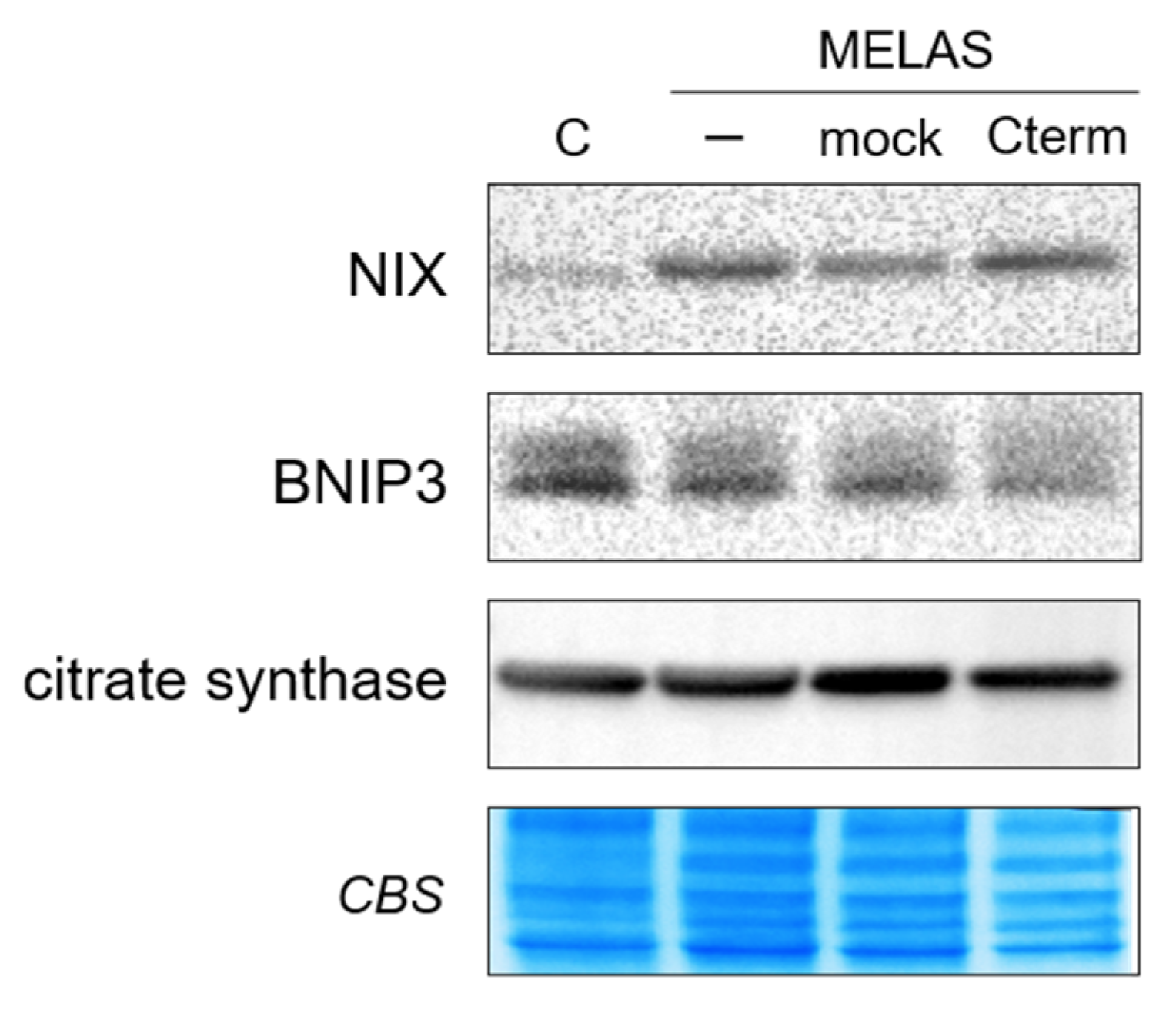
3.4. Cterm Does Not Affect Mitochondrial Bioenergetic Competence, Mitophagy and Mitochondrial Mass
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial Diseases. Nat. Rev. Dis. Primers 2016, 2, 16080. [Google Scholar] [CrossRef]
- Shokolenko, I.N.; Alexeyev, M.F. Mitochondrial Transcription in Mammalian Cells. Front. Biosci. 2017, 22, 835–853. [Google Scholar] [CrossRef]
- Pfanner, N.; Warscheid, B.; Wiedemann, N. Mitochondrial Proteins: From Biogenesis to Functional Networks. Nat. Rev. Mol. Cell Biol. 2019, 20, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Majamaa-Voltti, K.; Peuhkurinen, K.; Kortelainen, M.-L.; Hassinen, I.E.; Majamaa, K. Cardiac Abnormalities in Patients with Mitochondrial DNA Mutation 3243A>G. BMC Cardiovasc. Disord. 2002, 2, 12. [Google Scholar] [CrossRef]
- Bates, M.G.D.; Newman, J.H.; Jakovljevic, D.G.; Hollingsworth, K.G.; Alston, C.L.; Zalewski, P.; Klawe, J.J.; Blamire, A.M.; MacGowan, G.A.; Keavney, B.D.; et al. Defining Cardiac Adaptations and Safety of Endurance Training in Patients with m.3243A>G-Related Mitochondrial Disease. Int. J. Cardiol. 2013, 168, 3599–3608. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Adesina, A.M.; Jones, J.; Scaglia, F. MELAS Syndrome: Clinical Manifestations, Pathogenesis, and Treatment Options. Mol. Genet. Metab. 2015, 116, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Yasukawa, T.; Suzuki, T.; Suzuki, T.; Ueda, T.; Ohta, S.; Watanabe, K. Modification Defect at Anticodon Wobble Nucleotide of Mitochondrial TRNAs Leu (UUR) with Pathogenic Mutations of Mitochondrial Myopathy, Encephalopathy, Lactic Acidosis, and Stroke-like Episodes. J. Biol. Chem. 2000, 275, 4251–4257. [Google Scholar] [CrossRef] [PubMed]
- Kirino, Y.; Goto, Y.-I.; Campos, Y.; Arenas, J.; Suzuki, T. Specific Correlation between the Wobble Modification Deficiency in Mutant TRNAs and the Clinical Features of a Human Mitochondrial Disease. Proc. Natl. Acad. Sci. USA 2005, 102, 7127–7132. [Google Scholar] [CrossRef]
- King, M.P.; Koga, Y.; Davidson, M.; Schon, E.A. Defects in Mitochondrial Protein Synthesis and Respiratory Chain Activity Segregate with the TRNA(Leu(UUR)) Mutation Associated with Mitochondrial Myopathy, Encephalopathy, Lactic Acidosis, and Strokelike Episodes. Mol. Cell. Biol. 1992, 12, 480–490. [Google Scholar] [CrossRef] [PubMed]
- Sasarman, F.; Antonicka, H.; Shoubridge, E.A. The A3243G TRNALeu(UUR) MELAS Mutation Causes Amino Acid Misincorporation and a Combined Respiratory Chain Assembly Defect Partially Suppressed by Overexpression of EFTu and EFG2. Hum. Mol. Genet. 2008, 17, 3697–3707. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Miyauchi, K.; Suzuki, T.; Yokobori, S.; Shigi, N.; Kondow, A.; Takeuchi, N.; Yamagishi, A.; Watanabe, K. Taurine-Containing Uridine Modifications in TRNA Anticodons Are Required to Decipher Non-Universal Genetic Codes in Ascidian Mitochondria. J. Biol. Chem. 2011, 286, 35494–35498. [Google Scholar] [CrossRef]
- Rorbach, J.; Yusoff, A.A.; Tuppen, H.; Abg-Kamaludin, D.P.; Chrzanowska-Lightowlers, Z.M.A.; Taylor, R.W.; Turnbull, D.M.; McFarland, R.; Lightowlers, R.N. Overexpression of Human Mitochondrial Valyl TRNA Synthetase Can Partially Restore Levels of Cognate Mt-TRNAVal Carrying the Pathogenic C25U Mutation. Nucleic Acids Res. 2008, 36, 3065–3074. [Google Scholar] [CrossRef]
- Hornig-Do, H.T.; Montanari, A.; Rozanska, A.; Tuppen, H.A.; Almalki, A.A.; Abg-Kamaludin, D.P.; Frontali, L.; Francisci, S.; Lightowlers, R.N.; Chrzanowska-Lightowlers, Z.M. Human Mitochondrial Leucyl TRNA Synthetase Can Suppress Non Cognate Pathogenic Mt-TRNA Mutations. EMBO Mol. Med. 2014, 6, 183–193. [Google Scholar] [CrossRef]
- Perli, E.; Giordano, C.; Pisano, A.; Montanari, A.; Campese, A.F.; Reyes, A.; Ghezzi, D.; Nasca, A.; Tuppen, H.A.; Orlandi, M.; et al. The Isolated Carboxy-Terminal Domain of Human Mitochondrial Leucyl-TRNA Synthetase Rescues the Pathological Phenotype of Mitochondrial TRNA Mutations in Human Cells. EMBO Mol. Med. 2014, 6, 169–182. [Google Scholar] [CrossRef]
- Giordano, C.; Morea, V.; Perli, E.; d’Amati, G. The Phenotypic Expression of Mitochondrial TRNA-Mutations Can Be Modulated by Either Mitochondrial Leucyl-TRNA Synthetase or the C-Terminal Domain Thereof. Front. Genet. 2015, 6, 113. [Google Scholar] [CrossRef][Green Version]
- Perli, E.; Fiorillo, A.; Giordano, C.; Pisano, A.; Montanari, A.; Grazioli, P.; Campese, A.F.; Di Micco, P.; Tuppen, H.A.; Genovese, I.; et al. Short Peptides from Leucyl-TRNA Synthetase Rescue Disease-Causing Mitochondrial TRNA Point Mutations. Hum. Mol. Genet. 2016, 25, 903–915. [Google Scholar] [CrossRef]
- Perli, E.; Pisano, A.; Pignataro, M.G.; Campese, A.F.; Pelullo, M.; Genovese, I.; de Turris, V.; Ghelli, A.M.; Cerbelli, B.; Giordano, C.; et al. Exogenous Peptides Are Able to Penetrate Human Cell and Mitochondrial Membranes, Stabilize Mitochondrial TRNA Structures, and Rescue Severe Mitochondrial Defects. FASEB J. 2020, 34, 7675–7686. [Google Scholar] [CrossRef]
- Cámara, Y.; Asin-Cayuela, J.; Park, C.B.; Metodiev, M.D.; Shi, Y.; Ruzzenente, B.; Kukat, C.; Habermann, B.; Wibom, R.; Hultenby, K.; et al. MTERF4 Regulates Translation by Targeting the Methyltransferase NSUN4 to the Mammalian Mitochondrial Ribosome. Cell Metab. 2011, 13, 527–539. [Google Scholar] [CrossRef]
- Bruni, F.; Proctor-Kent, Y.; Lightowlers, R.N.; Chrzanowska-Lightowlers, Z.M. Messenger RNA Delivery to Mitoribosomes—Hints from a Bacterial Toxin. FEBS J. 2021, 288, 437–451. [Google Scholar] [CrossRef]
- Kim, S.W.; Li, Z.; Moore, P.S.; Monaghan, A.P.; Chang, Y.; Nichols, M.; John, B. A Sensitive Non-Radioactive Northern Blot Method to Detect Small RNAs. Nucleic Acids Res. 2010, 38, e98. [Google Scholar] [CrossRef]
- Spinazzi, M.; Casarin, A.; Pertegato, V.; Salviati, L.; Angelini, C. Assessment of Mitochondrial Respiratory Chain Enzymatic Activities on Tissues and Cultured Cells. Nat. Protoc. 2012, 7, 1235–1246. [Google Scholar] [CrossRef] [PubMed]
- Garin, S.; Levi, O.; Cohen, B.; Golani-Armon, A.; Arava, Y.S. Localization and RNA Binding of Mitochondrial Aminoacyl TRNA Synthetases. Genes 2020, 11, 1185. [Google Scholar] [CrossRef] [PubMed]
- Robinson, B.H.; Petrova-Benedict, R.; Buncic, J.R.; Wallace, D.C. Nonviability of Cells with Oxidative Defects in Galactose Medium: A Screening Test for Affected Patient Fibroblasts. Biochem. Med. Metab. Biol. 1992, 48, 122–126. [Google Scholar] [CrossRef]
- Chomyn, A.; Meola, G.; Bresolin, N.; Lai, S.T.; Scarlato, G.; Attardi, G. In Vitro Genetic Transfer of Protein Synthesis and Respiration Defects to Mitochondrial DNA-Less Cells with Myopathy-Patient Mitochondria. Mol. Cell Biol. 1991, 11, 2236–2244. [Google Scholar] [CrossRef][Green Version]
- Chomyn, A.; Enriquez, J.A.; Micol, V.; Fernandez-Silva, P.; Attardi, G. The Mitochondrial Myopathy, Encephalopathy, Lactic Acidosis, and Stroke-like Episode Syndrome-Associated Human Mitochondrial TRNA Leu(UUR) Mutation Causes Aminoacylation Deficiency and Concomitant Reduced Association of MRNA with Ribosomes. J. Biol. Chem. 2000, 275, 19198–19209. [Google Scholar] [CrossRef] [PubMed]
- Jaksch, M.; Kleinle, S.; Scharfe, C.; Klopstock, T.; Pongratz, D.; Müller-Höcker, J.; Gerbitz, K.-D.; Liechti-Gallati, S.; Lochmuller, H.; Horvath, R. Frequency of Mitochondrial Transfer RNA Mutations and Deletions in 225 Patients Presenting with Respiratory Chain Deficiencies. J. Med. Genet. 2001, 38, 665–673. [Google Scholar] [CrossRef]
- Cotán, D.; Cordero, M.D.; Garrido-Maraver, J.; Oropesa-Ávila, M.; Rodríguez-Hernández, A.; Gómez Izquierdo, L.; De la Mata, M.; De Miguel, M.; Lorite, J.B.; Infante, E.R.; et al. Secondary Coenzyme Q10 Deficiency Triggers Mitochondria Degradation by Mitophagy in MELAS Fibroblasts. FASEB J. 2011, 25, 2669–2687. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Maraver, J.; Paz, M.V.; Cordero, M.D.; Bautista-Lorite, J.; Oropesa-Ávila, M.; de la Mata, M.; Pavón, A.D.; de Lavera, I.; Alcocer-Gómez, E.; Galán, F.; et al. Critical Role of AMP-Activated Protein Kinase in the Balance between Mitophagy and Mitochondrial Biogenesis in MELAS Disease. Biochim. Biophys. Acta 2015, 1852, 2535–2553. [Google Scholar] [CrossRef]
- Goto, Y.; Nonaka, I.; Horai, S. A Mutation in the TRNA Leu(UUR) Gene Associated with the MELAS Subgroup of Mitochondrial Encephalomyopathies. Nature 1990, 348, 651–653. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Momoi, M.Y.; Tominaga, K.; Momoi, T.; Nihei, K.; Yanagisawa, M.; Kagawa, Y.; Ohta, S. A Point Mutation in the Mitochondrial TRNALeu(UUR) Gene in Melas (Mitochondrial Myopathy, Encephalopathy, Lactic Acidosis and Stroke-like Episodes). Biochem. Biophys. Res. Commun. 1990, 173, 816–822. [Google Scholar] [CrossRef]
- Schon, E.A.; Koga, Y.; Davidson, M.; Moraes, C.T.; King, M.P. The Mitochondrial TRNALeu(UUR) Mutation in MELAS: A Model for Pathogenesis. Biochim. Et Biophys. Acta BBA Bioenergy 1992, 1101, 206–209. [Google Scholar] [CrossRef]
- Park, H.; Davidson, E.; King, M.P. The Pathogenic A3243G Mutation in Human Mitochondrial TRNALeu(UUR) Decreases the Efficiency of Aminoacylation. Biochemistry 2003, 42, 958–964. [Google Scholar] [CrossRef]
- Kazuhito, T.; Wei, F.-Y. Posttranscriptional Modifications in Mitochondrial TRNA and Its Implication in Mitochondrial Translation and Disease. J. Biochem. 2020, 168, 435–444. [Google Scholar] [CrossRef]
- Francisci, S.; Montanari, A.; De Luca, C.; Frontali, L. Peptides from Aminoacyl-TRNA Synthetases Can Cure the Defects Due to Mutations in Mt TRNA Genes. Mitochondrion 2011, 11, 919–923. [Google Scholar] [CrossRef]
- Fender, A.; Gaudry, A.; Jühling, F.; Sissler, M.; Florentz, C. Adaptation of Aminoacylation Identity Rules to Mammalian Mitochondria. Biochimie 2012, 94, 1090–1097. [Google Scholar] [CrossRef]
- Kaufmann, P.; Shanske, S.; Hirano, M.; DiMauro, S.; King, M.P.; Koga, Y.; Schon, E.A. Mitochondrial DNA and RNA Processing in MELAS. Ann Neurol. 1996, 40, 172–180. [Google Scholar] [CrossRef]
- Maniura-Weber, K.; Helm, M.; Engemann, K.; Eckertz, S.; Möllers, M.; Schauen, M.; Hayrapetyan, A.; von Kleist-Retzow, J.-C.; Lightowlers, R.N.; Bindoff, L.A.; et al. Molecular Dysfunction Associated with the Human Mitochondrial 3302A>G Mutation in the MTTL1 (Mt-TRNALeu(UUR)) Gene. Nucleic Acids Res. 2006, 34, 6404–6415. [Google Scholar] [CrossRef]
- Nijtmans, L.G.; Klement, P.; Houstĕk, J.; van den Bogert, C. Assembly of Mitochondrial ATP Synthase in Cultured Human Cells: Implications for Mitochondrial Diseases. Biochim. Biophys. Acta 1995, 1272, 190–198. [Google Scholar] [CrossRef]
- Wallace, D.C.; Fan, W. Energetics, Epigenetics, Mitochondrial Genetics. Mitochondrion 2010, 10, 12–31. [Google Scholar] [CrossRef] [PubMed]
- Meseguer, S.; Navarro-González, C.; Panadero, J.; Villarroya, M.; Boutoual, R.; Sánchez-Alcázar, J.A.; Armengod, M.-E. The MELAS Mutation m.3243A>G Alters the Expression of Mitochondrial TRNA Fragments. Biochim. Biophys. Acta BBA Mol. Cell Res. 2019, 1866, 1433–1449. [Google Scholar] [CrossRef]
- Picard, M.; Zhang, J.; Hancock, S.; Derbeneva, O.; Golhar, R.; Golik, P.; O’Hearn, S.; Levy, S.; Potluri, P.; Lvova, M.; et al. Progressive Increase in MtDNA 3243A>G Heteroplasmy Causes Abrupt Transcriptional Reprogramming. Proc. Natl. Acad. Sci. USA 2014, 111, E4033–E4042. [Google Scholar] [CrossRef]
- Lu, M. Circular RNA: Functions, Applications and Prospects. ExRNA 2020, 2, 1. [Google Scholar] [CrossRef]
- Bravo, J.I.; Nozownik, S.; Danthi, P.S.; Benayoun, B.A. Transposable Elements, Circular RNAs and Mitochondrial Transcription in Age-Related Genomic Regulation. Development 2020, 147, dev175786. [Google Scholar] [CrossRef] [PubMed]
- Meseguer, S. MicroRNAs and TRNA-Derived Small Fragments: Key Messengers in Nuclear–Mitochondrial Communication. Front. Mol. Biosci. 2021, 8, 643575. [Google Scholar] [CrossRef]
- Tukalo, M.; Yaremchuk, A.; Fukunaga, R.; Yokoyama, S.; Cusack, S. The Crystal Structure of Leucyl-TRNA Synthetase Complexed with TRNALeu in the Post-Transfer-Editing Conformation. Nat. Struct. Mol. Biol. 2005, 12, 923–930. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, J.; Poruri, K.; Boniecki, M.T.; McTavish, K.K.; Martinis, S.A. Yeast Mitochondrial Leucyl-TRNA Synthetase CP1 Domain Has Functionally Diverged to Accommodate RNA Splicing at Expense of Hydrolytic Editing. J. Biol. Chem. 2012, 287, 14772–14781. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amplicon | Primer Sequence |
|---|---|
| mt-tRNALeu(UUR) | For: GTTAAGATGGCAGAGCCC |
| Rev: GAAGAGGAATTGAACCTCTGAC | |
| RNA19 | For: TATACCCACACCCACCCAAG |
| Rev: GCGATTAGAATGGGTACAAT | |
| mt-tRNALys | For: ATAGGGCCCGTATTTACCCTA |
| Rev: ATACGGTAGTATTTAGTTGG | |
| mt-tRNATyr | For: TGGTAAAAAGAGGCCTAACCC |
| Rev: ATGGCTGAGTGAAGCATTGG | |
| ND4 mRNA | For: CCATTCTCCTCCTATCCCTCAAC |
| Rev: CACAATCTGATGTTTTGGTTAAAC | |
| 12S rRNA | For: ACACTACGAGCCACAGCT |
| Rev: GCTACACCTTGACCTAACGTC | |
| Cterm | For: AAATTCCTGTGCCCCAACAA |
| Rev: CTACTTATCGTCGTCATCCT | |
| 18S rRNA | For: GTAACCCGTTGAACCCCATT |
| Rev: CCATCCAATCGGTAGTAGCG |
| Target | Probe Sequence |
|---|---|
| mt-tRNALeu(UUR) | TATGCGATTACCGGGCTCTGCCATCTTAAC |
| mt-tRNAGlu | TATTCTCGCACGGACTACAACCACGAC |
| 5S rRNA | GGGTGGTATGGCCGTAGAC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capriglia, F.; Rizzo, F.; Petrosillo, G.; Morea, V.; d’Amati, G.; Cantatore, P.; Roberti, M.; Loguercio Polosa, P.; Bruni, F. Exploring the Ability of LARS2 Carboxy-Terminal Domain in Rescuing the MELAS Phenotype. Life 2021, 11, 674. https://doi.org/10.3390/life11070674
Capriglia F, Rizzo F, Petrosillo G, Morea V, d’Amati G, Cantatore P, Roberti M, Loguercio Polosa P, Bruni F. Exploring the Ability of LARS2 Carboxy-Terminal Domain in Rescuing the MELAS Phenotype. Life. 2021; 11(7):674. https://doi.org/10.3390/life11070674
Chicago/Turabian StyleCapriglia, Francesco, Francesca Rizzo, Giuseppe Petrosillo, Veronica Morea, Giulia d’Amati, Palmiro Cantatore, Marina Roberti, Paola Loguercio Polosa, and Francesco Bruni. 2021. "Exploring the Ability of LARS2 Carboxy-Terminal Domain in Rescuing the MELAS Phenotype" Life 11, no. 7: 674. https://doi.org/10.3390/life11070674
APA StyleCapriglia, F., Rizzo, F., Petrosillo, G., Morea, V., d’Amati, G., Cantatore, P., Roberti, M., Loguercio Polosa, P., & Bruni, F. (2021). Exploring the Ability of LARS2 Carboxy-Terminal Domain in Rescuing the MELAS Phenotype. Life, 11(7), 674. https://doi.org/10.3390/life11070674