
Non-Coding RNAs and Splicing Activity in Testicular Germ Cell Tumors




Abstract
:1. Introduction
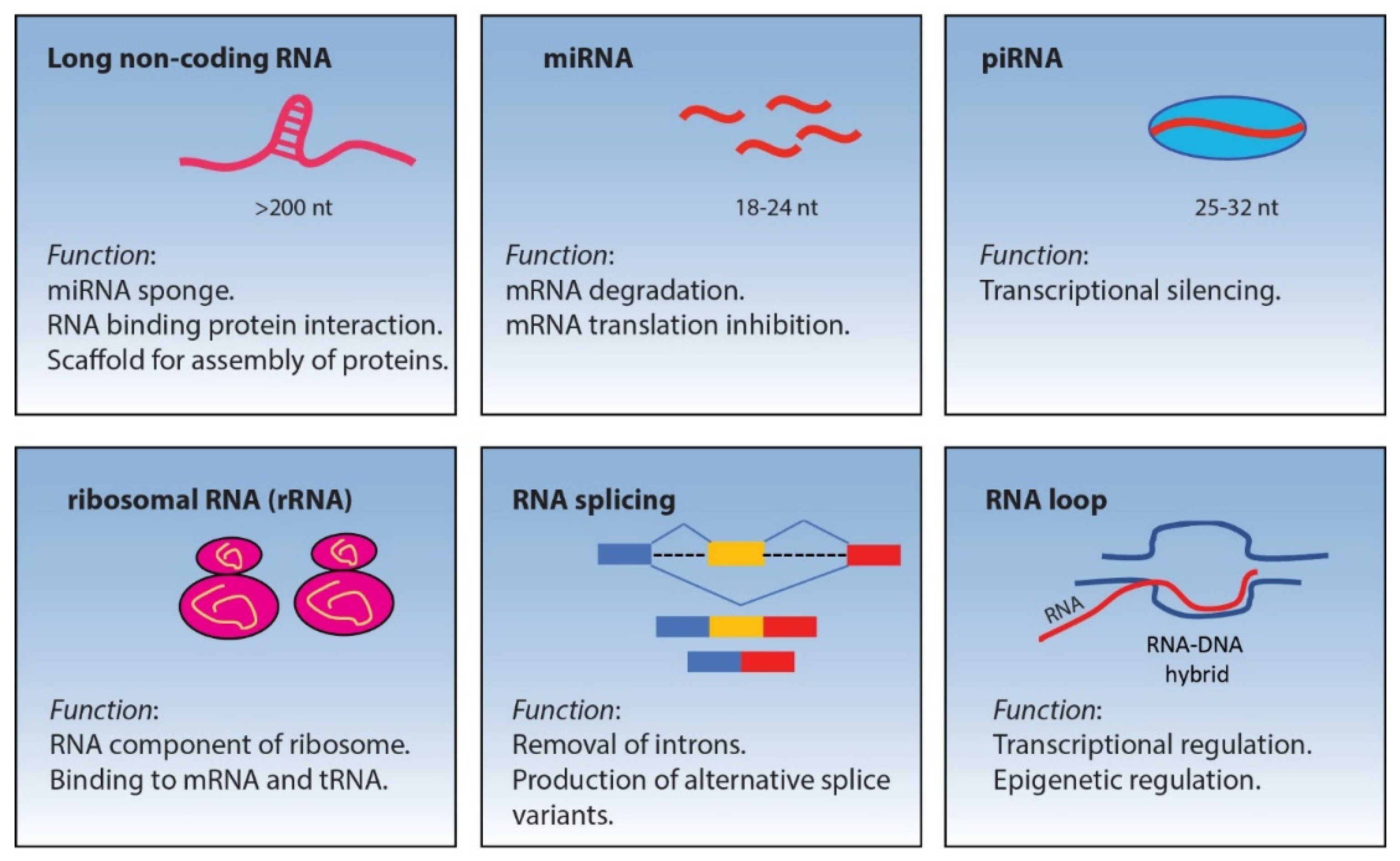
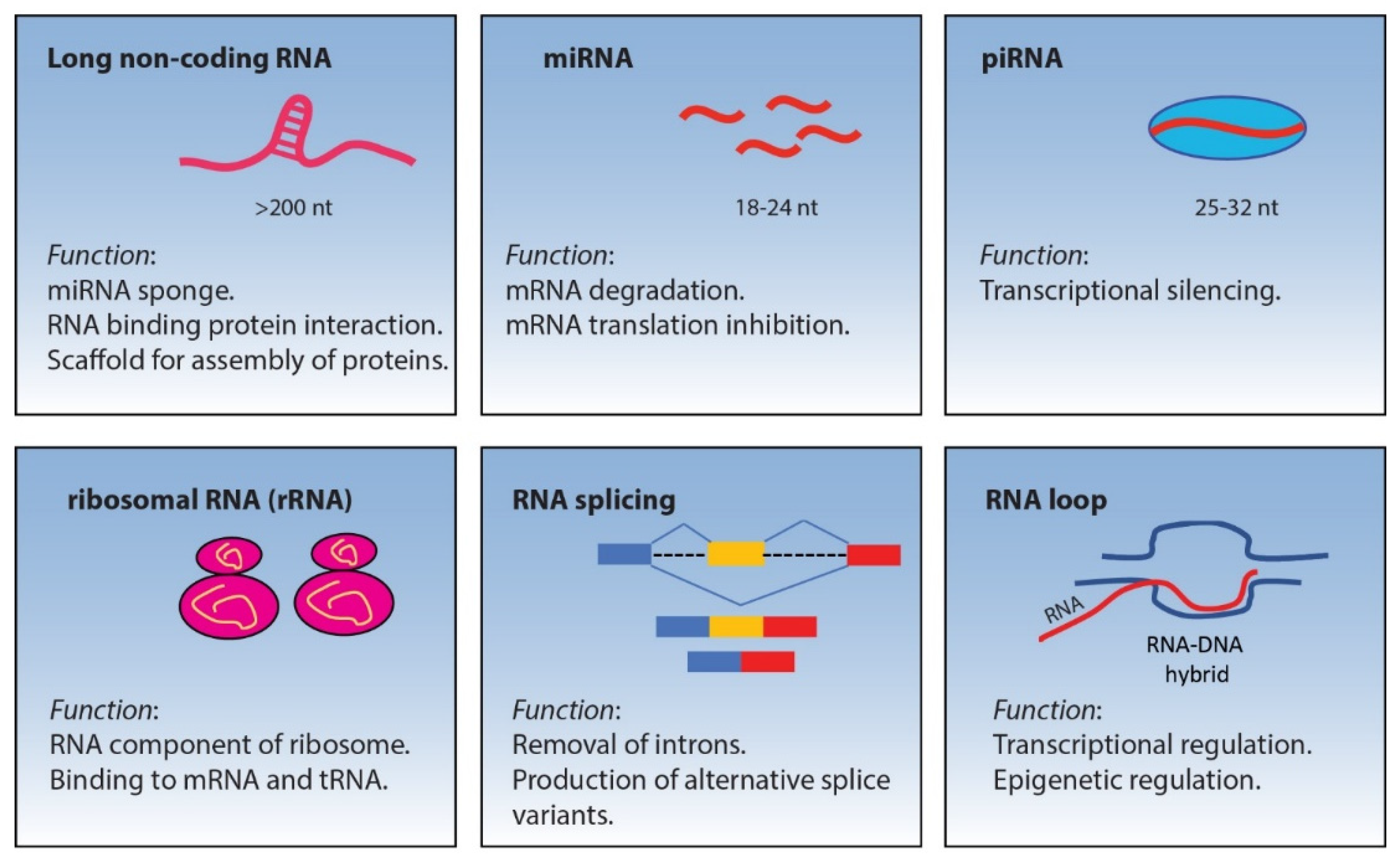
2. Non-Coding RNA
2.1. miRNA
2.2. piRNA
2.3. lncRNA
2.4. Ribosomal RNA (rRNA)
3. RNA Splicing
R-Loop
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Houldsworth, J.; Korkola, J.E.; Bosl, G.J.; Chaganti, R.S. Biology and genetics of adult male germ cell tumors. J. Clin. Oncol. 2006, 24, 5512–5518. [Google Scholar] [CrossRef]
- Huyghe, E.; Matsuda, T.; Thonneau, P. Increasing incidence of testicular cancer worldwide: A review. J. Urol. 2003, 170, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Looijenga, L.H.; Stoop, H.; Biermann, K. Testicular cancer: Biology and biomarkers. Virchows Arch. 2014, 464, 301–313. [Google Scholar] [CrossRef]
- Dieckmann, K.P.; Pichlmeier, U. Bilateral testicular germ cell tumors in Turkey: Increase in incidence in last decade and evaluation of risk factors in 30 patients. J. Urol. 2007, 178, 2222–2223. [Google Scholar] [CrossRef] [PubMed]
- De Felici, M.; Dolci, S. From testis to teratomas: A brief history of male germ cells in mammals. Int. J. Dev. Biol. 2013, 57, 115–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batool, A.; Karimi, N.; Wu, X.N.; Chen, S.R.; Liu, Y.X. Testicular germ cell tumor: A comprehensive review. Cell Mol. Life Sci. 2019, 76, 1713–1727. [Google Scholar] [CrossRef] [PubMed]
- Skakkebaek, N.E.; Berthelsen, J.G.; Giwercman, A.; Muller, J. Carcinoma-in-situ of the testis: Possible origin from gonocytes and precursor of all types of germ cell tumours except spermatocytoma. Int. J. Androl. 1987, 10, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Rajpert-De Meyts, E.; Bartkova, J.; Samson, M.; Hoei-Hansen, C.E.; Frydelund-Larsen, L.; Bartek, J.; Skakkebaek, N.E. The emerging phenotype of the testicular carcinoma in situ germ cell. APMIS 2003, 111, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Rajpert-De Meyts, E.; Jacobsen, G.K.; Bartkova, J.; Aubry, F.; Samson, M.; Bartek, J.; Skakkebaek, N.E. The immunohistochemical expression pattern of Chk2, p53, p19INK4d, MAGE-A4 and other selected antigens provides new evidence for the premeiotic origin of spermatocytic seminoma. Histopathology 2003, 42, 217–226. [Google Scholar] [CrossRef]
- Sesterhenn, I.A.; Davis, C.J., Jr. Pathology of germ cell tumors of the testis. Cancer Control 2004, 11, 374–387. [Google Scholar] [CrossRef] [PubMed]
- Banks, K.; Tuazon, E.; Berhane, K.; Koh, C.J.; De Filippo, R.E.; Chang, A.; Kim, S.S.; Daneshmand, S.; Davis-Dao, C.; Lewinger, J.P.; et al. Cryptorchidism and testicular germ cell tumors: Comprehensive meta-analysis reveals that association between these conditions diminished over time and is modified by clinical characteristics. Front. Endocrinol. 2012, 3, 182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petersen, P.M.; Giwercman, A.; Hansen, S.W.; Berthelsen, J.G.; Daugaard, G.; Rorth, M.; Skakkebaek, N.E. Impaired testicular function in patients with carcinoma-in-situ of the testis. J. Clin. Oncol. 1999, 17, 173–179. [Google Scholar] [CrossRef]
- Cools, M.; Honecker, F.; Stoop, H.; Veltman, J.D.; de Krijger, R.R.; Steyerberg, E.; Wolffenbuttel, K.P.; Bokemeyer, C.; Lau, Y.F.; Drop, S.L.; et al. Maturation delay of germ cells in fetuses with trisomy 21 results in increased risk for the development of testicular germ cell tumors. Hum. Pathol. 2006, 37, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Hasle, H.; Mellemgaard, A.; Nielsen, J.; Hansen, J. Cancer incidence in men with Klinefelter syndrome. Br. J. Cancer 1995, 71, 416–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verp, M.S.; Simpson, J.L. Abnormal sexual differentiation and neoplasia. Cancer Genet. Cytogenet. 1987, 25, 191–218. [Google Scholar] [CrossRef]
- Harries, L.W. RNA Biology Provides New Therapeutic Targets for Human Disease. Front. Genet. 2019, 10, 205. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Rinn, J.L. Modular regulatory principles of large non-coding RNAs. Nature 2012, 482, 339–346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotaja, N. MicroRNAs and spermatogenesis. Fertil. Steril. 2014, 101, 1552–1562. [Google Scholar] [CrossRef]
- Hong, S.H.; Kwon, J.T.; Kim, J.; Jeong, J.; Kim, J.; Lee, S.; Cho, C. Profiling of testis-specific long noncoding RNAs in mice. BMC Genom. 2018, 19, 539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pyle, L.C.; Nathanson, K.L. Genetic changes associated with testicular cancer susceptibility. Semin. Oncol. 2016, 43, 575–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sood, P.; Krek, A.; Zavolan, M.; Macino, G.; Rajewsky, N. Cell-type-specific signatures of microRNAs on target mRNA expression. Proc. Natl. Acad. Sci. USA 2006, 103, 2746–2751. [Google Scholar] [CrossRef] [Green Version]
- Esquela-Kerscher, A.; Slack, F.J. Oncomirs-microRNAs with a role in cancer. Nat. Rev. Cancer 2006, 6, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Palmer, R.D.; Murray, M.J.; Saini, H.K.; van Dongen, S.; Abreu-Goodger, C.; Muralidhar, B.; Pett, M.R.; Thornton, C.M.; Nicholson, J.C.; Enright, A.J.; et al. Malignant germ cell tumors display common microRNA profiles resulting in global changes in expression of messenger RNA targets. Cancer Res. 2010, 70, 2911–2923. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillis, A.J.; Stoop, H.J.; Hersmus, R.; Oosterhuis, J.W.; Sun, Y.; Chen, C.; Guenther, S.; Sherlock, J.; Veltman, I.; Baeten, J.; et al. High-throughput microRNAome analysis in human germ cell tumours. J. Pathol. 2007, 213, 319–328. [Google Scholar] [CrossRef]
- Voorhoeve, P.M.; le Sage, C.; Schrier, M.; Gillis, A.J.; Stoop, H.; Nagel, R.; Liu, Y.P.; van Duijse, J.; Drost, J.; Griekspoor, A.; et al. A genetic screen implicates miRNA-372 and miRNA-373 as oncogenes in testicular germ cell tumors. Cell 2006, 124, 1169–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyoshi, N.; Ishii, H.; Nagano, H.; Haraguchi, N.; Dewi, D.L.; Kano, Y.; Nishikawa, S.; Tanemura, M.; Mimori, K.; Tanaka, F.; et al. Reprogramming of mouse and human cells to pluripotency using mature microRNAs. Cell Stem Cell 2011, 8, 633–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laurent, L.C. MicroRNAs in embryonic stem cells and early embryonic development. J. Cell. Mol. Med. 2008, 12, 2181–2188. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Melton, C.; Li, Y.P.; Shenoy, A.; Zhang, X.X.; Subramanyam, D.; Blelloch, R. miR-294/miR-302 promotes proliferation, suppresses G1-S restriction point, and inhibits ESC differentiation through separable mechanisms. Cell Rep. 2013, 4, 99–109. [Google Scholar] [CrossRef] [Green Version]
- Das, M.K.; Evensen, H.S.F.; Furu, K.; Haugen, T.B. miRNA-302s may act as oncogenes in human testicular germ cell tumours. Sci. Rep. 2019, 9, 9189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, T.; Wang, W.; Zhang, S.; Stewart, R.A.; Yu, W. Identifying tumor suppressors in genetic mosaics: The Drosophila lats gene encodes a putative protein kinase. Development 1995, 121, 1053–1063. [Google Scholar] [CrossRef] [PubMed]
- McPherson, J.P.; Tamblyn, L.; Elia, A.; Migon, E.; Shehabeldin, A.; Matysiak-Zablocki, E.; Lemmers, B.; Salmena, L.; Hakem, A.; Fish, J.; et al. Lats2/Kpm is required for embryonic development, proliferation control and genomic integrity. EMBO J. 2004, 23, 3677–3688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimizu, K.; Nihira, N.T.; Inuzuka, H.; Wei, W. Physiological functions of FBW7 in cancer and metabolism. Cell Signal 2018, 46, 15–22. [Google Scholar] [CrossRef]
- Liu, J.; Shi, H.; Li, X.; Chen, G.; Larsson, C.; Lui, W.O. miR2233p regulates cell growth and apoptosis via FBXW7 suggesting an oncogenic role in human testicular germ cell tumors. Int. J. Oncol. 2017, 50, 356–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lize, M.; Pilarski, S.; Dobbelstein, M. E2F1-inducible microRNA 449a/b suppresses cell proliferation and promotes apoptosis. Cell Death Differ. 2010, 17, 452–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozata, D.M.; Li, X.; Lee, L.; Liu, J.; Warsito, D.; Hajeri, P.; Hultman, I.; Fotouhi, O.; Marklund, S.; Ahrlund-Richter, L.; et al. Loss of miR-514a-3p regulation of PEG3 activates the NF-kappa B pathway in human testicular germ cell tumors. Cell Death Dis. 2017, 8, e2759. [Google Scholar] [CrossRef] [PubMed]
- De Martino, M.; Esposito, F.; Pellecchia, S.; Cortez Cardoso Penha, R.; Botti, G.; Fusco, A.; Chieffi, P. HMGA1-Regulating microRNAs Let-7a and miR-26a are Downregulated in Human Seminomas. Int. J. Mol. Sci. 2020, 21, 3014. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Young, J.; Prabhala, H.; Pan, E.; Mestdagh, P.; Muth, D.; Teruya-Feldstein, J.; Reinhardt, F.; Onder, T.T.; Valastyan, S.; et al. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. Nat. Cell Biol. 2010, 12, 247–256. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Yang, P.; Sun, T.; Li, D.; Xu, X.; Rui, Y.; Li, C.; Chong, M.; Ibrahim, T.; Mercatali, L.; et al. miR-126 and miR-126* repress recruitment of mesenchymal stem cells and inflammatory monocytes to inhibit breast cancer metastasis. Nat. Cell Biol. 2013, 15, 284–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Yan, L.X.; Wu, Q.N.; Du, Z.M.; Chen, J.; Liao, D.Z.; Huang, M.Y.; Hou, J.H.; Wu, Q.L.; Zeng, M.S.; et al. miR-125b is methylated and functions as a tumor suppressor by regulating the ETS1 proto-oncogene in human invasive breast cancer. Cancer Res. 2011, 71, 3552–3562. [Google Scholar] [CrossRef] [Green Version]
- Batool, A.; Wang, Y.Q.; Hao, X.X.; Chen, S.R.; Liu, Y.X. A miR-125b/CSF1-CX3CL1/tumor-associated macrophage recruitment axis controls testicular germ cell tumor growth. Cell Death Dis. 2018, 9, 962. [Google Scholar] [CrossRef] [PubMed]
- Port, M.; Glaesener, S.; Ruf, C.; Riecke, A.; Bokemeyer, C.; Meineke, V.; Honecker, F.; Abend, M. Micro-RNA expression in cisplatin resistant germ cell tumor cell lines. Mol. Cancer 2011, 10, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almstrup, K.; Lobo, J.; Morup, N.; Belge, G.; Rajpert-De Meyts, E.; Looijenga, L.H.J.; Dieckmann, K.P. Application of miRNAs in the diagnosis and monitoring of testicular germ cell tumours. Nat. Rev. Urol. 2020, 17, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Rosenkranz, D.; Han, C.T.; Roovers, E.F.; Zischler, H.; Ketting, R.F. Piwi proteins and piRNAs in mammalian oocytes and early embryos: From sample to sequence. Genom. Data 2015, 5, 309–313. [Google Scholar] [CrossRef] [Green Version]
- Roovers, E.F.; Rosenkranz, D.; Mahdipour, M.; Han, C.T.; He, N.; Chuva de Sousa Lopes, S.M.; van der Westerlaken, L.A.; Zischler, H.; Butter, F.; Roelen, B.A.; et al. Piwi proteins and piRNAs in mammalian oocytes and early embryos. Cell Rep. 2015, 10, 2069–2082. [Google Scholar] [CrossRef] [Green Version]
- Akkouche, A.; Mugat, B.; Barckmann, B.; Varela-Chavez, C.; Li, B.; Raffel, R.; Pelisson, A.; Chambeyron, S. Piwi Is Required during Drosophila Embryogenesis to License Dual-Strand piRNA Clusters for Transposon Repression in Adult Ovaries. Mol. Cell 2017, 66, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Andersen, P.R.; Tirian, L.; Vunjak, M.; Brennecke, J. A heterochromatin-dependent transcription machinery drives piRNA expression. Nature 2017, 549, 54–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aravin, A.A.; Hannon, G.J.; Brennecke, J. The Piwi-piRNA pathway provides an adaptive defense in the transposon arms race. Science 2007, 318, 761–764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czech, B.; Munafo, M.; Ciabrelli, F.; Eastwood, E.L.; Fabry, M.H.; Kneuss, E.; Hannon, G.J. piRNA-Guided Genome Defense: From Biogenesis to Silencing. Annu. Rev. Genet. 2018, 52, 131–157. [Google Scholar] [CrossRef]
- Ferreira, H.J.; Heyn, H.; Garcia del Muro, X.; Vidal, A.; Larriba, S.; Munoz, C.; Villanueva, A.; Esteller, M. Epigenetic loss of the PIWI/piRNA machinery in human testicular tumorigenesis. Epigenetics 2014, 9, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Heyn, H.; Ferreira, H.J.; Bassas, L.; Bonache, S.; Sayols, S.; Sandoval, J.; Esteller, M.; Larriba, S. Epigenetic disruption of the PIWI pathway in human spermatogenic disorders. PLoS ONE 2012, 7, e47892. [Google Scholar] [CrossRef] [Green Version]
- Rounge, T.B.; Furu, K.; Skotheim, R.I.; Haugen, T.B.; Grotmol, T.; Enerly, E. Profiling of the small RNA populations in human testicular germ cell tumors shows global loss of piRNAs. Mol. Cancer 2015, 14, 153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morup, N.; Stakaitis, R.; Golubickaite, I.; Riera, M.; Dalgaard, M.D.; Schierup, M.H.; Jorgensen, N.; Daugaard, G.; Juul, A.; Almstrup, K. Small RNAs in Seminal Plasma as Novel Biomarkers for Germ Cell Tumors. Cancers 2021, 13, 2346. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitz, S.U.; Grote, P.; Herrmann, B.G. Mechanisms of long noncoding RNA function in development and disease. Cell Mol. Life Sci. 2016, 73, 2491–2509. [Google Scholar] [CrossRef] [Green Version]
- Prensner, J.R.; Chinnaiyan, A.M. The emergence of lncRNAs in cancer biology. Cancer Discov. 2011, 1, 391–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bresesti, C.; Vezzoli, V.; Cangiano, B.; Bonomi, M. Long Non-Coding RNAs: Role in Testicular Cancers. Front. Oncol. 2021, 11, 605606. [Google Scholar] [CrossRef] [PubMed]
- Looijenga, L.H.; Gillis, A.J.; van Gurp, R.J.; Verkerk, A.J.; Oosterhuis, J.W. X inactivation in human testicular tumors. XIST expression and androgen receptor methylation status. Am. J. Pathol. 1997, 151, 581–590. [Google Scholar] [PubMed]
- Lobo, J.; Nunes, S.P.; Gillis, A.J.M.; Barros-Silva, D.; Miranda-Goncalves, V.; Berg, A.V.D.; Cantante, M.; Guimaraes, R.; Henrique, R.; Jeronimo, C.; et al. XIST-Promoter Demethylation as Tissue Biomarker for Testicular Germ Cell Tumors and Spermatogenesis Quality. Cancers 2019, 11, 1385. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Gan, Y.; Tan, Z.; Zhou, J.; Kitazawa, R.; Jiang, X.; Tang, Y.; Yang, J. TDRG1 functions in testicular seminoma are dependent on the PI3K/Akt/mTOR signaling pathway. Onco Targets Ther. 2016, 9, 409–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, D.; Wei, J.; Gan, Y.; Yang, J.; Jiang, X.; Kitazawa, R.; Xiang, Y.; Dai, Y.; Tang, Y. Testis developmental related gene 1 regulates the chemosensitivity of seminoma TCam-2 cells to cisplatin via autophagy. J. Cell. Mol. Med. 2019, 23, 7773–7784. [Google Scholar] [CrossRef] [PubMed]
- Gan, Y.; Wang, Y.; Tan, Z.; Zhou, J.; Kitazawa, R.; Jiang, X.; Tang, Y.; Yang, J. TDRG1 regulates chemosensitivity of seminoma TCam-2 cells to cisplatin via PI3K/Akt/mTOR signaling pathway and mitochondria-mediated apoptotic pathway. Cancer Biol. Ther. 2016, 17, 741–750. [Google Scholar] [CrossRef] [Green Version]
- Gan, Y.U.; Yang, J.; Wang, Y.; Tan, Z.; Jiang, X.; Tang, Y. In vitro study on shRNA-mediated reduction of testis developmental related gene 1 expression and its effects on the proliferation, invasion and apoptosis of NTERA-2 cells. Oncol. Lett. 2015, 10, 61–66. [Google Scholar] [CrossRef] [Green Version]
- Nonomura, N.; Miki, T.; Nishimura, K.; Kanno, N.; Kojima, Y.; Okuyama, A. Altered imprinting of the H19 and insulin-like growth factor II genes in testicular tumors. J. Urol. 1997, 157, 1977–1979. [Google Scholar] [CrossRef]
- Mishina, M.; Ogawa, O.; Kinoshita, H.; Oka, H.; Okumura, K.; Mitsumori, K.; Kakehi, Y.; Reeve, A.E.; Yoshida, O. Equivalent parental distribution of frequently lost alleles and biallelic expression of the H19 gene in human testicular germ cell tumors. Jpn. J. Cancer Res. 1996, 87, 816–823. [Google Scholar] [CrossRef]
- Kawakami, T.; Zhang, C.; Okada, Y.; Okamoto, K. Erasure of methylation imprint at the promoter and CTCF-binding site upstream of H19 in human testicular germ cell tumors of adolescents indicate their fetal germ cell origin. Oncogene 2006, 25, 3225–3236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sievers, S.; Alemazkour, K.; Zahn, S.; Perlman, E.J.; Gillis, A.J.; Looijenga, L.H.; Gobel, U.; Schneider, D.T. IGF2/H19 imprinting analysis of human germ cell tumors (GCTs) using the methylation-sensitive single-nucleotide primer extension method reflects the origin of GCTs in different stages of primordial germ cell development. Genes Chromosomes Cancer 2005, 44, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Verkerk, A.J.; Ariel, I.; Dekker, M.C.; Schneider, T.; van Gurp, R.J.; de Groot, N.; Gillis, A.J.; Oosterhuis, J.W.; Hochberg, A.A.; Looijenga, L.H. Unique expression patterns of H19 in human testicular cancers of different etiology. Oncogene 1997, 14, 95–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, J.; Gan, Y.; Peng, D.; Jiang, X.; Kitazawa, R.; Xiang, Y.; Dai, Y.; Tang, Y.; Yang, J. Long non-coding RNA H19 promotes TDRG1 expression and cisplatin resistance by sequestering miRNA-106b-5p in seminoma. Cancer Med. 2018, 7, 6247–6257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazar, J.; Zhao, W.; Khalil, A.M.; Lee, B.; Shelley, J.; Govindarajan, S.S.; Yamamoto, F.; Ratnam, M.; Aftab, M.N.; Collins, S.; et al. The functional characterization of long noncoding RNA SPRY4-IT1 in human melanoma cells. Oncotarget 2014, 5, 8959–8969. [Google Scholar] [CrossRef] [Green Version]
- Das, M.K.; Furu, K.; Evensen, H.F.; Haugen, O.P.; Haugen, T.B. Knockdown of SPRY4 and SPRY4-IT1 inhibits cell growth and phosphorylation of Akt in human testicular germ cell tumours. Sci. Rep. 2018, 8, 2462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, M.; Tian, H.; Cao, Y.X.; He, X.; Chen, L.; Song, X.; Ping, P.; Huang, H.; Sun, F. Downregulation of miR-320a/383-sponge-like long non-coding RNA NLC1-C (narcolepsy candidate-region 1 genes) is associated with male infertility and promotes testicular embryonal carcinoma cell proliferation. Cell Death Dis. 2015, 6, e1960. [Google Scholar] [CrossRef] [Green Version]
- Ghafouri-Fard, S.; Dashti, S.; Taheri, M. The HOTTIP (HOXA transcript at the distal tip) lncRNA: Review of oncogenic roles in human. Biomed. Pharmacother. 2020, 127, 110158. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Zhou, L.L.; Zhang, Y.Q.; Ni, L.Y. Long noncoding RNA HOTTIP is associated with male infertility and promotes testicular embryonal carcinoma cell proliferation. Mol. Genet. Genom. Med. 2019, 7, e870. [Google Scholar] [CrossRef] [Green Version]
- Taoka, M.; Nobe, Y.; Yamaki, Y.; Sato, K.; Ishikawa, H.; Izumikawa, K.; Yamauchi, Y.; Hirota, K.; Nakayama, H.; Takahashi, N.; et al. Landscape of the complete RNA chemical modifications in the human 80S ribosome. Nucleic Acids Res. 2018, 46, 9289–9298. [Google Scholar] [CrossRef] [PubMed]
- Ruggero, D. Translational control in cancer etiology. Cold Spring Harb Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Ban, N.; Nissen, P.; Hansen, J.; Moore, P.B.; Steitz, T.A. The complete atomic structure of the large ribosomal subunit at 2.4 A resolution. Science 2000, 289, 905–920. [Google Scholar] [CrossRef] [PubMed]
- McStay, B. Nucleolar organizer regions: Genomic ‘dark matter’ requiring illumination. Genes Dev. 2016, 30, 1598–1610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arabi, A.; Wu, S.; Ridderstrale, K.; Bierhoff, H.; Shiue, C.; Fatyol, K.; Fahlen, S.; Hydbring, P.; Soderberg, O.; Grummt, I.; et al. c-Myc associates with ribosomal DNA and activates RNA polymerase I transcription. Nat. Cell Biol. 2005, 7, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Budde, A.; Grummt, I. p53 represses ribosomal gene transcription. Oncogene 1999, 18, 1119–1124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugimoto, M.; Kuo, M.L.; Roussel, M.F.; Sherr, C.J. Nucleolar Arf tumor suppressor inhibits ribosomal RNA processing. Mol. Cell 2003, 11, 415–424. [Google Scholar] [CrossRef]
- Cavanaugh, A.H.; Hempel, W.M.; Taylor, L.J.; Rogalsky, V.; Todorov, G.; Rothblum, L.I. Activity of RNA polymerase I transcription factor UBF blocked by Rb gene product. Nature 1995, 374, 177–180. [Google Scholar] [CrossRef]
- Neerman-Arbez, M.; DeLozier-Blanchet, C.D.; Bolle, J.F.; Rondez, R.; Morris, M. High incidence of ectopic nucleolar organizer regions in human testicular tumors. Cancer Genet. Cytogenet. 1993, 65, 58–63. [Google Scholar] [CrossRef]
- DeLozier-Blanchet, C.D.; Walt, H.; Engel, E. Ectopic nucleolus organizer regions (NORs) in human testicular tumors. Cytogenet. Genome Res. 1986, 41, 107–113. [Google Scholar] [CrossRef]
- Truitt, M.L.; Ruggero, D. New frontiers in translational control of the cancer genome. Nat. Rev. Cancer 2016, 16, 288–304. [Google Scholar] [CrossRef] [Green Version]
- Drygin, D.; Lin, A.; Bliesath, J.; Ho, C.B.; O’Brien, S.E.; Proffitt, C.; Omori, M.; Haddach, M.; Schwaebe, M.K.; Siddiqui-Jain, A.; et al. Targeting RNA polymerase I with an oral small molecule CX-5461 inhibits ribosomal RNA synthesis and solid tumor growth. Cancer Res. 2011, 71, 1418–1430. [Google Scholar] [CrossRef] [Green Version]
- Peltonen, K.; Colis, L.; Liu, H.; Jaamaa, S.; Moore, H.M.; Enback, J.; Laakkonen, P.; Vaahtokari, A.; Jones, R.J.; af Hallstrom, T.M.; et al. Identification of novel p53 pathway activating small-molecule compounds reveals unexpected similarities with known therapeutic agents. PLoS ONE 2010, 5, e12996. [Google Scholar] [CrossRef] [PubMed]
- Sasiela, C.A.; Stewart, D.H.; Kitagaki, J.; Safiran, Y.J.; Yang, Y.; Weissman, A.M.; Oberoi, P.; Davydov, I.V.; Goncharova, E.; Beutler, J.A.; et al. Identification of inhibitors for MDM2 ubiquitin ligase activity from natural product extracts by a novel high-throughput electrochemiluminescent screen. J. Biomol. Screen. 2008, 13, 229–237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caggiano, C.; Guida, E.; Todaro, F.; Bielli, P.; Mori, M.; Ghirga, F.; Quaglio, D.; Botta, B.; Moretti, F.; Grimaldi, P.; et al. Sempervirine inhibits RNA polymerase I transcription independently from p53 in tumor cells. Cell Death Discov. 2020, 6, 111. [Google Scholar] [CrossRef] [PubMed]
- Dvinge, H. Regulation of alternative mRNA splicing: Old players and new perspectives. FEBS Lett. 2018, 592, 2987–3006. [Google Scholar] [CrossRef]
- Baralle, F.E.; Giudice, J. Alternative splicing as a regulator of development and tissue identity. Nat. Rev. Mol. Cell Biol. 2017, 18, 437–451. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Park, J.Y.; Zheng, D.; Hoque, M.; Yehia, G.; Tian, B. Alternative cleavage and polyadenylation in spermatogenesis connects chromatin regulation with post-transcriptional control. BMC Biol. 2016, 14, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naro, C.; Pellegrini, L.; Jolly, A.; Farini, D.; Cesari, E.; Bielli, P.; de la Grange, P.; Sette, C. Functional Interaction between U1snRNP and Sam68 Insures Proper 3’ End Pre-mRNA Processing during Germ Cell Differentiation. Cell Rep. 2019, 26, 2929–2941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naro, C.; Jolly, A.; Di Persio, S.; Bielli, P.; Setterblad, N.; Alberdi, A.J.; Vicini, E.; Geremia, R.; De la Grange, P.; Sette, C. An Orchestrated Intron Retention Program in Meiosis Controls Timely Usage of Transcripts during Germ Cell Differentiation. Dev. Cell 2017, 41, 82–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazin, P.V.; Khaitovich, P.; Cardoso-Moreira, M.; Kaessmann, H. Alternative splicing during mammalian organ development. Nat. Genet. 2021, 53, 925–934. [Google Scholar] [CrossRef] [PubMed]
- Naro, C.; Cesari, E.; Sette, C. Splicing regulation in brain and testis: Common themes for highly specialized organs. Cell Cycle 2021, 20, 480–489. [Google Scholar] [CrossRef] [PubMed]
- Zhu, R.; Heaney, J.; Nadeau, J.H.; Ali, S.; Matin, A. Deficiency of splicing factor 1 suppresses the occurrence of testicular germ cell tumors. Cancer Res. 2010, 70, 7264–7272. [Google Scholar] [CrossRef] [Green Version]
- Novikov, L.; Park, J.W.; Chen, H.; Klerman, H.; Jalloh, A.S.; Gamble, M.J. QKI-mediated alternative splicing of the histone variant MacroH2A1 regulates cancer cell proliferation. Mol. Cell Biol. 2011, 31, 4244–4255. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Gray, D.R.; Robbins, A.K.; Crowgey, E.L.; Chanock, S.J.; Greene, M.H.; McGlynn, K.A.; Nathanson, K.; Turnbull, C.; Wang, Z.; et al. Subphenotype meta-analysis of testicular cancer genome-wide association study data suggests a role for RBFOX family genes in cryptorchidism susceptibility. Hum. Reprod. 2018, 33, 967–977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.J.; Li, Z.T.; Shen, K.J.; Chen, L.; Xu, D.F.; Gao, Y. Characterization of progression-related alternative splicing events in testicular germ cell tumors. Asian J. Androl. 2021, 23, 259–265. [Google Scholar] [CrossRef] [PubMed]
- Kempkensteffen, C.; Hinz, S.; Krause, H.; Jager, T.; Kollermann, J.; Weikert, S.; Christoph, F.; Schostak, M.; Miller, K.; Schrader, M. Expression of splicing variants of the inhibitor of apoptosis livin in testicular germ cell tumors. Tumour Biol. 2008, 29, 76–82. [Google Scholar] [CrossRef]
- Gainetdinov, I.V.; Skvortsova, Y.V.; Stukacheva, E.A.; Bychenko, O.S.; Kondratieva, S.A.; Zinovieva, M.V.; Azhikina, T.L. Expression profiles of PIWIL2 short isoforms differ in testicular germ cell tumors of various differentiation subtypes. PLoS ONE 2014, 9, e112528. [Google Scholar] [CrossRef] [Green Version]
- Naro, C.; Barbagallo, F.; Chieffi, P.; Bourgeois, C.F.; Paronetto, M.P.; Sette, C. The centrosomal kinase NEK2 is a novel splicing factor kinase involved in cell survival. Nucleic Acids Res. 2014, 42, 3218–3227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rhee, K.; Wolgemuth, D.J. The NIMA-related kinase 2, Nek2, is expressed in specific stages of the meiotic cell cycle and associates with meiotic chromosomes. Development 1997, 124, 2167–2177. [Google Scholar] [CrossRef]
- Barbagallo, F.; Paronetto, M.P.; Franco, R.; Chieffi, P.; Dolci, S.; Fry, A.M.; Geremia, R.; Sette, C. Increased expression and nuclear localization of the centrosomal kinase Nek2 in human testicular seminomas. J. Pathol. 2009, 217, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Di Agostino, S.; Rossi, P.; Geremia, R.; Sette, C. The MAPK pathway triggers activation of Nek2 during chromosome condensation in mouse spermatocytes. Development 2002, 129, 1715–1727. [Google Scholar] [CrossRef] [PubMed]
- Di Agostino, S.; Fedele, M.; Chieffi, P.; Fusco, A.; Rossi, P.; Geremia, R.; Sette, C. Phosphorylation of high-mobility group protein A2 by Nek2 kinase during the first meiotic division in mouse spermatocytes. Mol. Biol. Cell 2004, 15, 1224–1232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fung, M.K.; Cheung, H.W.; Wong, H.L.; Yuen, H.F.; Ling, M.T.; Chan, K.W.; Wong, Y.C.; Cheung, A.L.; Wang, X. MAD2 expression and its significance in mitotic checkpoint control in testicular germ cell tumour. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2007, 1773, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Yin, F.; Du, Y.; Hu, W.; Qiao, T.; Ding, J.; Wu, K.; Liu, Z.; Fan, D. Mad2beta, an alternative variant of Mad2 reducing mitotic arrest and apoptosis induced by adriamycin in gastric cancer cells. Life Sci. 2006, 78, 1277–1286. [Google Scholar] [CrossRef]
- Lopez-Saavedra, A.; Ramirez-Otero, M.; Diaz-Chavez, J.; Caceres-Gutierrez, R.; Justo-Garrido, M.; Andonegui, M.A.; Mendoza, J.; Downie-Ruiz, A.; Cortes-Gonzalez, C.; Reynoso, N.; et al. MAD2gamma, a novel MAD2 isoform, reduces mitotic arrest and is associated with resistance in testicular germ cell tumors. Cell Cycle 2016, 15, 2066–2076. [Google Scholar] [CrossRef]
- Liu, Q.; Hirohashi, Y.; Du, X.; Greene, M.I.; Wang, Q. Nek2 targets the mitotic checkpoint proteins Mad2 and Cdc20: A mechanism for aneuploidy in cancer. Exp. Mol. Pathol. 2010, 88, 225–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bates, M.; Furlong, F.; Gallagher, M.F.; Spillane, C.D.; McCann, A.; O’Toole, S.; O’Leary, J.J. Too MAD or not MAD enough: The duplicitous role of the spindle assembly checkpoint protein MAD2 in cancer. Cancer Lett. 2020, 469, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, A.V.; Singh, A.; Bousbaa, H.; Ferreira, D.; Sarmento, B.; Amiji, M.M. Overcoming cisplatin resistance in non-small cell lung cancer with Mad2 silencing siRNA delivered systemically using EGFR-targeted chitosan nanoparticles. Acta Biomater. 2017, 47, 71–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vucic, D.; Stennicke, H.R.; Pisabarro, M.T.; Salvesen, G.S.; Dixit, V.M. ML-IAP, a novel inhibitor of apoptosis that is preferentially expressed in human melanomas. Curr. Biol. 2000, 10, 1359–1366. [Google Scholar] [CrossRef] [Green Version]
- Ashhab, Y.; Alian, A.; Polliack, A.; Panet, A.; Ben Yehuda, D. Two splicing variants of a new inhibitor of apoptosis gene with different biological properties and tissue distribution pattern. FEBS Lett. 2001, 495, 56–60. [Google Scholar] [CrossRef] [Green Version]
- Schmollinger, J.C.; Dranoff, G. Targeting melanoma inhibitor of apoptosis protein with cancer immunotherapy. Apoptosis 2004, 9, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koslowski, M.; Tureci, O.; Bell, C.; Krause, P.; Lehr, H.A.; Brunner, J.; Seitz, G.; Nestle, F.O.; Huber, C.; Sahin, U. Multiple splice variants of lactate dehydrogenase C selectively expressed in human cancer. Cancer Res. 2002, 62, 6750–6755. [Google Scholar] [PubMed]
- Bonatelli, M.; Silva, E.C.A.; Carcano, F.M.; Zaia, M.G.; Lopes, L.F.; Scapulatempo-Neto, C.; Pinheiro, C. The Warburg Effect Is Associated With Tumor Aggressiveness in Testicular Germ Cell Tumors. Front. Endocrinol. 2019, 10, 417. [Google Scholar] [CrossRef]
- Kozlovski, I.; Siegfried, Z.; Amar-Schwartz, A.; Karni, R. The role of RNA alternative splicing in regulating cancer metabolism. Hum. Genet. 2017, 136, 1113–1127. [Google Scholar] [CrossRef]
- Courtens, J.L.; Ploen, L. Improvement of spermatogenesis in adult cryptorchid rat testis by intratesticular infusion of lactate. Biol. Reprod. 1999, 61, 154–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erkkila, K.; Aito, H.; Aalto, K.; Pentikainen, V.; Dunkel, L. Lactate inhibits germ cell apoptosis in the human testis. Mol. Hum. Reprod. 2002, 8, 109–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.H.; Lin, T.T.; Wu, Y.P.; Li, X.D.; Chen, S.H.; Xue, X.Y.; Wei, Y.; Zheng, Q.S.; Huang, J.B.; Xu, N. Identification of key genes and pathways in seminoma by bioinformatics analysis. Onco Targets Ther. 2019, 12, 3683–3693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Muse, T.; Aguilera, A. R Loops: From Physiological to Pathological Roles. Cell 2019, 179, 604–618. [Google Scholar] [CrossRef]
- Brambati, A.; Zardoni, L.; Nardini, E.; Pellicioli, A.; Liberi, G. The dark side of RNA:DNA hybrids. Mutat. Res. Rev. Mutat. Res. 2020, 784, 108300. [Google Scholar] [CrossRef] [PubMed]
- Niehrs, C.; Luke, B. Regulatory R-loops as facilitators of gene expression and genome stability. Nat. Rev. Mol. Cell Biol. 2020, 21, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Sanz, L.A.; Chedin, F. High-resolution, strand-specific R-loop mapping via S9.6-based DNA-RNA immunoprecipitation and high-throughput sequencing. Nat. Protoc. 2019, 14, 1734–1755. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.X.; Grunseich, C.; Fox, J.; Burdick, J.; Zhu, Z.; Ravazian, N.; Hafner, M.; Cheung, V.G. Human proteins that interact with RNA/DNA hybrids. Genome Res. 2018, 28, 1405–1414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boros-Olah, B.; Dobos, N.; Hornyak, L.; Szabo, Z.; Karanyi, Z.; Halmos, G.; Roszik, J.; Szekvolgyi, L. Drugging the R-loop interactome: RNA-DNA hybrid binding proteins as targets for cancer therapy. DNA Repair 2019, 84, 102642. [Google Scholar] [CrossRef] [PubMed]
- Groh, M.; Albulescu, L.O.; Cristini, A.; Gromak, N. Senataxin: Genome Guardian at the Interface of Transcription and Neurodegeneration. J. Mol. Biol. 2017, 429, 3181–3195. [Google Scholar] [CrossRef] [PubMed]
- Yeo, A.J.; Becherel, O.J.; Luff, J.E.; Cullen, J.K.; Wongsurawat, T.; Jenjaroenpun, P.; Kuznetsov, V.A.; McKinnon, P.J.; Lavin, M.F. R-loops in proliferating cells but not in the brain: Implications for AOA2 and other autosomal recessive ataxias. PLoS ONE 2014, 9, e90219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catford, S.R.; O’Bryan, M.K.; McLachlan, R.I.; Delatycki, M.B.; Rombauts, L. Germ cell arrest associated with aSETX mutation in ataxia oculomotor apraxia type 2. Reprod. Biomed. Online 2019, 38, 961–965. [Google Scholar] [CrossRef] [PubMed]
- Becherel, O.J.; Yeo, A.J.; Stellati, A.; Heng, E.Y.; Luff, J.; Suraweera, A.M.; Woods, R.; Fleming, J.; Carrie, D.; McKinney, K.; et al. Senataxin plays an essential role with DNA damage response proteins in meiotic recombination and gene silencing. PLoS Genet. 2013, 9, e1003435. [Google Scholar] [CrossRef] [Green Version]
- Yuce, O.; West, S.C. Senataxin, defective in the neurodegenerative disorder ataxia with oculomotor apraxia 2, lies at the interface of transcription and the DNA damage response. Mol. Cell Biol. 2013, 33, 406–417. [Google Scholar] [CrossRef] [Green Version]
- Skourti-Stathaki, K.; Proudfoot, N.J.; Gromak, N. Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol. Cell 2011, 42, 794–805. [Google Scholar] [CrossRef] [PubMed]
- Gromak, N.; West, S.; Proudfoot, N.J. Pause sites promote transcriptional termination of mammalian RNA polymerase II. Mol. Cell Biol. 2006, 26, 3986–3996. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barlow, C.; Liyanage, M.; Moens, P.B.; Tarsounas, M.; Nagashima, K.; Brown, K.; Rottinghaus, S.; Jackson, S.P.; Tagle, D.; Ried, T.; et al. Atm deficiency results in severe meiotic disruption as early as leptonema of prophase I. Development 1998, 125, 4007–4017. [Google Scholar] [CrossRef] [PubMed]
- Barchi, M.; Roig, I.; Di Giacomo, M.; de Rooij, D.G.; Keeney, S.; Jasin, M. ATM promotes the obligate XY crossover and both crossover control and chromosome axis integrity on autosomes. PLoS Genet. 2008, 4, e1000076. [Google Scholar] [CrossRef] [Green Version]
- Katyal, S.; el-Khamisy, S.F.; Russell, H.R.; Li, Y.; Ju, L.; Caldecott, K.W.; McKinnon, P.J. TDP1 facilitates chromosomal single-strand break repair in neurons and is neuroprotective in vivo. EMBO J. 2007, 26, 4720–4731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatia, V.; Barroso, S.I.; Garcia-Rubio, M.L.; Tumini, E.; Herrera-Moyano, E.; Aguilera, A. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature 2014, 511, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Hatchi, E.; Skourti-Stathaki, K.; Ventz, S.; Pinello, L.; Yen, A.; Kamieniarz-Gdula, K.; Dimitrov, S.; Pathania, S.; McKinney, K.M.; Eaton, M.L.; et al. BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol. Cell 2015, 57, 636–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sollier, J.; Stork, C.T.; Garcia-Rubio, M.L.; Paulsen, R.D.; Aguilera, A.; Cimprich, K.A. Transcription-coupled nucleotide excision repair factors promote R-loop-induced genome instability. Mol. Cell 2014, 56, 777–785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Rubio, M.L.; Perez-Calero, C.; Barroso, S.I.; Tumini, E.; Herrera-Moyano, E.; Rosado, I.V.; Aguilera, A. The Fanconi Anemia Pathway Protects Genome Integrity from R-loops. PLoS Genet. 2015, 11, e1005674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwab, R.A.; Nieminuszczy, J.; Shah, F.; Langton, J.; Lopez Martinez, D.; Liang, C.C.; Cohn, M.A.; Gibbons, R.J.; Deans, A.J.; Niedzwiedz, W. The Fanconi Anemia Pathway Maintains Genome Stability by Coordinating Replication and Transcription. Mol. Cell 2015, 60, 351–361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatia, V.; Herrera-Moyano, E.; Aguilera, A.; Gomez-Gonzalez, B. The Role of Replication-Associated Repair Factors on R-Loops. Genes 2017, 8, 171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinaldi, C.; Pizzul, P.; Longhese, M.P.; Bonetti, D. Sensing R-Loop-Associated DNA Damage to Safeguard Genome Stability. Front. Cell Dev. Biol. 2020, 8, 618157. [Google Scholar] [CrossRef] [PubMed]
- Cavallo, F.; Graziani, G.; Antinozzi, C.; Feldman, D.R.; Houldsworth, J.; Bosl, G.J.; Chaganti, R.S.; Moynahan, M.E.; Jasin, M.; Barchi, M. Reduced proficiency in homologous recombination underlies the high sensitivity of embryonal carcinoma testicular germ cell tumors to Cisplatin and poly (adp-ribose) polymerase inhibition. PLoS ONE 2012, 7, e51563. [Google Scholar] [CrossRef] [Green Version]
- Welsh, C.; Day, R.; McGurk, C.; Masters, J.R.; Wood, R.D.; Koberle, B. Reduced levels of XPA, ERCC1 and XPF DNA repair proteins in testis tumor cell lines. Int. J. Cancer 2004, 110, 352–361. [Google Scholar] [CrossRef]
- Caggiano, C.; Cavallo, F.; Giannattasio, T.; Cappelletti, G.; Rossi, P.; Grimaldi, P.; Feldman, D.R.; Jasin, M.; Barchi, M. Testicular Germ Cell Tumors Acquire Cisplatin Resistance by Rebalancing the Usage of DNA Repair Pathways. Cancers 2021, 13, 787. [Google Scholar] [CrossRef]
- Cavallo, F.; Feldman, D.R.; Barchi, M. Revisiting DNA damage repair, p53-mediated apoptosis and cisplatin sensitivity in germ cell tumors. Int. J. Dev. Biol. 2013, 57, 273–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bartkova, J.; Rajpert-De Meyts, E.; Skakkebaek, N.E.; Lukas, J.; Bartek, J. DNA damage response in human testes and testicular germ cell tumours: Biology and implications for therapy. Int. J. Androl. 2007, 30, 282–291. [Google Scholar] [CrossRef]
- Gan, W.; Guan, Z.; Liu, J.; Gui, T.; Shen, K.; Manley, J.L.; Li, X. R-loop-mediated genomic instability is caused by impairment of replication fork progression. Genes Dev. 2011, 25, 2041–2056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schenk, P.W.; Stoop, H.; Bokemeyer, C.; Mayer, F.; Stoter, G.; Oosterhuis, J.W.; Wiemer, E.; Looijenga, L.H.; Nooter, K. Resistance to platinum-containing chemotherapy in testicular germ cell tumors is associated with downregulation of the protein kinase SRPK1. Neoplasia 2004, 6, 297–301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koizumi, J.; Okamoto, Y.; Onogi, H.; Mayeda, A.; Krainer, A.R.; Hagiwara, M. The subcellular localization of SF2/ASF is regulated by direct interaction with SR protein kinases (SRPKs). J. Biol. Chem. 1999, 274, 11125–11131. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Yang, D.; Ding, J.H.; Wang, W.; Chu, P.H.; Dalton, N.D.; Wang, H.Y.; Bermingham, J.R., Jr.; Ye, Z.; Liu, F.; et al. ASF/SF2-regulated CaMKIIdelta alternative splicing temporally reprograms excitation-contraction coupling in cardiac muscle. Cell 2005, 120, 59–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
| RNA Molecule | Expression Level | Function/s | References |
|---|---|---|---|
| miRNA | |||
| miR302/367 | Upregulated | OncomiR (upregulation of SPRY4 expression and MAPK/ERK pathway) | [29] |
| miR371/3 | Upregulated | OncomiR (downregulation of tumor suppressor LATS2) | [25] |
| miR223-3p | Upregulated | OncomiR (downregulation of tumor suppressor FBXW7) | [33] |
| miR449a-b | Downregulated | Tumor suppressor (upregulation of CDK6) | [34] |
| miR125b | Downregulated | Tumor suppressor (upregulation of tumor-derived chemokine CSF1 and CX3CL1; macrophage recruitment) | [40] |
| miR506/14 | Downregulated | Tumor suppressor (upregulation of PEG and NF-kB pathway) | [35] |
| Let7/26a | Downregulated | Tumor suppressor (HMGA1 upregulation) | [36] |
| piRNA | |||
| piRNAs | downregulated | [51] | |
| lncRNA | |||
| XIST | Upregulated in seminoma | [58] | |
| TDRG1 | Upregulated in seminoma | Onco-lncRNA | [59,60,61,62] |
| H19 | upregulated | Onco-lncRNA | [63,64,65,66,67] |
| SPRY4-IT1 | upregulated | Onco-lncRNA | [70] |
| NLC1-C | accumulated in the nucleus | Onco-lncRNA | [71] |
| HOTTIP | upregulated | Onco-lncRNA | [73] |
| RNA splicing variant | |||
| MAD2β and MAD2γ | upregulated | Cell cycle progression | [108,109] |
| Livin α and β | Upregulated in seminoma | Cell survival and apoptosis | [100] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barchi, M.; Bielli, P.; Dolci, S.; Rossi, P.; Grimaldi, P. Non-Coding RNAs and Splicing Activity in Testicular Germ Cell Tumors. Life 2021, 11, 736. https://doi.org/10.3390/life11080736
Barchi M, Bielli P, Dolci S, Rossi P, Grimaldi P. Non-Coding RNAs and Splicing Activity in Testicular Germ Cell Tumors. Life. 2021; 11(8):736. https://doi.org/10.3390/life11080736
Chicago/Turabian StyleBarchi, Marco, Pamela Bielli, Susanna Dolci, Pellegrino Rossi, and Paola Grimaldi. 2021. "Non-Coding RNAs and Splicing Activity in Testicular Germ Cell Tumors" Life 11, no. 8: 736. https://doi.org/10.3390/life11080736
APA StyleBarchi, M., Bielli, P., Dolci, S., Rossi, P., & Grimaldi, P. (2021). Non-Coding RNAs and Splicing Activity in Testicular Germ Cell Tumors. Life, 11(8), 736. https://doi.org/10.3390/life11080736