
Influencing Factors and Molecular Pathogenesis of Sarcopenia and Osteosarcopenia in Chronic Liver Disease



Abstract
1. Introduction
2. Materials and Methods Sarcopenia in Chronic Liver Disease
2.1. Assessment Criteria for Sarcopenia
2.2. Prevalence and Clinical Significance of Sarcopenia in Patients with Chronic Liver Disease
2.3. Pathogenic Mechanism of Sarcopenia
2.3.1. Imbalance of Protein Anabolism and Catabolism
2.3.2. Suppressed Muscle Growth by Decreased IGF-1 Levels
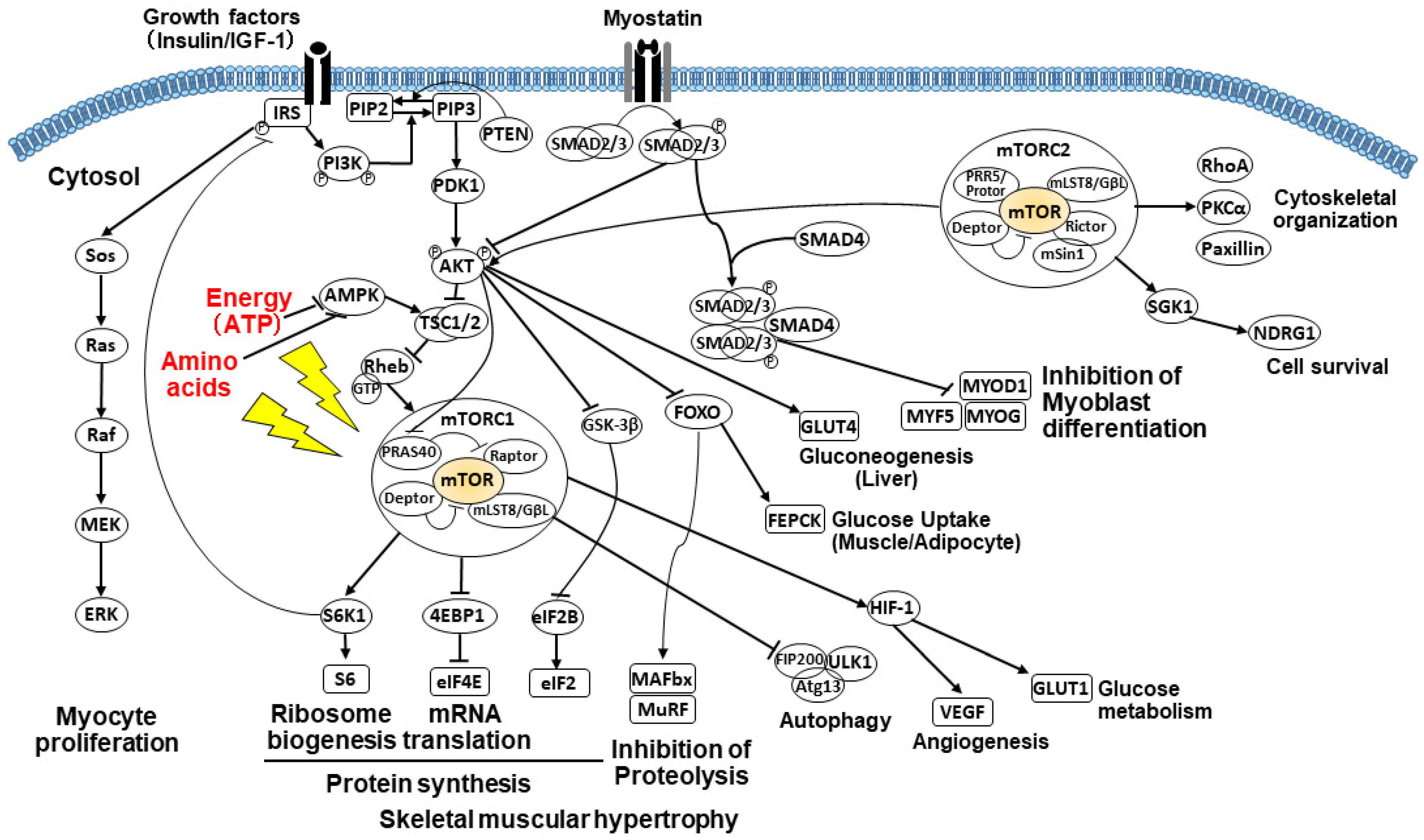
2.3.3. PI3K/AKT/mTOR Signaling Pathway
2.3.4. Impact of Amino Acids on the mTOR Pathway
2.3.5. Impact of Energy on the mTOR Pathway
2.3.6. Alterations of Glucose Metabolism and Gluconeogenesis
2.3.7. Suppressed Muscle Growth by Elevated Myostatin and Increased Autophagy
2.3.8. Impaired Molecular Functions via Hyperammonemia
2.3.9. Impact of Exercise on Muscle Anabolism
2.3.10. Decreased Anabolic Hormone Testosterone
2.3.11. Other Factors Influencing Sarcopenia
3. Osteosarcopenia in Chronic Liver Disease
3.1. Prevalence and Clinical Significance of Osteosarcopenia in Patients with Chronic Liver Disease
3.2. Pathogenic Mechanisms of Osteosarcopenia: Relationship between Muscle and Bone
3.2.1. Mechanical Factors
3.2.2. Genetic Factors
3.2.3. Chronic Inflammation
3.2.4. Myokines
3.2.5. Osteokines
3.2.6. Vitamin D
3.3. Treatment for Osteosarcopenia
3.3.1. Physical Exercise
3.3.2. Nutritional Interventions
3.3.3. Pharmacological Treatment
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cruz-Jentoft, A.J.; Baeyens, J.P.; Bauer, J.M.; Boirie, Y.; Cederholm, T.; Landi, F.; Martin, F.C.; Michel, J.P.; Rolland, Y.; Schneider, S.M.; et al. Sarcopenia: European consensus on definition and diagnosis: Report of the European Working Group on Sarcopenia in Older People. Age Ageing 2010, 39, 412–423. [Google Scholar] [CrossRef]
- Cruz-Jentoft, A.J.; Bahat, G.; Bauer, J.; Boirie, Y.; Bruyère, O.; Cederholm, T.; Cooper, C.; Landi, F.; Rolland, Y.; Sayer, A.A.; et al. Sarcopenia: Revised European consensus on definition and diagnosis. Age Ageing 2019, 48, 16–31. [Google Scholar] [CrossRef]
- Chen, L.K.; Liu, L.K.; Woo, J.; Assantachai, P.; Auyeung, T.W.; Bahyah, K.S.; Chou, M.Y.; Chen, L.Y.; Hsu, P.S.; Krairit, O.; et al. Sarcopenia in Asia: Consensus report of the Asian Working Group for Sarcopenia. J. Am. Med. Dir. Assoc. 2014, 15, 95–101. [Google Scholar] [CrossRef]
- Chen, L.K.; Woo, J.; Assantachai, P.; Auyeung, T.W.; Chou, M.Y.; Iijima, K.; Jang, H.C.; Kang, L.; Kim, M.; Kim, S.; et al. Asian Working Group for Sarcopenia: 2019 Consensus Update on Sarcopenia Diagnosis and Treatment. J. Am. Med. Dir. Assoc. 2020, 21, 300–307. [Google Scholar] [CrossRef]
- Nishikawa, H.; Shiraki, M.; Hiramatsu, A.; Moriya, K.; Hino, K.; Nishiguchi, S. Japan Society of Hepatology guidelines for sarcopenia in liver disease (1st edition): Recommendation from the working group for creation of sarcopenia assessment criteria. Hepatol. Res. 2016, 46, 951–963. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, I.H. Summary comments: Epidemiological and methodological problems in determining nutritional status of older persons. Am. J. Clin. Nutr. 1989, 50, 1231–1233. [Google Scholar] [CrossRef]
- Kim, Y. Emerging Treatment Options for Sarcopenia in Chronic Liver Disease. Life 2021, 11, 250. [Google Scholar] [CrossRef]
- Dasarathy, S.; Merli, M. Sarcopenia from mechanism to diagnosis and treatment in liver disease. J. Hepatol. 2016, 65, 1232–1244. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Kang, S.H.; Kim, M.Y.; Baik, S.K. Prognostic value of sarcopenia in patients with liver cirrhosis: A systematic review and meta-analysis. PLoS ONE 2017, 12, e0186990. [Google Scholar] [CrossRef]
- Hanai, T.; Shiraki, M.; Nishimura, K.; Ohnishi, S.; Imai, K.; Suetsugu, A.; Takai, K.; Shimizu, M.; Moriwaki, H. Sarcopenia impairs prognosis of patients with liver cirrhosis. Nutrition 2015, 31, 193–199. [Google Scholar] [CrossRef]
- Kobayashi, K.; Imagama, S.; Ando, K.; Machino, M.; Ota, K.; Tanaka, S.; Morozumi, M.; Kanbara, S.; Ito, S.; Ishiguro, N.; et al. Epidemiology and effect on physical function of osteosarcopenia in community-dwelling elderly people in Japan. Mod. Rheumatol. 2020, 30, 592–597. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.J.; Wang, Y.; Zhan, J.K.; Tang, Z.Y.; He, J.Y.; Tan, P.; Deng, H.Q.; Huang, W.; Liu, Y.S. Sarco-Osteoporosis: Prevalence and Association with Frailty in Chinese Community-Dwelling Older Adults. Int. J. Endocrinol. 2015, 2015, 482940. [Google Scholar] [CrossRef] [PubMed]
- Park, K.S.; Lee, G.Y.; Seo, Y.M.; Seo, S.H.; Yoo, J.I. Disability, Frailty and Depression in the community-dwelling older adults with Osteosarcopenia. BMC. Geriatr. 2021, 21, 69. [Google Scholar] [CrossRef]
- Binkley, N.; Buehring, B. Beyond FRAX: ItSrs time to consider “sarco-osteopenia”. J. Clin. Densitom. 2009, 12, 413–416. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Zhang, L.; Wang, D.; AIQudsy, L.; Jiang, J.X.; Xu, H.; Shang, P. Muscle-bone crosstalk and potential therapies for sarco-osteoporosis. J. Cell. Biochem. 2019, 120, 14262–14273. [Google Scholar] [CrossRef]
- Brotto, M.; Bonewald, L. Bone and muscle: Interactions beyond mechanical. Bone 2015, 80, 109–114. [Google Scholar] [CrossRef]
- Kawao, N.; Kaji, H. Interactions between muscle tissues and bone metabolism. J. Cell. Biochem. 2015, 116, 687–695. [Google Scholar] [CrossRef]
- Yang, Y.J.; Kim, D.J. An Overview of the Molecular Mechanisms Contributing to Musculoskeletal Disorders in Chronic Liver Disease: Osteoporosis, Sarcopenia, and Osteoporotic Sarcopenia. Int. J. Mol. Sci. 2021, 22, 2604. [Google Scholar] [CrossRef]
- Hirschfeld, H.P.; Kinsella, R.; Duque, G. Osteosarcopenia: Where bone, muscle, and fat collide. Osteoporos. Int. 2017, 28, 2781–2790. [Google Scholar] [CrossRef]
- Saeki, C.; Takano, K.; Oikawa, T.; Aoki, Y.; Kanai, T.; Takakura, K.; Nakano, M.; Torisu, Y.; Sasaki, N.; Abo, M.; et al. Comparative assessment of sarcopenia using the JSH, AWGS, and EWGSOP2 criteria and the relationship between sarcopenia, osteoporosis, and osteosarcopenia in patients with liver cirrhosis. BMC. Musculoskelet. Disord. 2019, 20, 615. [Google Scholar] [CrossRef]
- Saeki, C.; Oikawa, T.; Kanai, T.; Nakano, M.; Torisu, Y.; Sasaki, N.; Saruta, M.; Tsubota, A. Relationship between osteoporosis, sarcopenia, vertebral fracture, and osteosarcopenia in patients with primary biliary cholangitis. Eur. J. Gastroenterol. Hepatol. 2021, 33, 731–737. [Google Scholar] [CrossRef]
- Saeki, C.; Kanai, T.; Nakano, M.; Oikawa, T.; Torisu, Y.; Abo, M.; Saruta, M.; Tsubota, A. Relationship between Osteosarcopenia and Frailty in Patients with Chronic Liver Disease. J. Clin. Med. 2020, 9, 2381. [Google Scholar] [CrossRef] [PubMed]
- Malmstrom, T.K.; Morley, J.E. SARC-F: A simple questionnaire to rapidly diagnose sarcopenia. J. Am. Med. Dir. Assoc. 2013, 14, 531–532. [Google Scholar] [CrossRef] [PubMed]
- Woo, J.; Leung, J.; Morley, J.E. Validating the SARC-F: A suitable community screening tool for sarcopenia? J. Am. Med. Dir. Assoc. 2014, 15, 630–634. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, H.; Fukunishi, S.; Asai, A.; Nishiguchi, S.; Higuchi, K. Sarcopenia and Frailty in Liver Cirrhosis. Life 2021, 11, 399. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, Y.; Kaido, T.; Okumura, S.; Kobayashi, A.; Hammad, A.; Tamai, Y.; Inagaki, N.; Uemoto, S. Proposal for new diagnostic criteria for low skeletal muscle mass based on computed tomography imaging in Asian adults. Nutrition 2016, 32, 1200–1205. [Google Scholar] [CrossRef] [PubMed]
- Eslamparast, T.; Montano-Loza, A.J.; Raman, M.; Tandon, P. Sarcopenic obesity in cirrhosis-The confluence of 2 prognostic titans. Liver. Int. 2018, 38, 1706–1717. [Google Scholar] [CrossRef]
- Bhanji, R.A.; Moctezuma-Velazquez, C.; Duarte-Rojo, A.; Ebadi, M.; Ghosh, S.; Rose, C.; Montano-Loza, A.J. Myosteatosis and sarcopenia are associated with hepatic encephalopathy in patients with cirrhosis. Hepatol. Int. 2018, 12, 377–386. [Google Scholar] [CrossRef]
- Benjamin, J.; Shasthry, V.; Kaal, C.R.; Anand, L.; Bhardwaj, A.; Pandit, V.; Arora, A.; Rajesh, S.; Pamecha, V.; Jain, V.; et al. Characterization of body composition and definition of sarcopenia in patients with alcoholic cirrhosis: A computed tomography based study. Liver. Int. 2017, 37, 1668–1674. [Google Scholar] [CrossRef]
- Saeki, C.; Kanai, T.; Nakano, M.; Oikawa, T.; Torisu, Y.; Saruta, M.; Tsubota, A. Clinical characteristics of sarcopenia in patients with alcoholic liver cirrhosis. JGH Open 2021, 5, 763–769. [Google Scholar] [CrossRef]
- Hanai, T.; Shiraki, M.; Ohnishi, S.; Miyazaki, T.; Ideta, T.; Kochi, T.; Imai, K.; Suetsugu, A.; Takai, K.; Moriwaki, H.; et al. Rapid skeletal muscle wasting predicts worse survival in patients with liver cirrhosis. Hepatol. Res. 2016, 46, 743–751. [Google Scholar] [CrossRef]
- Yoshida, D.; Suzuki, T.; Shimada, H.; Park, H.; Makizako, H.; Doi, T.; Anan, Y.; Tsutsumimoto, K.; Uemura, K.; Ito, T.; et al. Using two different algorithms to determine the prevalence of sarcopenia. Geriatr. Gerontol. Int. 2014, 14, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, N.; Muraki, S.; Oka, H.; Iidaka, T.; Kodama, R.; Kawaguchi, H.; Nakamura, K.; Tanaka, S.; Akune, T. Is osteoporosis a predictor for future sarcopenia or vice versa? Four-year observations between the second and third ROAD study surveys. Osteoporos. Int. 2017, 28, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, H.; Yoh, K.; Enomoto, H.; Nishimura, T.; Nishiguchi, S.; Iijima, H. Combined grip strength and calf circumference as a useful prognostic system in patients with liver diseases: A large cohort study. Ann. Transl. Med. 2021, 9, 624. [Google Scholar] [CrossRef]
- Nishikawa, H.; Shiraki, M.; Hiramatsu, A.; Hara, N.; Moriya, K.; Hino, K.; Koike, K. Reduced Handgrip Strength Predicts Poorer Survival in Chronic Liver Diseases: A Large Multi-Center Study in Japan. Hepatol. Res. 2021. online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Glass, D.J. Skeletal muscle hypertrophy and atrophy signaling pathways. Int. J. Biochem. Cell. Biol. 2005, 37, 1974–1984. [Google Scholar] [CrossRef] [PubMed]
- Foster, K.G.; Fingar, D.C. Mammalian target of rapamycin (mTOR): Conducting the cellular signaling symphony. J. Biol. Chem. 2010, 285, 14071–14077. [Google Scholar] [CrossRef]
- Periyalwar, P.; Dasarathy, S. Malnutrition in cirrhosis: Contribution and consequences of sarcopenia on metabolic and clinical responses. Clin. Liver. Dis. 2012, 16, 95–131. [Google Scholar] [CrossRef]
- Sinclair, M.; Gow, P.J.; Grossmann, M.; Angus, P.W. Review article: Sarcopenia in cirrhosis etiology, implications, and potential therapeutic interventions. Aliment. Pharmacol. Ther. 2016, 43, 765–777. [Google Scholar] [CrossRef] [PubMed]
- Anand, A.C. Nutrition and Muscle in Cirrhosis. J. Clin. Exp. Hepatol. 2017, 7, 340–357. [Google Scholar] [CrossRef]
- Meyer, F.; Bannert, K.; Wiese, M.; Esau, S.; Sautter, L.F.; Ehlers, L.; Aghdassi, A.A.; Metges, C.C.; Garbe, L.A.; Jaster, R.; et al. Molecular Mechanism Contributing to Malnutrition and Sarcopenia in Patients with Liver Cirrhosis. Int. J. Mol. Sci. 2020, 21, 5357. [Google Scholar] [CrossRef]
- Tandon, P.; Montano-Loza, A.J.; Lai, J.C.; Dasarathy, S.; Merli, M. Sarcopenia and frailty in decompensated cirrhosis. J. Hepatol. 2021, 75 (Suppl. S1), S147–S162. [Google Scholar] [CrossRef] [PubMed]
- Saeki, C.; Kanai, T.; Nakano, M.; Oikawa, T.; Torisu, Y.; Saruta, M.; Tsubota, A. Low Serum Branched-Chain Amino Acid and Insulin-Like Growth Factor-1 Levels Are Associated with Sarcopenia and Slow Gait Speed in Patients with Liver Cirrhosis. J. Clin. Med. 2020, 9, 3239. [Google Scholar] [CrossRef] [PubMed]
- Retamales, A.; Zuloaga, R.; Valenzuela, C.A.; Gallardo-Escarate, C.; Molina, A.; Valdés, J.A. Insulin-like growth factor-1 suppresses the Myostatin signaling pathway during myogenic differentiation. Biochem. Biophys. Res. Commun. 2015, 464, 596–602. [Google Scholar] [CrossRef]
- Drummond, M.J.; Dreyer, H.C.; Fry, C.S.; Glynn, E.L.; Rasmussen, B.B. Nutritional and contractile regulation of human skeletal muscle protein synthesis and mTORC1 signaling. J. Appl. Physiol. 2009, 106, 1374–1384. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zeng, X.; Ren, M.; Mao, X.; Qiao, S. Novel metabolic and physiological functions of branched chain amino acids: A review. J. Anim. Sci. Biotechnol. 2017, 8, 10. [Google Scholar] [CrossRef]
- Kalyani, R.R.; Corriere, M.; Ferrucci, L. Age-related and disease-related muscle loss: The effect of diabetes, obesity, and other diseases. Lancet Diabetes Endocrinol. 2014, 2, 819–829. [Google Scholar] [CrossRef]
- Calmet, F.; Martin, P.; Pearlman, M. Nutrition in Patients with Cirrhosis. Gastroenterol. Hepatol. 2019, 15, 248–254. [Google Scholar]
- Reiling, J.H.; Sabatini, D.M. Stress and mTORture signaling. Oncogene 2006, 25, 6373–6383. [Google Scholar] [CrossRef]
- Wang, X.; Hu, Z.; Hu, J.; Du, J.; Mitch, W.E. Insulin Resistance Accelerates Muscle Protein Degradation: Activation of the Ubiquitin-Proteasome Pathway by Defects in Muscle Cell Signaling. Endocrinology 2006, 147, 4160–4168. [Google Scholar] [CrossRef]
- MacKrell, J.G.; Yaden, B.C.; Bullock, H.; Chen, K.; Shetler, P.; Bryant, H.U.; Krishnan, V. Molecular targets of androgen signaling that characterize skeletal muscle recovery and regeneration. Nucl. Recept. Signal. 2015, 13, e005. [Google Scholar] [CrossRef]
- Kovacheva, E.L.; Hikim, A.P.; Shen, R.; Sinha, I.; Sinha-Hikim, I. Testosterone supplementation reverses sarcopenia in aging through regulation of myostatin, c-Jun NH2-terminal kinase, Notch, and Akt signaling pathways. Endocrinology 2010, 151, 628–638. [Google Scholar] [CrossRef]
- Han, H.Q.; Zhou, X.; Mitch, W.E.; Goldberg, A.L. Myostatin/activin pathway antagonism: Molecular basis and therapeutic potential. Int. J. Biochem. Cell Biol. 2013, 45, 2333–2347. [Google Scholar] [CrossRef]
- Sandri, M.; Sandri, C.; Gilbert, A.; Skurk, C.; Calabria, E.; Picard, A.; Walsh, K.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 2004, 117, 399–412. [Google Scholar] [CrossRef]
- Langley, B.; Thomas, M.; Bishop, A.; Sharma, M.; Gilmour, S.; Kambadur, R. Myostatin inhibits myoblast differentiation by down-regulating MyoD expression. J. Biol. Chem. 2002, 277, 49831–49840. [Google Scholar] [CrossRef]
- Qiu, J.; Tsien, C.; Thapalaya, S.; Narayanan, A.; Weihl, C.C.; Ching, J.K.; Eghtesad, B.; Singh, K.; Fu, X.; Dubyak, G.R.; et al. Hyperammonemia-mediated autophagy in skeletal muscle contributes to sarcopenia of cirrhosis. Am. J. Physiol. Metab. 2012, 303, E983–E993. [Google Scholar] [CrossRef]
- Campos, F.; Abrigo, J.; Aguirre, F.; Garcés, B.; Arrese, M.; Karpen, S.; Cabrera, D.; Andia, M.E.; Simon, F.; Cabello-Verrugio, C. Sarcopenia in a mice model of chronic liver disease: Role of the ubiquitin–proteasome system and oxidative stress. Pflug. Arch. 2018, 470, 1503–1519. [Google Scholar] [CrossRef]
- Milan, G.; Romanello, V.; Pescatore, F.; Armani, A.; Paik, J.H.; Frasson, L.; Seydel, A.; Zhao, J.; Abraham, R.; Goldberg, A.L.; et al. Regulation of autophagy and the ubiquitin-proteasome system by the FoxO transcriptional network during muscle atrophy. Nat. Commun. 2015, 6, 6670. [Google Scholar] [CrossRef]
- Wilkes, J.J.; Lloyd, D.J.; Gekakis, N. Loss-of-function mutation in myostatin reduces tumor necrosis factor alpha production and protects liver against obesity induced insulin resistance. Diabetes 2009, 58, 1133–1143. [Google Scholar] [CrossRef] [PubMed]
- Kellum, E.; Starr, H.; Arounleut, P.; Immel, D.; Fulzele, S.; Wenger, K.; Hamrick, M.W. Myostatin (GDF-8) deficiency increases fracture callus size, Sox-5expression, and callus bone volume. Bone 2009, 44, 17–23. [Google Scholar] [CrossRef]
- Dasarathy, S.; Hatzoglou, M. Hyperammonemia and proteostasis in cirrhosis. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Cadavid, N.F.; Taylor, W.E.; Yarasheski, K.; Sinha-Hikim, I.; Ma, K.; Ezzat, S.; Shen, R.; Lalani, R.; Asa, S.; Mamita, M.; et al. Organization of the human myostatin gene and expression in healthy men and HIV-infected men with muscle wasting. Proc. Natl. Acad. Sci. USA 1998, 95, 14938–14943. [Google Scholar] [CrossRef]
- Baracco, E.E.; Castoldi, F.; Durand, S.; Enot, D.P.; Tadic, J.; Kainz, K.; Madeo, F.; Chery, A.; Izzo, V.; Maiuri, M.C.; et al. Alpha-Ketoglutarate inhibits autophagy. Aging 2019, 11, 3418–3431. [Google Scholar] [CrossRef]
- Davuluri, G.; Giusto, M.; Chandel, R.; Welch, N.; Alsabbagh, K.; Kant, S.; Kumar, A.; Kim, A.; Gangadhariah, M.; Ghosh, P.K.; et al. Impaired ribosomal biogenesis by noncanonical degradation of β-catenin during hyperammonemia. Mol. Cell. Biol. 2019, 39, e00451-18. [Google Scholar] [CrossRef]
- Hulmi, J.J.; Ahtiainen, J.P.; Kaasalainen, T.; Pöllänen, E.; Häkkinen, K.; Alen, M.; Selänne, H.; Kovanen, V.; Mero, A.A. Postexercise myostatin and activin IIb mRNA levels: Effects of strength training. Med. Sci. Sports. Exerc. 2007, 39, 289–297. [Google Scholar] [CrossRef]
- Allen, D.L.; Hittel, D.S.; McPherron, A.C. Expression and function of myostatin in obesity, diabetes, and exercise adaptation. Med. Sci. Sports Exerc. 2011, 43, 1828–1835. [Google Scholar] [CrossRef]
- Kraemer, W.J.; Ratamess, N.A.; Hymer, W.C.; Nindl, B.C.; Fragala, M.S. Growth Hormone(s), testosterone, insulin-like growth factors, and cortisol: Roles and integration for cellular development and growth with exercise. Front. Endocrinol. 2020, 11, 33. [Google Scholar] [CrossRef]
- Gharahdaghi, N.; Phillips, B.E.; Szewczyk, N.J.; Smith, K.; Wilkinson, D.J.; Atherton, P.J. Links Between Testosterone, Oestrogen, and the Growth Hormone/Insulin-Like Growth Factor Axis and Resistance Exercise Muscle Adaptations. Front. Physiol. 2021, 11, 621226. [Google Scholar] [CrossRef]
- White, J.P.; Gao, S.; Puppa, M.J.; Sato, S.; Welle, S.L.; Carson, J.A. Testosterone regulation of Akt/mTORC1/FoxO3a signaling in skeletal muscle. Mol. Cell. Endocrinol. 2013, 365, 174–186. [Google Scholar] [CrossRef]
- Basualto-Alarcón, C.; Jorquera, G.; Altamirano, F.; Jaimovich, E.; Estrada, M. Testosterone signals through mTOR and androgen receptor to induce muscle hypertrophy. Med. Sci. Sports Exerc. 2013, 45, 1712–1720. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Yin, L.; Lin, X.; Lu, J.; Wang, X. Effects of Cyclic Mechanical Stretch on the Proliferation of L6 Myoblasts and Its Mechanisms: PI3K/Akt and MAPK Signal Pathways Regulated by IGF-1 Receptor. Int. J. Mol. Sci. 2018, 19, 1649. [Google Scholar] [CrossRef] [PubMed]
- Dent, J.R.; Fletcher, D.K.; McGuigan, M.R. Evidence for a non-genomic action of testosterone in skeletal muscle which may improve athletic performance: Implications for the female athlete. J. Sports Sci. Med. 2012, 11, 363–370. [Google Scholar]
- Sinclair, M.; Grossmann, M.; Gow, P.J.; Angus, P.W. Testosterone in men with advanced liver disease: Abnormalities and implications. J. Gastroenterol. Hepatol. 2015, 30, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Deng, N.; Mallepally, N.; Peng, F.B.; Kanji, A.; Marcelli, M.; Hernaez, R. Serum testosterone levels and testosterone supplementation in cirrhosis: A systematic review. Liver Int. 2021. [Google Scholar] [CrossRef]
- Tandon, P.; Raman, M.; Mourtzakis, M.; Merli, M. A practical approach to nutritional screening and assessment in cirrhosis. Hepatology 2017, 65, 1044–1057. [Google Scholar] [CrossRef]
- Ockenga, J.; Bischo, S.C.; Tillmann, H.L.; Rifai, K.; Widjaja, A.; Böker, K.H.; Manns, M.P.; Brabant, G. Elevated bound leptin correlates with energy expenditure in cirrhotics. Gastroenterology 2000, 119, 1656–1662. [Google Scholar] [CrossRef]
- Kalaitzakis, E.; Bosaeus, I.; Öhman, L.; Björnsson, E. Altered postprandial glucose, insulin, leptin, and ghrelin in liver cirrhosis: Correlations with energy intake and resting energy expenditure. Am. J. Clin. Nutr. 2007, 85, 808–815. [Google Scholar] [CrossRef] [PubMed]
- Freire-Regatillo, A.; Argente-Arizón, P.; Argente, J.; Garcia-Segura, L.M.; Chowen, J. Non-Neuronal Cells in the Hypothalamic Adaptation to Metabolic Signals. Front. Endocrinol. 2017, 8, 51. [Google Scholar] [CrossRef]
- Moreno, M.; Chaves, J.F.; Sancho-Bru, P.; Ramalho, F.; Ramalho, L.N.; Mansego, M.L.; Ivorra, C.; Dominguez, M.; Conde, L.; Millán, C.; et al. Ghrelin attenuates hepatocellular injury and liver fibrogenesis in rodents and influences fibrosis progression in humans. Hepatology 2010, 51, 974–985. [Google Scholar] [CrossRef]
- Grüngreiff, K.; Reinhold, D.; Wedemeyer, H. The role of zinc in liver cirrhosis. Ann. Hepatol. 2016, 15, 7–16. [Google Scholar] [CrossRef]
- Kirk, B.; Zanker, J.; Duque, G. Osteosarcopenia: Epidemiology, diagnosis, and treatment-facts and numbers. J. Cachexia Sarcopenia Muscle 2020, 11, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Salech, F.; Marquez, C.; Lera, L.; Angel, B.; Saguez, R.; Albala, C. Osteosarcopenia Predicts Falls, Fractures, and Mortality in Chilean Community-Dwelling Older Adults. J. Am. Med. Dir. Assoc. 2021, 22, 853–858. [Google Scholar] [CrossRef]
- Yoo, J.I.; Kim, H.; Ha, Y.C.; Kwon, H.B.; Koo, K.H. Osteosarcopenia in Patients with Hip Fracture Is Related with High Mortality. J. Korean. Med. Sci. 2018, 33, e27. [Google Scholar] [CrossRef]
- Teng, Z.; Zhu, Y.; Teng, Y.; Long, Q.; Hao, Q.; Yu, X.; Yang, L.; Lv, Y.; Liu, J.; Zeng, Y.; et al. The analysis of osteosarcopenia as a risk factor for fractures, mortality, and falls. Osteoporos. Int. 2021. online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Abe, K.; Fujita, M.; Okai, K.; Takahashi, A.; Ohira, H. Association between sarcopenia and osteoporosis in chronic liver disease. Hepatol. Res. 2018, 48, 893–904. [Google Scholar] [CrossRef] [PubMed]
- Bering, T.; Diniz, K.G.D.; Coelho, M.P.P.; Vieira, D.A.; Soares, M.M.S.; Kakehasi, A.M.; Correia, M.I.T.D.; Teixeira, R.; Queiroz, D.M.M.; Rocha, G.A.; et al. Association between pre-sarcopenia, sarcopenia, and bone mineral density in patients with chronic hepatitis C. J. Cachexia Sarcopenia Muscle 2018, 9, 255–268. [Google Scholar] [CrossRef] [PubMed]
- Sepúlveda-Loyola, W.; Phu, S.; Bani Hassan, E.; Brennan-Olsen, S.L.; Zanker, J.; Vogrin, S.; Santos, N.Q.D.; Nascimento, J.R.A.D., Jr. The Joint Occurrence of Osteoporosis and Sarcopenia (Osteosarcopenia): Definitions and Characteristics. J. Am. Med. Dir. Assoc. 2020, 21, 220–225. [Google Scholar] [CrossRef]
- Wong, R.M.Y.; Wong, H.; Zhang, N.; Chow, S.K.H.; Chau, W.W.; Wang, J.; Chim, Y.N.; Leung, K.S.; Cheung, W.H. The relationship between sarcopenia and fragility fracture-a systematic review. Osteoporos. Int. 2019, 30, 541–553. [Google Scholar] [CrossRef]
- Inoue, T.; Maeda, K.; Nagano, A.; Shimizu, A.; Ueshima, J.; Murotani, K.; Sato, K.; Tsubaki, A. Undernutrition, Sarcopenia, and Frailty in Fragility Hip Fracture: Advanced Strategies for Improving Clinical Outcomes. Nutrients 2020, 12, 3743. [Google Scholar] [CrossRef]
- Goodman, C.A.; Hornberger, T.A.; Robling, A.G. Bone and skeletal muscle: Key players in mechanotransduction and potential overlapping mechanisms. Bone 2015, 80, 24–36. [Google Scholar] [CrossRef]
- Karasik, D.; Kiel, D.P. Evidence for pleiotropic factors in genetics of the musculoskeletal system. Bone 2010, 46, 1226–1237. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.F.; Zhang, L.S.; Liu, Y.J.; Hu, H.G.; Li, J.; Tian, Q.; Yu, P.; Zhang, F.; Yang, T.L.; Guo, Y.; et al. Suggestion of GLYAT gene underlying variation of bone size and body lean mass as revealed by a bivariate genome-wide association study. Hum. Genet. 2013, 132, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Hsu, Y.H.; Mo, C.; Abreu, E.; Kiel, D.P.; Bonewald, L.F.; Brotto, M.; Karasik, D. METTL21C is a potential pleiotropic gene for osteoporosis and sarcopenia acting through the modulation of the NF-κB signaling pathway. J. Bone Miner. Res. 2014, 29, 1531–1540. [Google Scholar] [CrossRef]
- Karasik, D.; Cohen-Zinder, M. The genetic pleiotropy of musculoskeletal aging. Front. Physiol. 2012, 3, 303. [Google Scholar] [CrossRef] [PubMed]
- Trajanoska, K.; Rivadeneira, F.; Kiel, D.P.; Karasik, D. Genetics of Bone and Muscle Interactions in Humans. Curr. Osteoporos. Rep. 2019, 17, 86–95. [Google Scholar] [CrossRef]
- Kramer, I.; Baertschi, S.; Halleux, C.; Keller, H.; Kneissel, M. Mef2c deletion in osteocytes results in increased bone mass. J. Bone Miner. Res. 2012, 27, 360–373. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.; Seto, J.T.; MacArthur, D.G.; Yang, N.; North, K.N.; Head, S.I. A gene for speed: Contractile properties of isolated whole EDL muscle from an alpha-actinin-3 knockout mouse. Am. J. Physiol. Cell. Physiol. 2008, 295, C897–C904. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Schindeler, A.; McDonald, M.M.; Seto, J.T.; Houweling, P.J.; Lek, M.; Hogarth, M.; Morse, A.R.; Raftery, J.M.; Balasuriya, D.; et al. α-Actinin-3 deficiency is associated with reduced bone mass in human and mouse. Bone 2011, 49, 790–798. [Google Scholar] [CrossRef]
- Kitada, T.; Seki, S.; Iwai, S.; Yamada, T.; Sakaguchi, H.; Wakasa, K. In situ detection of oxidative DNA damage, 8-hydroxydeoxyguanosine, in chronic human liver disease. J. Hepatol. 2001, 35, 613–618. [Google Scholar] [CrossRef]
- Tilg, H.; Wilmer, A.; Vogel, W.; Herold, M.; Nölchen, B.; Judmaier, G.; Huber, C. Serum levels of cytokines in chronic liver dis eases. Gastroenterology 1992, 103, 264–274. [Google Scholar] [CrossRef]
- Saito, M.; Marumo, K. Collagen cross-links as a determinant of bone quality: A possible explanation for bone fragility in aging, osteoporosis, and diabetes mellitus. Osteoporos. Int. 2010, 21, 195–214. [Google Scholar] [CrossRef]
- Tanaka, K.; Kanazawa, I.; Sugimoto, T. Elevated Serum Pentosidine and Decreased Serum IGF-I Levels are Associated with Loss of Muscle Mass in Postmenopausal Women with Type 2 Diabetes Mellitus. Exp. Clin. Endocrinol. Diabetes 2016, 124, 163–166. [Google Scholar] [CrossRef] [PubMed]
- Saeki, C.; Saito, M.; Kanai, T.; Nakano, M.; Oikawa, T.; Torisu, Y.; Saruta, M.; Tsubota, A. Plasma pentosidine levels are associated with prevalent fractures in patients with chronic liver disease. PLoS ONE 2021, 16, e0249728. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Han, T.; Werner, H.; Rosen, C.J.; Schaffler, M.B.; Yakar, S. Reduced Serum IGF-1 Associated with Hepatic Osteodystrophy Is a Main Determinant of Low Cortical but Not Trabecular Bone Mass. J. Bone Miner. Res. 2018, 33, 123–136. [Google Scholar] [CrossRef]
- George, J.; Ganesh, H.K.; Acharya, S.; Bandgar, T.R.; Shivane, V.; Karvat, A.; Bhatia, S.J.; Shah, S.; Menon, P.S.; Shah, N. Bone mineral density and disorders of mineral metabolism in chronic liver disease. World J. Gastroenterol. 2009, 15, 3516–3522. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, H.; Enomoto, H.; Ishii, A.; Iwata, Y.; Miyamoto, Y.; Ishii, N.; Yuri, Y.; Hasegawa, K.; Nakano, C.; Nishimura, T.; et al. Elevated serum myostatin level is associated with worse survival in patients with liver cirrhosis. J. Cachexia Sarcopenia Muscle 2017, 8, 915–925. [Google Scholar] [CrossRef]
- García, P.S.; Cabbabe, A.; Kambadur, R.; Nicholas, G.; Csete, M. Brief-reports: Elevated myostatin levels in patients with liver disease: A potential contributor to skeletal muscle wasting. Anesth. Analg. 2010, 111, 707–709. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Yi, Q.; Sun, W.; Huang, D.; Zhang, H.; Duan, L.; Shang, H.; Wang, D.; Xiong, J. Molecular basis and therapeutic potential of myostatin on bone formation and metabolism in orthopedic disease. Biofactors 2020. online ahead of print. [Google Scholar] [CrossRef]
- Puolakkainen, T.; Ma, H.; Kainulainen, H.; Pasternack, A.; Rantalainen, T.; Ritvos, O.; Heikinheimo, K.; Hulmi, J.J.; Kiviranta, R. Treatment with soluble activin type IIB-receptor improves bone mass and strength in a mouse model of Duchenne muscular dystrophy. BMC. Musculoskelet. Disord. 2017, 18, 20. [Google Scholar] [CrossRef]
- Colaianni, G.; Cinti, S.; Colucci, S.; Grano, M. Irisin and musculoskeletal health. Ann. N. Y. Acad. Sci. 2017, 1402, 5–9. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Zhou, X.; Yuan, C.; Li, R.; Ma, Y.; Tang, X. Association between serum irisin concentrations and sarcopenia in patients with liver cirrhosis: A cross-sectional study. Sci. Rep. 2020, 10, 16093. [Google Scholar] [CrossRef]
- Colaianni, G.; Cuscito, C.; Mongelli, T.; Pignataro, P.; Buccoliero, C.; Liu, P.; Lu, P.; Sartini, L.; Di Comite, M.; Mori, G.; et al. The myokine irisin increases cortical bone mass. Proc. Natl. Acad. Sci. USA 2015, 112, 12157–12162. [Google Scholar] [CrossRef]
- Aydin, S.; Kuloglu, T.; Aydin, S.; Kalayci, M.; Yilmaz, M.; Cakmak, T.; Albayrak, S.; Gungor, S.; Colakoglu, N.; Ozercan, I.H. A comprehensive immunohistochemical examination of the distribution of the fat-burning protein irisin in biological tissues. Peptides 2014, 61, 130–136. [Google Scholar] [CrossRef]
- Liu, T.Y.; Xiong, X.Q.; Ren, X.S.; Zhao, M.X.; Shi, C.X.; Wang, J.J.; Zhou, Y.B.; Zhang, F.; Han, Y.; Gao, X.Y.; et al. FNDC5 Alleviates Hepatosteatosis by Restoring AMPK/mTOR-Mediated Autophagy, Fatty Acid Oxidation, and Lipogenesis in Mice. Diabetes 2016, 65, 3262–3275. [Google Scholar] [CrossRef] [PubMed]
- Kitase, Y.; Vallejo, J.A.; Gutheil, W.; Vemula, H.; Jähn, K.; Yi, J.; Zhou, J.; Brotto, M.; Bonewald, L.F. beta-aminoisobutyric Acid, l-BAIBA, Is a Muscle-Derived Osteocyte Survival Factor. Cell. Rep. 2018, 22, 1531–1544. [Google Scholar] [CrossRef]
- Komori, T. Functions of Osteocalcin in Bone, Pancreas, Testis, and Muscle. Int. J. Mol. Sci. 2020, 21, 7513. [Google Scholar] [CrossRef]
- Mera, P.; Laue, K.; Wei, J.; Berger, J.M.; Karsenty, G. Osteocalcin is necessary and sufficient to maintain muscle mass in older mice. Mol. Metab. 2016, 5, 1042–1047. [Google Scholar] [CrossRef] [PubMed]
- Ducy, P.; Desbois, C.; Boyce, B.; Pinero, G.; Story, B.; Dunstan, C.; Smith, E.; Bonadio, J.; Goldstein, S.; Gundberg, C.; et al. Increased bone formation in osteocalcin-deficient mice. Nature 1996, 382, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Moriishi, T.; Ozasa, R.; Ishimoto, T.; Nakano, T.; Hasegawa, T.; Miyazaki, T.; Liu, W.; Fukuyama, R.; Wang, Y.; Komori, H.; et al. Osteocalcin is necessary for the alignment of apatite crystallites, but not glucose metabolism, testosterone synthesis, or muscle mass. PLoS Genet. 2020, 16, e1008586. [Google Scholar] [CrossRef]
- Maeda, K.; Kobayashi, Y.; Koide, M.; Uehara, S.; Okamoto, M.; Ishihara, A.; Kayama, T.; Saito, M.; Marumo, K. The Regulation of Bone Metabolism and Disorders by Wnt Signaling. Int. J. Mol. Sci. 2019, 20, 5525. [Google Scholar] [CrossRef]
- Cisternas, P.; Henriquez, J.P.; Brandan, E.; Inestrosa, N.C. Wnt signaling in skeletal muscle dynamics: Myogenesis, neuromuscular synapse and fibrosis. Mol. Neurobiol. 2014, 49, 574–589. [Google Scholar] [CrossRef]
- Delgado-Calle, J.; Sato, A.Y.; Bellido, T. Role and mechanism of action of sclerostin in bone. Bone 2017, 96, 29–37. [Google Scholar] [CrossRef]
- Guañabens, N.; Ruiz-Gaspà, S.; Gifre, L.; Miquel, R.; Peris, P.; Monegal, A.; Dubrueil, M.; Arias, A.; Parés, A. Sclerostin Expression in Bile Ducts of Patients with Chronic Cholestasis May Influence the Bone Disease in Primary Biliary Cirrhosis. J. Bone Miner. Res. 2016, 31, 1725–1733. [Google Scholar] [CrossRef]
- Kim, J.A.; Roh, E.; Hong, S.H.; Lee, Y.B.; Kim, N.H.; Yoo, H.J.; Seo, J.A.; Kim, N.H.; Kim, S.G.; Baik, S.H.; et al. Association of serum sclerostin levels with low skeletal muscle mass: The Korean Sarcopenic Obesity Study (KSOS). Bone 2019, 128, 115053. [Google Scholar] [CrossRef]
- Krause, A.; Speacht, T.; Govey, P.; Zhang, Y.; Steiner, J.; Lang, C.; Donahue, H. Sarcopenia and increased body fat in sclerostin deficient mice. J. Bone Miner. Res. 2014, 29, S8–S9. [Google Scholar]
- Tang, Y.; Wu, X.; Lei, W.; Pang, L.; Wan, C.; Shi, Z.; Zhao, L.; Nagy, T.R.; Peng, X.; Hu, J.; et al. TGF-beta1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat. Med. 2009, 15, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Mendias, C.L.; Gumucio, J.P.; Davis, M.E.; Bromley, C.W.; Davis, C.S.; Brooks, S.V. Transforming growth factor-beta induces skeletal muscle atrophy and fibrosis through the induction of atrogin-1 and scleraxis. Muscle Nerve 2012, 45, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Waning, D.L.; Mohammad, K.S.; Reiken, S.; Xie, W.; Andersson, D.C.; John, S.; Chiechi, A.; Wright, L.E.; Umanskaya, A.; Niewolna, M.; et al. Excess TGF-β mediates muscle weakness associated with bone metastases in mice. Nat. Med. 2015, 21, 1262–1271. [Google Scholar] [CrossRef] [PubMed]
- Agostini, D.; Zeppa Donati, S.; Lucertini, F.; Annibalini, G.; Gervasi, M.; Ferri Marini, C.; Piccoli, G.; Stocchi, V.; Barbieri, E.; Sestili, P. Muscle and Bone Health in Postmenopausal Women: Role of Protein and Vitamin D Supplementation Combined with Exercise Training. Nutrients 2018, 10, 1103. [Google Scholar] [CrossRef]
- Wintermeyer, E.; Ihle, C.; Ehnert, S.; Stöckle, U.; Ochs, G.; de Zwart, P.; Flesch, I.; Bahrs, C.; Nussler, A.K. Crucial Role of Vitamin D in the Musculoskeletal System. Nutrients 2016, 8, 319. [Google Scholar] [CrossRef] [PubMed]
- Garcia, L.A.; King, K.K.; Ferrini, M.G.; Norris, K.C.; Artaza, J.N. 1,25(OH)2vitamin D3 stimulates myogenic differentiation by inhibiting cell proliferation and modulating the expression of promyogenic growth factors and myostatin in C2C12 skeletal muscle cells. Endocrinology 2011, 152, 2976–2986. [Google Scholar] [CrossRef] [PubMed]
- Girgis, C.M.; Cha, K.M.; Houweling, P.J.; Rao, R.; Mokbel, N.; Lin, M.; Clifton-Bligh, R.J.; Gunton, J.E. Vitamin D Receptor Ablation and Vitamin D Deficiency Result in Reduced Grip Strength, Altered Muscle Fibers, and Increased Myostatin in Mice. Calcif. Tissue Int. 2015, 97, 602–610. [Google Scholar] [CrossRef] [PubMed]
- Saeki, C.; Kanai, T.; Nakano, M.; Oikawa, T.; Torisu, Y.; Saruta, M.; Tsubota, A. Low Serum 25-Hydroxyvitamin D Levels Are Related to Frailty and Sarcopenia in Patients with Chronic Liver Disease. Nutrients 2020, 12, 3810. [Google Scholar] [CrossRef]
- Okubo, T.; Atsukawa, M.; Tsubota, A.; Yoshida, Y.; Arai, T.; Iwashita, A.N.; Itokawa, N.; Kondo, C.; Iwakiri, K. Relationship between serum vitamin D level and sarcopenia in chronic liver disease. Hepatol. Res. 2020, 50, 588–597. [Google Scholar] [CrossRef]
- Daly, R.M.; Gianoudis, J.; Kersh, M.E.; Bailey, C.A.; Ebeling, P.R.; Krug, R.; Nowson, C.A.; Hill, K.; Sanders, K.M. Effects of a 12-Month Supervised, Community-Based, Multimodal Exercise Program Followed by a 6-Month Research-to-Practice Transition on Bone Mineral Density, Trabecular Microarchitecture, and Physical Function in Older Adults: A Randomized Controlled Trial. J. Bone Miner. Res. 2020, 35, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Kemmler, W.; Kohl, M.; Fröhlich, M.; Jakob, F.; Engelke, K.; von Stengel, S.; Schoene, D. Effects of High-Intensity Resistance Training on Osteopenia and Sarcopenia Parameters in Older Men with Osteosarcopenia-One-Year Results of the Randomized Controlled Franconian Osteopenia and Sarcopenia Trial (FrOST). J. Bone Miner. Res. 2020, 35, 1634–1644. [Google Scholar] [CrossRef] [PubMed]
- Román, E.; García-Galcerán, C.; Torrades, T.; Herrera, S.; Marín, A.; Doñate, M.; Alvarado-Tapias, E.; Malouf, J.; Nácher, L.; Serra-Grima, R.; et al. Effects of an Exercise Programme on Functional Capacity, Body Composition and Risk of Falls in Patients with Cirrhosis: A Randomized Clinical Trial. PLoS ONE 2016, 11, e0151652. [Google Scholar] [CrossRef]
- Kruger, C.; McNeely, M.L.; Bailey, R.J.; Yavari, M.; Abraldes, J.G.; Carbonneau, M.; Newnham, K.; DenHeyer, V.; Ma, M.; Thompson, R.; et al. Home Exercise Training Improves Exercise Capacity in Cirrhosis Patients: Role of Exercise Adherence. Sci. Rep. 2018, 8, 99. [Google Scholar] [CrossRef] [PubMed]
- Naseer, M.; Turse, E.P.; Syed, A.; Dailey, F.E.; Zatreh, M.; Tahan, V. Interventions to improve sarcopenia in cirrhosis: A systematic review. World J. Clin. Cases 2019, 7, 156–170. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Arnau, F.M.; Fonfría-Vivas, R.; Cauli, O. Beneficial Effects of Leucine Supplementation on Criteria for Sarcopenia: A Systematic Review. Nutrients 2019, 11, 2504. [Google Scholar] [CrossRef]
- Kitajima, Y.; Takahashi, H.; Akiyama, T.; Murayama, K.; Iwane, S.; Kuwashiro, T.; Tanaka, K.; Kawazoe, S.; Ono, N.; Eguchi, T.; et al. Supplementation with branched-chain amino acids ameliorates hypoalbuminemia, prevents sarcopenia, and reduces fat accumulation in the skeletal muscles of patients with liver cirrhosis. J. Gastroenterol. 2018, 53, 427–437. [Google Scholar] [CrossRef]
- Tsien, C.; Davuluri, G.; Singh, D.; Allawy, A.; Ten Have, G.A.; Thapaliya, S.; Schulze, J.M.; Barnes, D.; McCullough, A.J.; Engelen, M.P.; et al. Metabolic and molecular responses to leucine-enriched branched chain amino acid supplementation in the skeletal muscle of alcoholic cirrhosis. Hepatology 2015, 61, 2018–2029. [Google Scholar] [CrossRef]
- Ohara, M.; Ogawa, K.; Suda, G.; Kimura, M.; Maehara, O.; Shimazaki, T.; Suzuki, K.; Nakamura, A.; Umemura, M.; Izumi, T.; et al. L-Carnitine Suppresses Loss of Skeletal Muscle Mass in Patients with Liver Cirrhosis. Hepatol. Commun. 2018, 2, 906–918. [Google Scholar] [CrossRef]
- Hiramatsu, A.; Aikata, H.; Uchikawa, S.; Ohya, K.; Kodama, K.; Nishida, Y.; Daijo, K.; Osawa, M.; Teraoka, Y.; Honda, F.; et al. Levocarnitine Use Is Associated with Improvement in Sarcopenia in Patients With Liver Cirrhosis. Hepatol. Commun. 2019, 3, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Hanai, T.; Shiraki, M.; Imai, K.; Suetugu, A.; Takai, K.; Shimizu, M. Usefulness of Carnitine Supplementation for the Complications of Liver Cirrhosis. Nutrients 2020, 12, 1915. [Google Scholar] [CrossRef] [PubMed]
- Reid, I.R.; Bolland, M.J.; Grey, A. Effects of vitamin D supplements on bone mineral density: A systematic review and meta-analysis. Lancet 2014, 383, 146–155. [Google Scholar] [CrossRef]
- Bolland, M.J.; Grey, A.; Avenell, A. Effects of vitamin D supplementation on musculoskeletal health: A systematic review, meta-analysis, and trial sequential analysis. Lancet Diabetes Endocrinol. 2018, 6, 847–858. [Google Scholar] [CrossRef]
- Macdonald, H.M.; Reid, I.R.; Gamble, G.D.; Fraser, W.D.; Tang, J.C.; Wood, A.D. 25-Hydroxyvitamin D Threshold for the Effects of Vitamin D Supplements on Bone Density: Secondary Analysis of a Randomized Controlled Trial. J. Bone Miner. Res. 2018, 33, 1464–1469. [Google Scholar] [CrossRef]
- Reid, I.R.; Horne, A.M.; Mihov, B.; Gamble, G.D.; Al-Abuwsi, F.; Singh, M.; Taylor, L.; Fenwick, S.; Camargo, C.A.; Stewart, A.W.; et al. Effect of monthly high-dose vitamin D on bone density in community-dwelling older adults substudy of a randomized controlled trial. J. Intern. Med. 2017, 282, 452–460. [Google Scholar] [CrossRef]
- Ceglia, L.; Niramitmahapanya, S.; da Silva Morais, M.; Rivas, D.A.; Harris, S.S.; Bischoff-Ferrari, H.; Fielding, R.A.; Dawson-Hughes, B. A randomized study on the effect of vitamin D3 supplementation on skeletal muscle morphology and vitamin D receptor concentration in older women. J. Clin. Endocrinol. Metab. 2013, 98, E1927–E1935. [Google Scholar] [CrossRef]
- Abshirini, M.; Mozaffari, H.; Kord-Varkaneh, H.; Omidian, M.; Kruger, M.C. The effects of vitamin D supplementation on muscle strength and mobility in postmenopausal women: A systematic review and meta-anaalysis of randomized controlled trials. J. Hum. Nutr. Diet. 2020, 33, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Okubo, T.; Atsukawa, M.; Tsubota, A.; Ono, H.; Kawano, T.; Yoshida, Y.; Arai, T.; Hayama, K.; Itokawa, N.; Kondo, C.; et al. Effect of Vitamin D Supplementation on Skeletal Muscle Volume and Strength in Patients with Decompensated Liver Cirrhosis Undergoing Branched Chain Amino Acids Supplementation: A Prospective, Randomized, Controlled Pilot Trial. Nutrients 2021, 13, 1874. [Google Scholar] [CrossRef] [PubMed]
- Ukon, Y.; Makino, T.; Kodama, J.; Tsukazaki, H.; Tateiwa, D.; Yoshikawa, H.; Kaito, T. Molecular-Based Treatment Strategies for Osteoporosis: A Literature Review. Int. J. Mol. Sci. 2019, 20, 2557. [Google Scholar] [CrossRef] [PubMed]
- Ono, T.; Hayashi, M.; Sasaki, F.; Nakashima, T. RANKL biology: Bone metabolism, the immune system, and beyond. Inflamm. Regen. 2020, 40, 2. [Google Scholar] [CrossRef]
- Saeki, C.; Saito, M.; Oikawa, T.; Nakano, M.; Torisu, Y.; Saruta, M.; Tsubota, A. Effects of denosumab treatment in chronic liver disease patients with osteoporosis. World. J. Gastroenterol. 2020, 26, 4960–4971. [Google Scholar] [CrossRef]
- Dufresne, S.S.; Dumont, N.A.; Boulanger-Piette, A.; Fajardo, V.A.; Gamu, D.; Kake-Guena, S.A.; David, R.O.; Bouchard, P.; Lavergne, É.; Penninger, J.M.; et al. Muscle RANK is a key regulator of Ca2+ storage, SERCA activity, and function of fast-twitch skeletal muscles. Am. J. Physiol. Cell. Physiol. 2016, 310, C663–C672. [Google Scholar] [CrossRef] [PubMed]
- Miedany, Y.E.; Gaafary, M.E.; Toth, M.; Hegazi, M.O.; Aroussy, N.E.; Hassan, W.; Almedany, S.; Nasr, A.; Bahlas, S.; Galal, S. Is there a potential dual effect of denosumab for treatment of osteoporosis and sarcopenia? Clin. Rheumatol. 2021, 1–8, online ahead of print. [Google Scholar] [CrossRef]
- Chotiyarnwong, P.; McCloskey, E.; Eastell, R.; McClung, M.R.; Gielen, E.; Gostage, J.; McDermott, M.; Chines, A.; Huang, S.; Cummings, S.R. A Pooled Analysis of Fall Incidence from Placebo-Controlled Trials of Denosumab. J. Bone Miner. Res. 2020, 35, 1014–1021. [Google Scholar] [CrossRef]
- Attie, K.M.; Borgstein, N.G.; Yang, Y.; Condon, C.H.; Wilson, D.M.; Pearsall, A.E.; Kumar, R.; Willins, D.A.; Seehra, J.S.; Sherman, M.L. A single ascending-dose study of muscle regulator ACE-031 in healthy volunteers. Muscle Nerve 2013, 47, 416–423. [Google Scholar] [CrossRef]
- Becker, C.; Lord, S.R.; Studenski, S.A.; Warden, S.J.; Fielding, R.A.; Recknor, C.P.; Hochberg, M.C.; Ferrari, S.L.; Blain, H.; Binder, E.F.; et al. Myostatin antibody (LY2495655) in older weak fallers: A proof-of-concept, randomised, phase 2 trial. Lancet Diabetes Endocrinol. 2015, 3, 948–957. [Google Scholar] [CrossRef]
- Campbell, C.; McMillan, H.J.; Mah, J.K.; Tarnopolsky, M.; Selby, K.; McClure, T.; Wilson, D.M.; Sherman, M.L.; Escolar, D.; Attie, K.M. Myostatin inhibitor ACE-031 treatment of ambulatory boys with Duchenne muscular dystrophy: Results of a randomized, placebo-controlled clinical trial. Muscle Nerve 2017, 55, 458–464. [Google Scholar] [CrossRef]
- Ozawa, T.; Morikawa, M.; Morishita, Y.; Ogikubo, K.; Itoh, F.; Koinuma, D.; Nygren, P.Å.; Miyazono, K. Systemic administration of monovalent follistatin-like 3-Fc-fusion protein increases muscle mass in mice. iScience 2021, 24, 102488. [Google Scholar] [CrossRef]
- Parise, G.; Snijders, T. Myostatin inhibition for treatment of sarcopenia. Lancet Diabetes Endocrinol. 2015, 3, 917–918. [Google Scholar] [CrossRef]


{kind=link}
| Measurement Method | JSH | EWGSOP | EWGSOP2 | AWGS | AWGS 2019 | |
|---|---|---|---|---|---|---|
| Muscle mass | DEXA | M: YAM − 2SD | M: 7.0 kg/m2 | M: 7.0 kg/m2 | M: 7.0 kg/m2 | |
| F: YAM − 2SD | F: 5.5 kg/m2 | F: 5.4 kg/m2 | F: 5.4 kg/m2 | |||
| BIA | M: 7.0 kg/m2 | M: 7.0 kg/m2 | M: 7.0 kg/m2 | |||
| F: 5.7 kg/m2 | F: 5.7 kg/m2 | F: 5.7 kg/m2 | ||||
| CT (L3 level) | M: 42 cm2/m2 | |||||
| F: 38 cm2/m2 | ||||||
| Muscle strength or Physical performance | Handgrip strength | M: 26 kg | M: 30kg | M: 27 kg | M: 26 kg | M: 28 kg |
| F: 18 kg | F: 20kg | F: 16 kg | F: 18 kg | F: 18 kg | ||
| Chair stand test (5-time) | 15 s | 12 s | ||||
| Gait speed | 0.8 m/s | 0.8 m/s | 0.8 m/s | 1.0 m/s | ||
| SPPB | 8 points | 8 points | 9 points | 9 points |
| Authors (Year, Country) [Reference] | Patients Characteristics | Prevalence of Osteosarcopenia | Diagnostic Method for Osteosarcopenia (Criteria for Sarcopenia) | Main Findings |
|---|---|---|---|---|
| Hayashi et al. (2018, Japan) [85] | 112 patients with CLD (LC, 36.0%) | 7.1% | DEXA and BIA (JSH) | Sarcopenia and LC were significantly associated with the BMD. Sarcopenia (OR, 6.16) and LC (OR, 15.8) were independent risk factors for osteoporosis. |
| Bering et al. (2018, Brazil) [86] | 104 patients with CHC | ― | DEXA (EWGSOP) | Low BMD, low muscle strength, pre-sarcopenia, and sarcopenia were noticed in 34.6%, 27.9%, 14.4%, and 8.7% of subjects, respectively. Appendicular skeletal muscle mass was an independent predictor of BMD. Sarcopenia was independently related to bone mineral content. |
| Saeki et al. (2020, Japan) [21] | 117 patients with PBC (LC, 9.4%) | 15.4% | DEXA and BIA (JSH) | The SMI and handgrip strength were significantly correlated with the BMD. Patients with osteosarcopenia had a higher prevalence of vertebral fracture (55.6%) than those without both sarcopenia and osteoporosis (6.7%). |
| Saeki et al. (2020, Japan) [22] | 291 patients with CLD (LC, 51.9%) | LC 20.5% Non-LC 12.9% | DEXA and BIA (JSH) | Frailty was an independent risk factor associated with osteosarcopenia (OR, 9.837), and vice versa (OR, 10.069). The prevalence of frailty and vertebral fracture was significantly higher in patients with osteosarcopenia than in those without osteosarcopenia (79.6% vs. 17.4% and 59.2% vs. 20.2%, respectively). |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saeki, C.; Tsubota, A. Influencing Factors and Molecular Pathogenesis of Sarcopenia and Osteosarcopenia in Chronic Liver Disease. Life 2021, 11, 899. https://doi.org/10.3390/life11090899
Saeki C, Tsubota A. Influencing Factors and Molecular Pathogenesis of Sarcopenia and Osteosarcopenia in Chronic Liver Disease. Life. 2021; 11(9):899. https://doi.org/10.3390/life11090899
Chicago/Turabian StyleSaeki, Chisato, and Akihito Tsubota. 2021. "Influencing Factors and Molecular Pathogenesis of Sarcopenia and Osteosarcopenia in Chronic Liver Disease" Life 11, no. 9: 899. https://doi.org/10.3390/life11090899
APA StyleSaeki, C., & Tsubota, A. (2021). Influencing Factors and Molecular Pathogenesis of Sarcopenia and Osteosarcopenia in Chronic Liver Disease. Life, 11(9), 899. https://doi.org/10.3390/life11090899