
Identification of Immune Function-Related Subtypes in Cutaneous Melanoma

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients and Samples
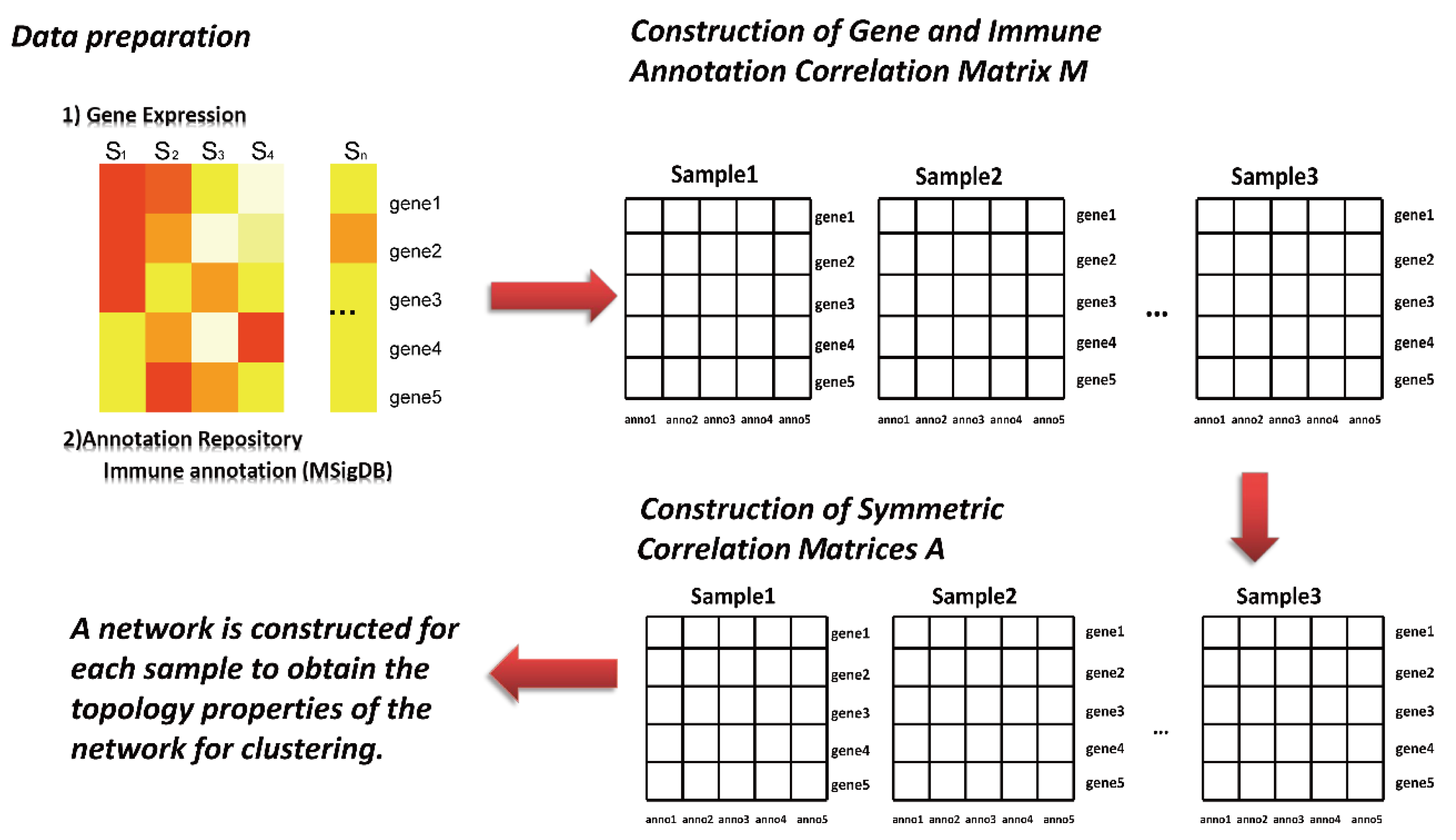
2.2. Sample-Specific Networks Were Constructed with the SING Method
- (1)
- The high-dimensional nature of gene expression data makes it necessary to reduce dimensionality before analysis. For each gene data set, the top 500 genes were selected according to the order of their total variance and included in subsequent analysis and calculation.
- (2)
- Construction of gene and immune annotation correlation matrix
- (3)
- is the correlation matrix between the gene and immune annotation, in which corresponds to the sample, corresponds to the first gene, and corresponds to the first annotation information in the immune annotation database. The corresponding element in the matrix (line , column ) is the expression value of ; otherwise, it is recorded as 0.
- (4)
- Development of sample-specific network
- (5)
- The symmetric correlation matrix is further generated by the matrix . Each element in the matrix corresponds to the Euclidean distance between two annotation pieces of information and . Afterward, to transform into a discrete network, this study further selected the edge of the top 10% weight in the symmetric correlation matrix to construct the adjacency matrix.
- (6)
- Computing network topology information.
2.3. Clustering Algorithm
2.4. Assessment of Immune and Stromal Cell Infiltration
2.5. Cytolytic Activity
2.6. Gene Differential Expression Analysis
2.7. Differential Gene Enrichment Analysis
2.8. Recognition of Cancer Mutation-Driven Genes and Calculation of Tumour Mutation Load (TMB)
2.9. DNA Methylation Differential Analysis
2.10. Survival Analyses
3. Results
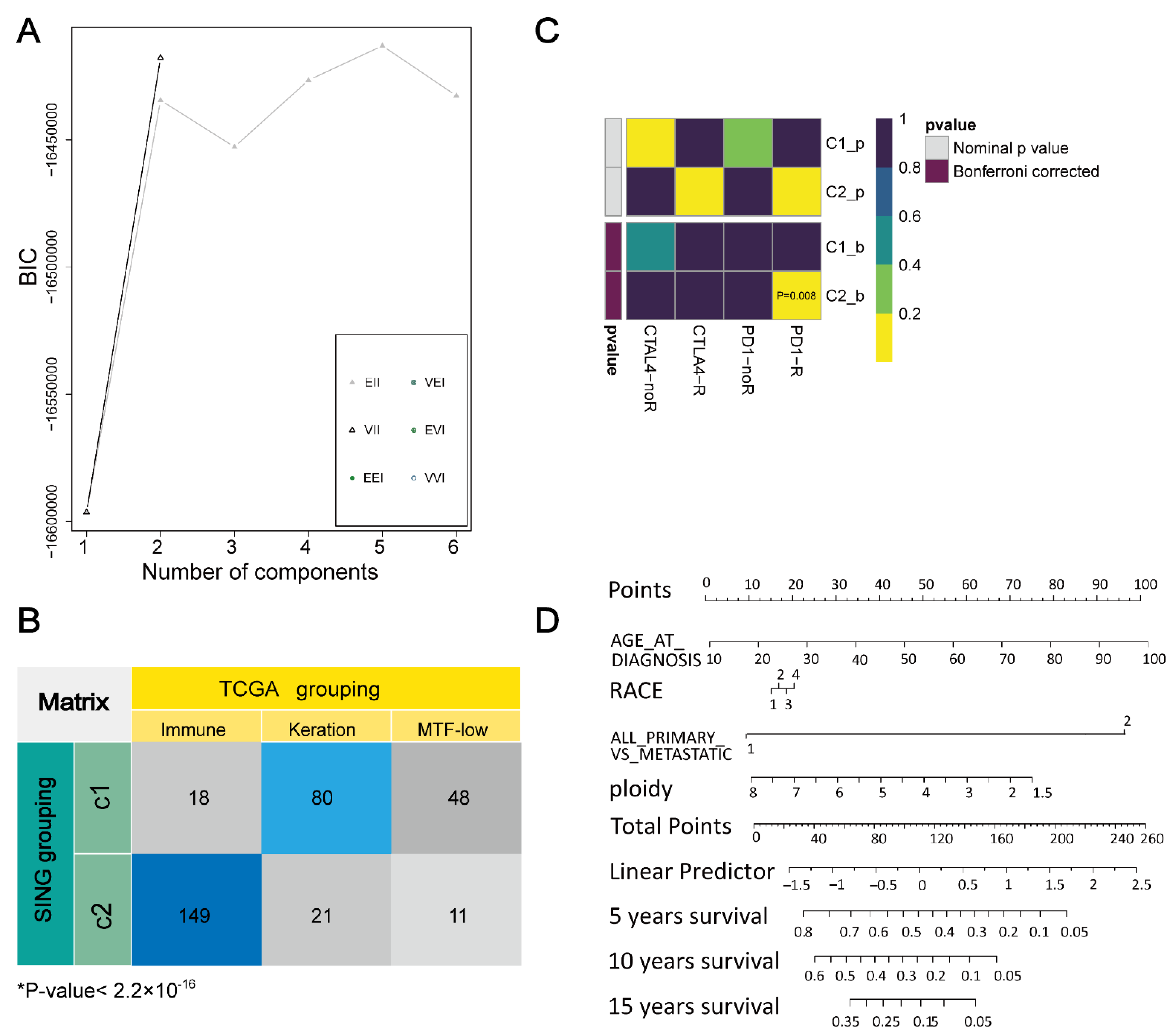
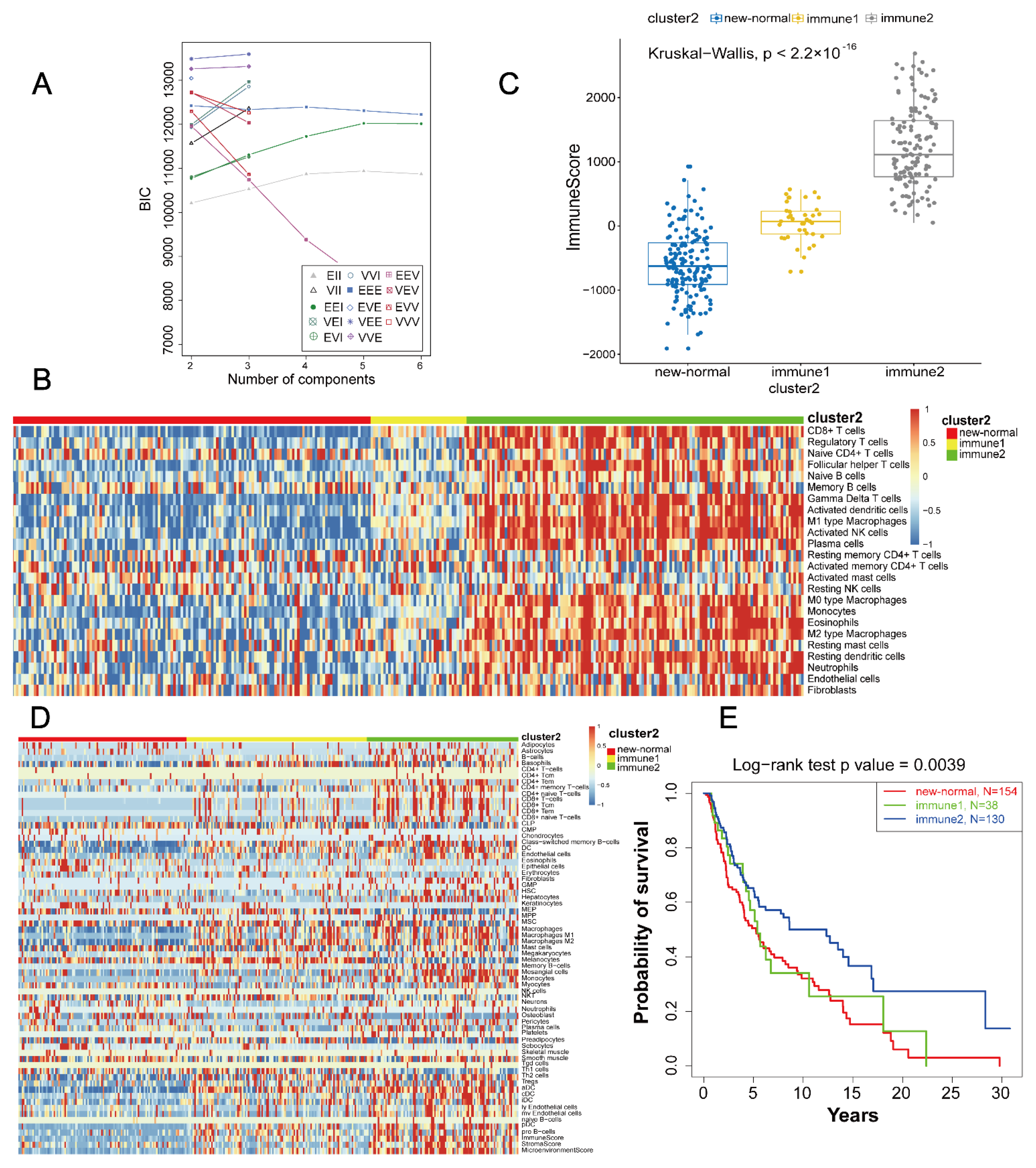
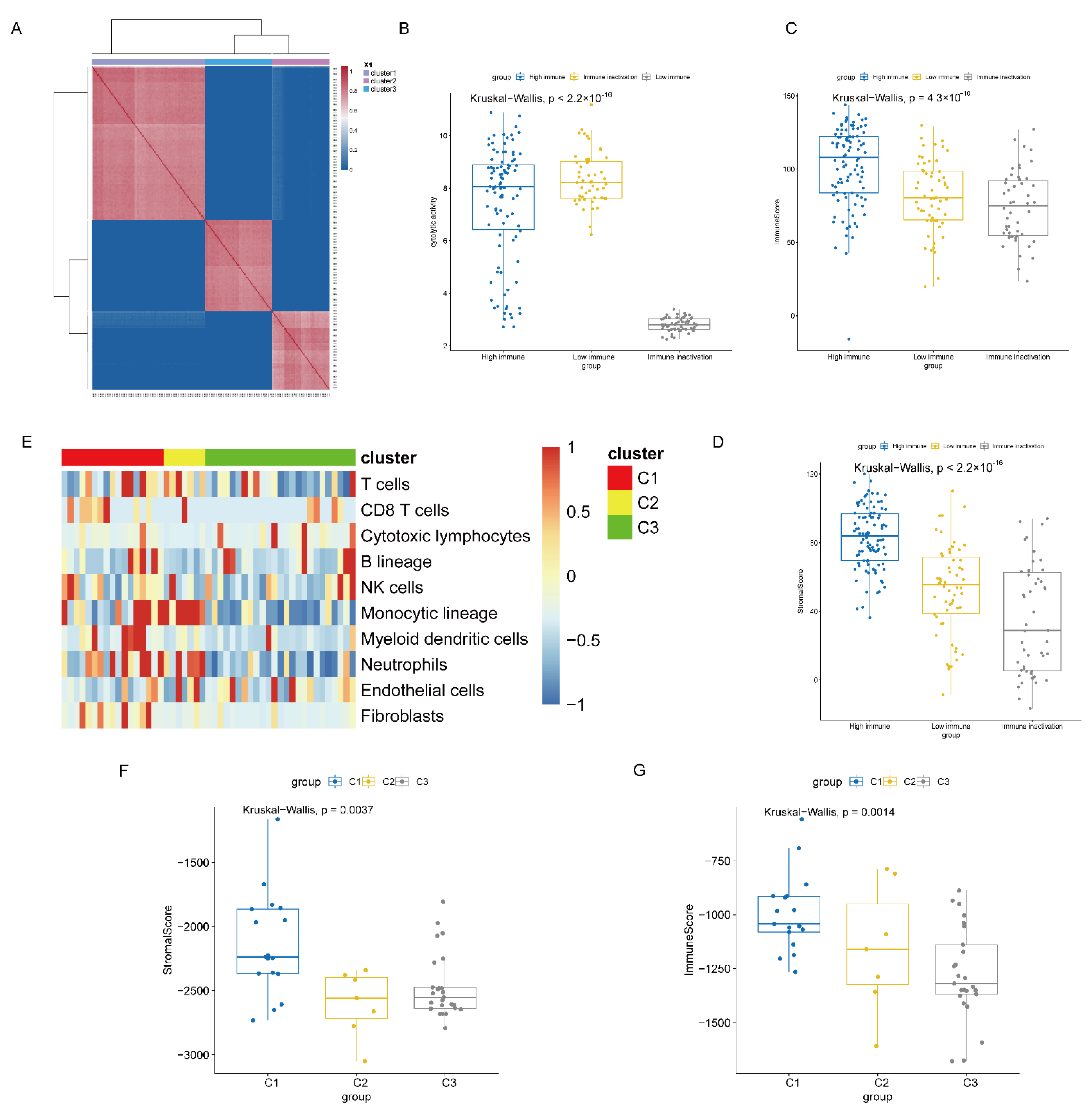
3.1. Determination Aand Verification of Cluster Number
3.2. Definition and Extraction of Immune Subtypes
3.3. Heterogeneity Analysis between High and Low Immune Subtypes
3.4. Immunity Subtype Validation
3.5. Specific Pathological and Prognostic Features of Immune Subtypes
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dai, X.; Li, T.; Bai, Z.; Yang, Y.; Liu, X.; Zhan, J.; Shi, B. Breast cancer intrinsic subtype classification, clinical use and future trends. Am. J. Cancer Res. 2015, 5, 2929–2943. [Google Scholar]
- Gendoo, D.M.; Ratanasirigulchai, N.; Schroder, M.S.; Pare, L.; Parker, J.S.; Prat, A.; Haibe-Kains, B. Genefu: An R/Bioconductor package for computation of gene expression-based signatures in breast cancer. Bioinformatics 2016, 32, 1097–1099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beattie, J.; Yarmus, L.; Wahidi, M.; Rivera, M.P.; Gilbert, C.; Maldonado, F.; Czarnecka, K.; Argento, A.; Chen, A.; Herth, F.; et al. The Immune Landscape of Non-Small-Cell Lung Cancer. Utility of Cytologic and Histologic Samples Obtained through Minimally Invasive Pulmonary Procedures. Am. J. Respir. Crit. Care Med. 2018, 198, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Dagogo-Jack, I.; Shaw, A.T. Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 2018, 15, 81–94. [Google Scholar] [CrossRef]
- Guo, W.F.; Zhang, S.W.; Liu, L.L.; Liu, F.; Shi, Q.Q.; Zhang, L.; Tang, Y.; Zeng, T.; Chen, L. Discovering personalized driver mutation profiles of single samples in cancer by network control strategy. Bioinformatics 2018, 34, 1893–1903. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, Q.; Zhao, W.; Qi, L.; Gu, Y.; Li, P.; Zhang, M.; Li, Y.; Liu, S.L.; Guo, Z. Individual-level analysis of differential expression of genes and pathways for personalized medicine. Bioinformatics 2015, 31, 62–68. [Google Scholar] [CrossRef]
- Yu, X.; Zhang, J.; Sun, S.; Zhou, X.; Zeng, T.; Chen, L. Individual-specific edge-network analysis for disease prediction. Nucleic Acids Res. 2017, 45, e170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gnant, M.; Harbeck, N.; Thomssen, C. St. Gallen/Vienna 2017: A Brief Summary of the Consensus Discussion about Escalation and De-Escalation of Primary Breast Cancer Treatment. Breast Care 2017, 12, 102–107. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.P.; Wang, Y.T.; Ji, H.B.; Aihara, K.; Chen, L.N. Personalized characterization of diseases using sample-specific networks. Nucleic Acids Res. 2016, 44, e164. [Google Scholar] [CrossRef]
- Wayteck, L.; Breckpot, K.; Demeester, J.; De Smedt, S.C.; Raemdonck, K. A personalized view on cancer immunotherapy. Cancer Lett. 2014, 352, 113–125. [Google Scholar] [CrossRef] [Green Version]
- Balaguer, M.A.D.; Fisher, A.P.; Clark, N.M.; Fernandez-Espinosa, M.G.; Moller, B.K.; Weijers, D.; Lohmann, J.U.; Williams, C.; Lorenzo, O.; Sozzani, R. Predicting gene regulatory networks by combining spatial and temporal gene expression data in Arabidopsis root stem cells. Proc. Natl. Acad. Sci. USA 2017, 114, E7632–E7640. [Google Scholar] [CrossRef] [Green Version]
- Qiu, X.; Rahimzamani, A.; Wang, L.; Mao, Q.; Durham, T.; McFaline-Figueroa, J.L.; Saunders, L.; Trapnell, C.; Kannan, S. Towards inferring causal gene regulatory networks from single cell expression Measurements. bioRxiv 2018, 10, 426981. [Google Scholar] [CrossRef]
- Liu, Z.P.; Wu, H.; Zhu, J.; Miao, H. Systematic identification of transcriptional and post-transcriptional regulations in human respiratory epithelial cells during influenza A virus infection. BMC Bioinform. 2014, 15, 336. [Google Scholar] [CrossRef] [Green Version]
- Borgwardt, K.M.; Kriegel, H.P.; Vishwanathan, S.V.; Schraudolph, N.N. Graph kernels for disease outcome prediction from protein-protein interaction networks. In Biocomputing 2007; World Scientific: Singapore, 2006; pp. 4–15. [Google Scholar] [CrossRef] [Green Version]
- Kuijjer, M.L.; Tung, M.G.; Yuan, G.; Quackenbush, J.; Glass, K. Estimating Sample-Specific Regulatory Networks. iScience 2019, 14, 226–240. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.; Lee, S.C.; Quinn, T.P.; Truong, B.; Li, X.; Tran, T.; Venkatesh, S.; Le, T.D. PAN: Personalized Annotation-based Networks for the Prediction of Breast Cancer Relapse. In IEEE/ACM Transactions on Computational Biology and Bioinformatics; IEEE: Piscataway, NJ, USA, 2021. [Google Scholar] [CrossRef]
- Chi, Z.; Li, S.; Sheng, X.; Si, L.; Cui, C.; Han, M.; Guo, J. Clinical presentation, histology, and prognoses of malignant melanoma in ethnic Chinese: A study of 522 consecutive cases. BMC Cancer 2011, 11, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonardi, G.C.; Falzone, L.; Salemi, R.; Zanghi, A.; Spandidos, D.A.; McCubrey, J.A.; Candido, S.; Libra, M. Cutaneous melanoma: From pathogenesis to therapy (Review). Int. J. Oncol. 2018, 52, 1071–1080. [Google Scholar] [CrossRef] [Green Version]
- Davis, L.E.; Shalin, S.C.; Tackett, A.J. Current state of melanoma diagnosis and treatment. Cancer Biol. Ther. 2019, 20, 1366–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erdag, G.; Schaefer, J.T.; Smolkin, M.E.; Deacon, D.H.; Shea, S.M.; Dengel, L.T.; Patterson, J.W.; Slingluff, C.L., Jr. Immunotype and immunohistologic characteristics of tumor-infiltrating immune cells are associated with clinical outcome in metastatic melanoma. Cancer Res. 2012, 72, 1070–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marra, A.; Ferrone, C.R.; Fusciello, C.; Scognamiglio, G.; Ferrone, S.; Pepe, S.; Perri, F.; Sabbatino, F. Translational Research in Cutaneous Melanoma: New Therapeutic Perspectives. Anticancer. Agents Med. Chem. 2018, 18, 166–181. [Google Scholar] [CrossRef]
- Riker, A.I.; Enkemann, S.A.; Fodstad, O.; Liu, S.H.; Ren, S.P.; Morris, C.; Xi, Y.G.; Howell, P.; Metge, B.; Samant, R.S.; et al. The gene expression profiles of primary and metastatic melanoma yields a transition point of tumor progression and metastasis. BMC Med. Genom. 2008, 1, 13. [Google Scholar] [CrossRef] [Green Version]
- Holtan, S.G.; Mansfield, A.S.; Creedon, D.J.; Nevala, W.K.; Haluska, P.; Leontovich, A.A.; Markovic, S.N. An organ system based approach to prognosis in advanced melanoma. Front. Biosci. Elite. Ed. 2012, 4, 2723–2733. Available online: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE23376 (accessed on 1 July 2021).
- Bogunovic, D.; O’Neill, D.W.; Belitskaya-Levy, I.; Vacic, V.; Yu, Y.L.; Adams, S.; Darvishian, F.; Berman, R.; Shapiro, R.; Pavlick, A.C.; et al. Immune profile and mitotic index of metastatic melanoma lesions enhance clinical staging in predicting patient survival. Proc. Natl. Acad. Sci. USA 2009, 106, 20429–20434. [Google Scholar] [CrossRef] [Green Version]
- Raskin, L.; Fullen, D.R.; Giordano, T.J.; Thomas, D.G.; Frohm, M.L.; Cha, K.B.; Ahn, J.; Mukherjee, B.; Johnson, T.M.; Gruber, S.B. Transcriptome Profiling Identifies HMGA2 as a Biomarker of Melanoma Progression and Prognosis. J. Investig. Derm. 2013, 133, 2585–2592. [Google Scholar] [CrossRef] [Green Version]
- Ghandi, M.; Huang, F.W.; Jane-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R., 3rd; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef]
- The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014, 507, 315–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becht, E.; Giraldo, N.A.; Lacroix, L.; Buttard, B.; Elarouci, N.; Petitprez, F.; Selves, J.; Laurent-Puig, P.; Sautes-Fridman, C.; Fridman, W.H.; et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016, 17, 218. [Google Scholar] [CrossRef]
- Aran, D.; Hu, Z.; Butte, A.J. xCell: Digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017, 18, 220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshihara, K.; Shahmoradgoli, M.; Martinez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Trevino, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef] [PubMed]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and Genetic Properties of Tumors Associated with Local Immune Cytolytic Activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef] [Green Version]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [Green Version]
- Dennis, G., Jr.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, r60. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Yu, G.C.; Wang, L.G.; Han, Y.Y.; He, Q.Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. Omics A J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Hanzelmann, S.; Castelo, R.; Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-Seq data. BMC Bioinform. 2013, 14, 7. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Steuer, C.E.; Ramalingam, S.S. Tumor Mutation Burden: Leading Immunotherapy to the Era of Precision Medicine? J. Clin. Oncol. 2018, 36, 631–632. [Google Scholar] [CrossRef] [PubMed]
- Morris, T.J.; Butcher, L.M.; Feber, A.; Teschendorff, A.E.; Chakravarthy, A.R.; Wojdacz, T.K.; Beck, S. ChAMP: 450k Chip Analysis Methylation Pipeline. Bioinformatics 2014, 30, 428–430. [Google Scholar] [CrossRef] [PubMed]
- Bland, J.M.; Altman, D.G. Statistics notes–Survival probabilities (the Kaplan-Meier method). Bri. Med. J. 1998, 317, 1572. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.J. Cox proportional hazards regression models for survival data in cancer research. Cancer Treat. Res. 2002, 113, 59–70. [Google Scholar] [CrossRef]
- Roemeling, S.; Roobol, M.J.; Kattan, M.W.; van der Kwast, T.H.; Steyerberg, E.W.; Schroder, F.H. Nomogram use for the prediction of indolent prostate cancer–Impact on screen-detected populations. Cancer Am. Cancer Soc. 2007, 110, 2218–2221. [Google Scholar] [CrossRef]
- Huang, R.; Mao, M.; Lu, Y.; Yu, Q.; Liao, L. A novel immune-related genes prognosis biomarker for melanoma: Associated with tumor microenvironment. Aging 2020, 12, 6966–6980. [Google Scholar] [CrossRef]
- Hu, B.; Wei, Q.; Zhou, C.; Ju, M.; Wang, L.; Chen, L.; Li, Z.; Wei, M.; He, M.; Zhao, L. Analysis of immune subtypes based on immunogenomic profiling identifies prognostic signature for cutaneous melanoma. Int. Immunopharmacol. 2020, 89, 107162. [Google Scholar] [CrossRef] [PubMed]
- Roh, W.; Chen, P.L.; Reuben, A.; Spencer, C.N.; Prieto, P.A.; Miller, J.P.; Gopalakrishnan, V.; Wang, F.; Cooper, Z.A.; Reddy, S.M.; et al. Integrated molecular analysis of tumor biopsies on sequential CTLA-4 and PD-1 blockade reveals markers of response and resistance. Sci. Transl. Med. 2017, 9, eaah3560. [Google Scholar] [CrossRef] [Green Version]
- Zeng, H.L.; Jorapur, A.; Shain, A.H.; Lang, U.E.; Torres, R.; Zhang, Y.T.; McNeal, A.S.; Botton, T.; Lin, J.; Donne, M.; et al. Bi-allelic Loss of CDKN2A Initiates Melanoma Invasion via BRN2 Activation. Cancer Cell 2018, 34, 56–68. [Google Scholar] [CrossRef]
- Chen, W.Z.; Cheng, P.; Jiang, J.X.; Ren, Y.Q.; Wu, D.; Xue, D. Epigenomic and genomic analysis of transcriptome modulation in skin cutaneous melanoma. Aging 2020, 12, 12703–12725. [Google Scholar] [CrossRef]
- Lee, W.; Huang, D.S.; Han, K. Constructing cancer patient-specific and group-specific gene networks with multi-omics data. BMC Med. Genom. 2020, 13, 81. [Google Scholar] [CrossRef]
- Rodig, S.J.; Gusenleitner, D.; Jackson, D.G.; Gjini, E.; Giobbie-Hurder, A.; Jin, C.; Chang, H.; Lovitch, S.B.; Horak, C.; Weber, J.S.; et al. MHC proteins confer differential sensitivity to CTLA-4 and PD-1 blockade in untreated metastatic melanoma. Sci. Transl. Med. 2018, 10, aar3342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Gu, J. Efficacy and safety of PD-1 inhibitors for treating advanced melanoma: A systematic review and meta-analysis. Immunotherapy 2018, 10, 1293–1302. [Google Scholar] [CrossRef]
- Gellrich, F.F.; Schmitz, M.; Beissert, S.; Meier, F. Anti-PD-1 and Novel Combinations in the Treatment of Melanoma-An Update. J. Clin. Med. 2020, 9, 223. [Google Scholar] [CrossRef] [Green Version]
- Sana, G.; Madigan, J.P.; Gartner, J.J.; Fourrez, M.; Lin, J.; Qutob, N.; Narayan, J.; Shukla, S.; Ambudkar, S.V.; Xia, D.; et al. Exome Sequencing of ABCB5 Identifies Recurrent Melanoma Mutations that Result in Increased Proliferative and Invasive Capacities. J. Investig. Dermatol. 2019, 139, 1985–1992. [Google Scholar] [CrossRef] [Green Version]
- Batlle, E.; Massague, J. Transforming Growth Factor-beta Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Zaiatz-Bittencourt, V.; Finlay, D.K.; Gardiner, C.M. Canonical TGF-beta Signaling Pathway Represses Human NK Cell Metabolism. J. Immunol. 2018, 200, 3934–3941. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Pang, Y.; Moses, H.L. TGF-beta and immune cells: An important regulatory axis in the tumor microenvironment and progression. Trends Immunol. 2010, 31, 220–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Travis, M.A.; Sheppard, D. TGF-beta activation and function in immunity. Annu Rev. Immunol. 2014, 32, 51–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.Y.; Chung, C.L.; Hu, T.H.; Chen, J.J.; Liu, P.F.; Chen, C.L. Recent progress in TGF-beta inhibitors for cancer therapy. Biomed. Pharm. 2021, 134, 111046. [Google Scholar] [CrossRef]
- Morris, J.C.; Tan, A.R.; Olencki, T.E.; Shapiro, G.I.; Dezube, B.J.; Reiss, M.; Hsu, F.J.; Berzofsky, J.A.; Lawrence, D.P. Phase I study of GC1008 (fresolimumab): A human anti-transforming growth factor-beta (TGFbeta) monoclonal antibody in patients with advanced malignant melanoma or renal cell carcinoma. PLoS ONE 2014, 9, e90353. [Google Scholar] [CrossRef]
- Kaczorowski, M.; Biecek, P.; Donizy, P.; Pieniazek, M.; Matkowski, R.; Halon, A. SMAD7 is a novel independent predictor of survival in patients with cutaneous melanoma. Transl. Res. 2019, 204, 72–81. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, X.L.; Wang, G.Y.; Cui, X.C. Triple Combination Therapy With PD-1/PD-L1, BRAF, and MEK Inhibitor for Stage III-IV Melanoma: A Systematic Review and Meta-Analysis. Front. Oncol. 2021, 11, 2088. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| KERRYPNX | FRE (%) | Specific Immune Subtype | p-Value | |||
|---|---|---|---|---|---|---|
| Immune in Activation Subtype | Low Immune Subtype | High Immune Subtype | ||||
| Age | ≤60 | 179 (55) | 79 | 23 | 77 | 0.64 |
| >60 | 144 (44) | 75 | 15 | 54 | ||
| NA | 4 (1) | 2 | 0 | 2 | ||
| TNM Stage | 0 + I + II | 163 (50) | 89 | 21 | 53 | 0.05 |
| III + IV | 137 (42) | 57 | 14 | 66 | ||
| NA | 27 (8) | 10 | 3 | 14 | ||
| Race | White | 318 (97) | 150 | 37 | 131 | 0.43 |
| Asians | 7 (2) | 5 | 1 | 1 | ||
| Black or African American | 1 (0) | 0 | 0 | 1 | ||
| NA | 1 (0) | 1 | 0 | 0 | ||
| BMI | ≤25 | 46 (14) | 27 | 5 | 14 | 0.52 |
| >25 | 90 (28) | 42 | 12 | 36 | ||
| NA | 191 (58) | 87 | 21 | 83 | ||
| Tumour Status | Primary | 62 (19) | 49 | 5 | 8 | 6.214 × 10−8 * |
| Metastatic | 265 (81) | 107 | 33 | 125 | ||
| UV Exposure Rate | ≤0.8 | 83 (25) | 47 | 11 | 25 | 0.07 |
| >0.8 | 244 (75) | 109 | 27 | 108 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, L.; Zhu, J.; Jin, T.; Huang, M.; Chen, Y.; Xu, L.; Chen, W.; Jiang, B.; Yan, F. Identification of Immune Function-Related Subtypes in Cutaneous Melanoma. Life 2021, 11, 925. https://doi.org/10.3390/life11090925
Liu L, Zhu J, Jin T, Huang M, Chen Y, Xu L, Chen W, Jiang B, Yan F. Identification of Immune Function-Related Subtypes in Cutaneous Melanoma. Life. 2021; 11(9):925. https://doi.org/10.3390/life11090925
Chicago/Turabian StyleLiu, Lin, Junkai Zhu, Tong Jin, Mengjia Huang, Yi Chen, Li Xu, Wenxuan Chen, Bo Jiang, and Fangrong Yan. 2021. "Identification of Immune Function-Related Subtypes in Cutaneous Melanoma" Life 11, no. 9: 925. https://doi.org/10.3390/life11090925