
Distinct Mechanisms of Human Retinal Endothelial Barrier Modulation In Vitro by Mediators of Diabetes and Uveitis



Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
2.2. Western Blot Analysis
2.3. Measurement of Endothelial-Barrier Resistance
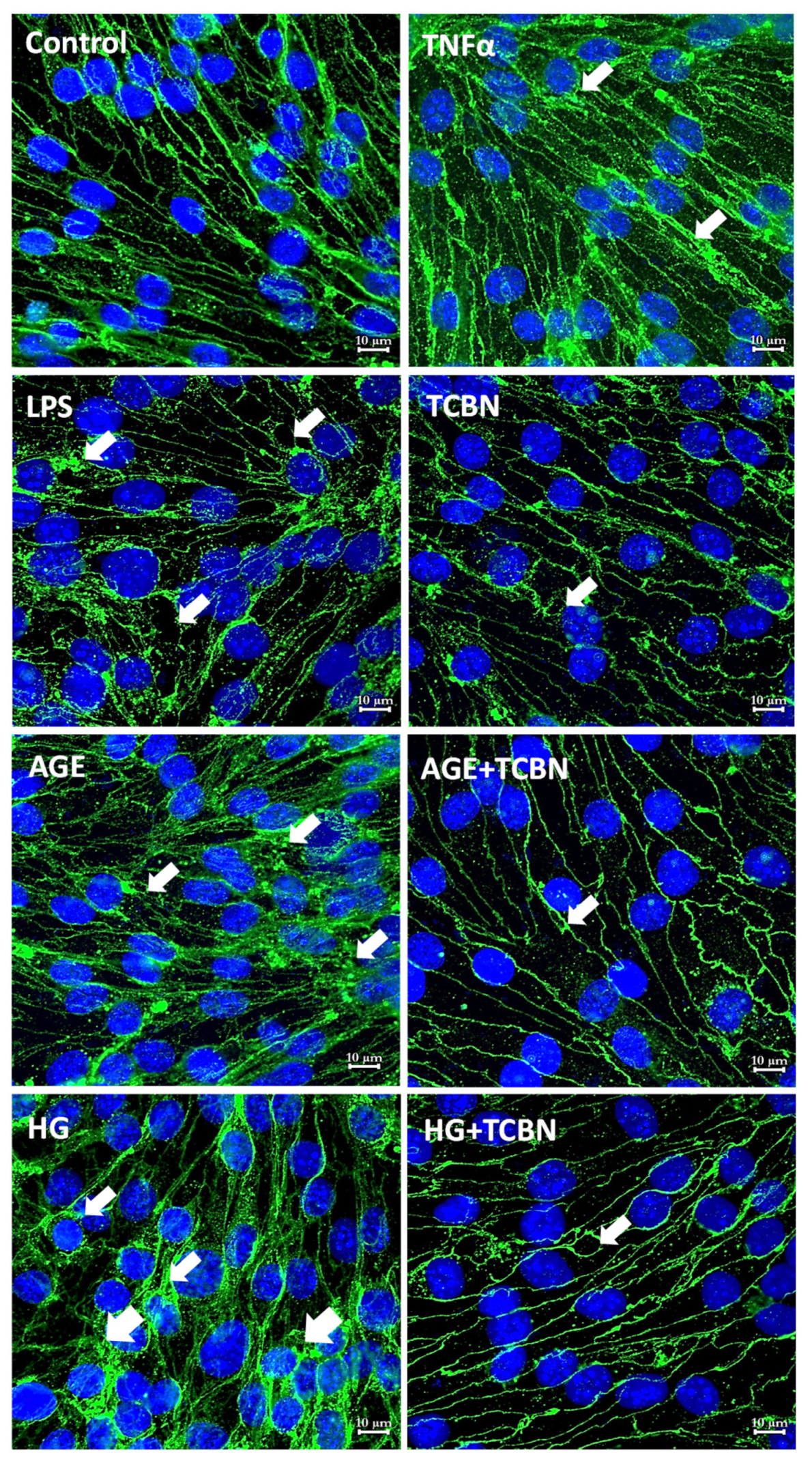
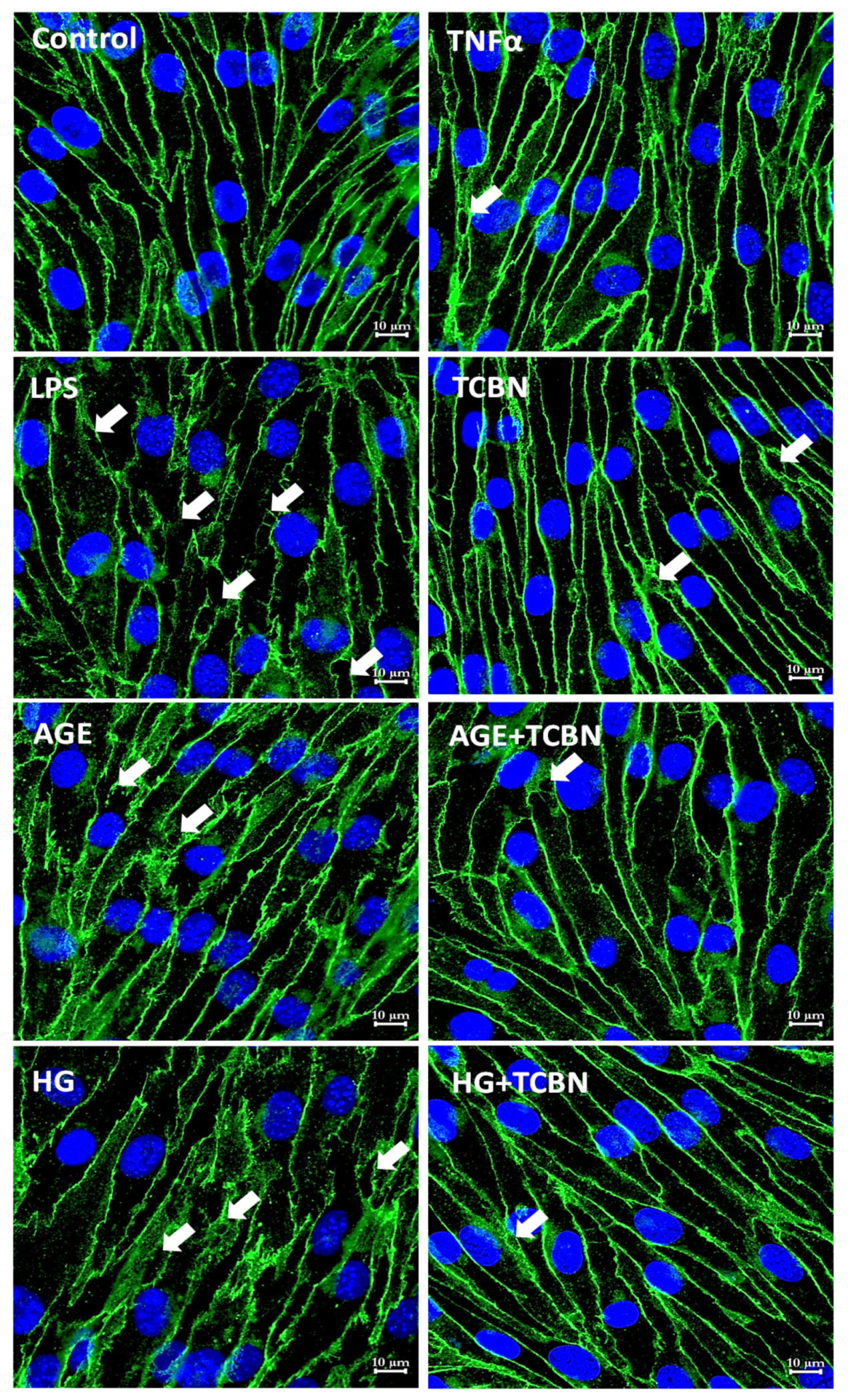
2.4. Immunofluorescence Staining and Confocal Imaging
2.5. Statistical Analysis
3. Results
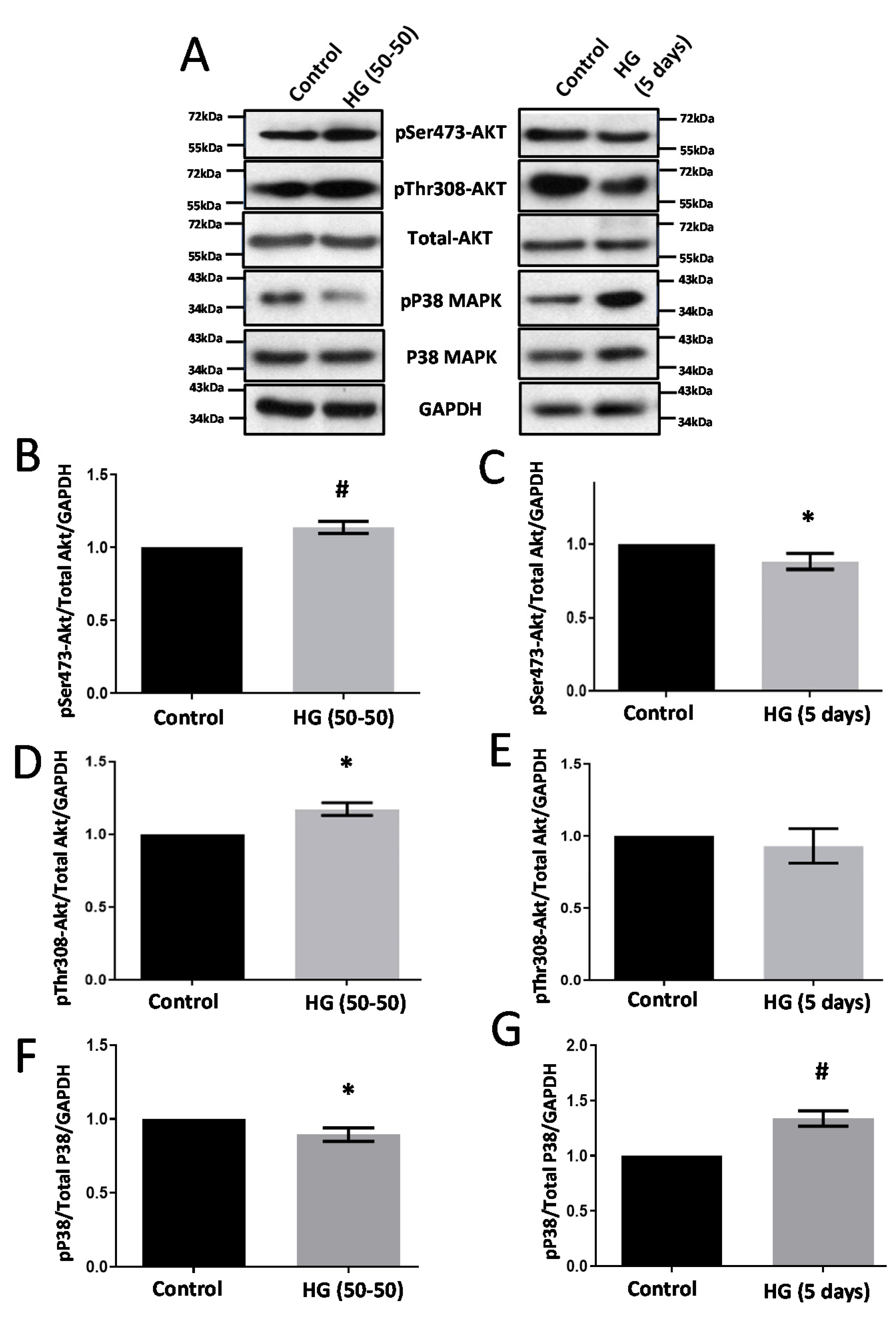
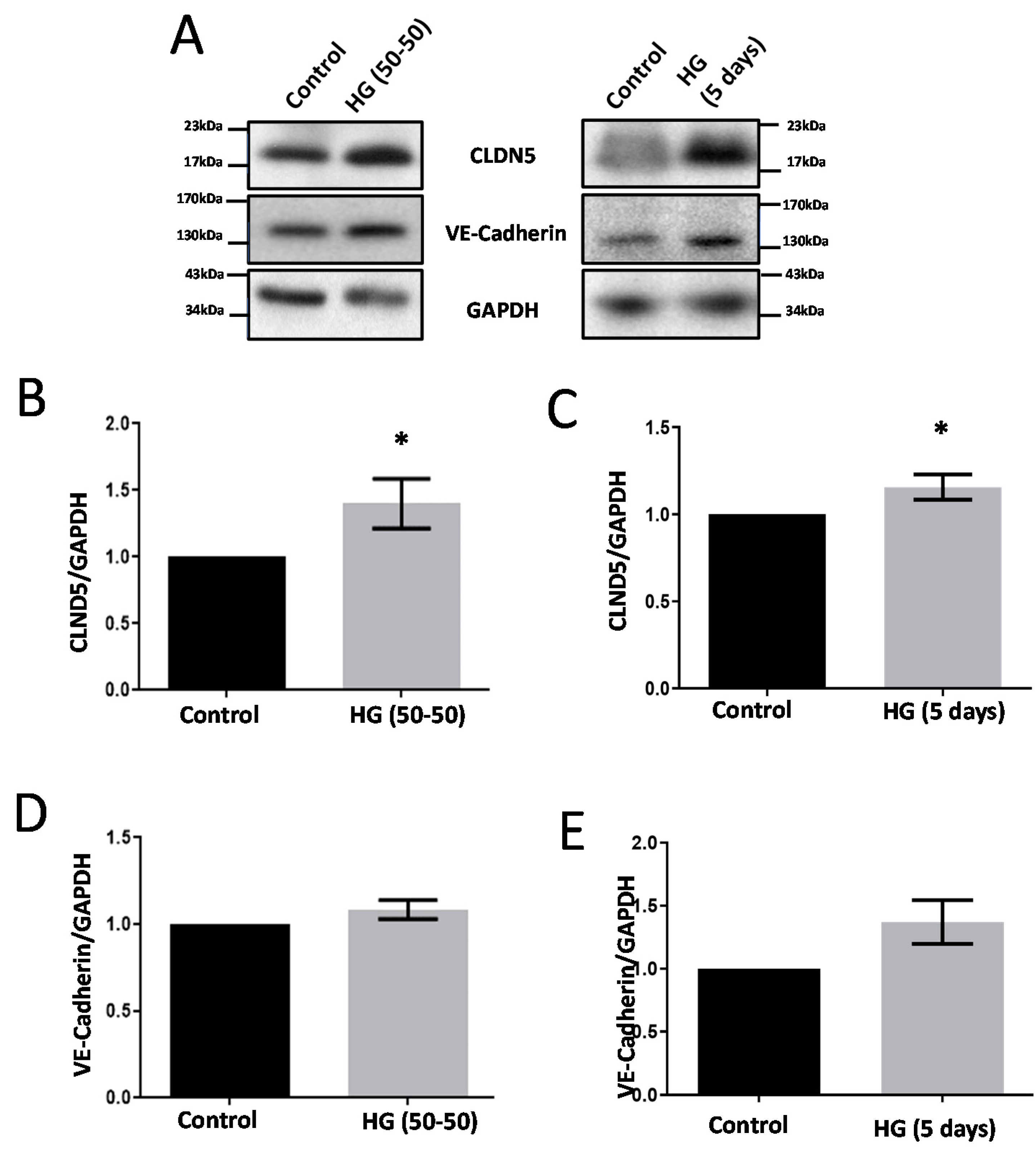
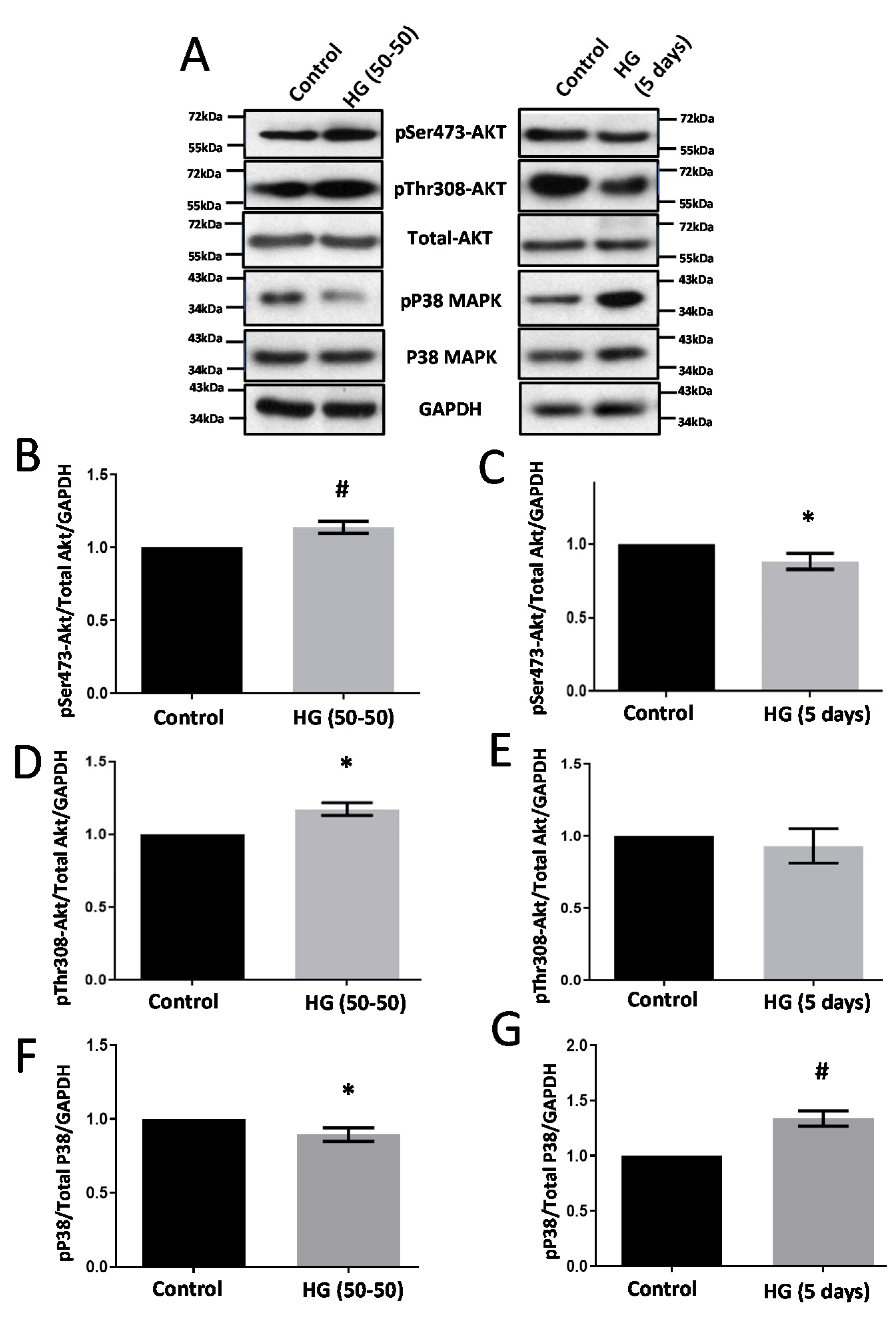
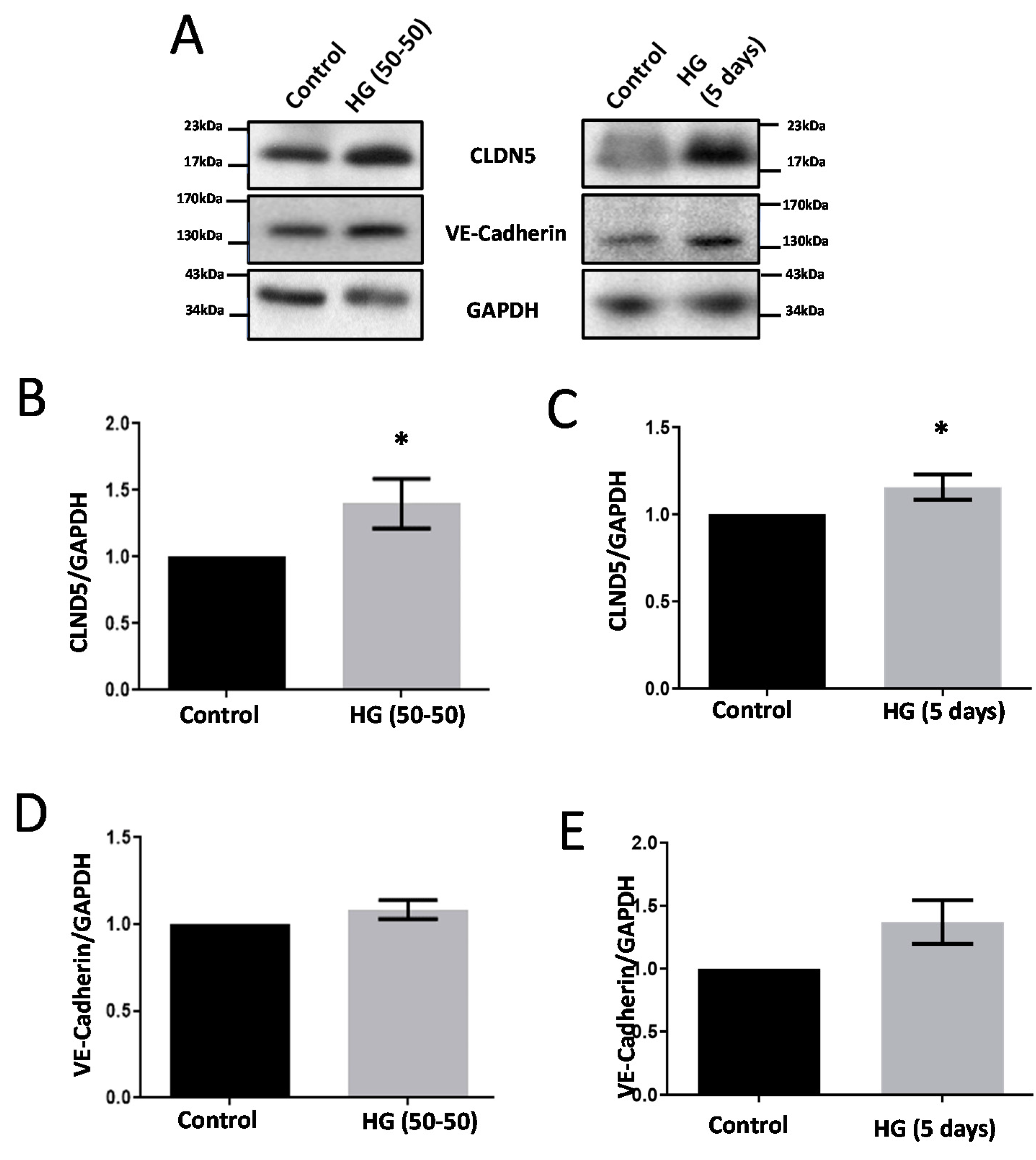
3.1. Treatment with HG Increases CLDN5 Expression
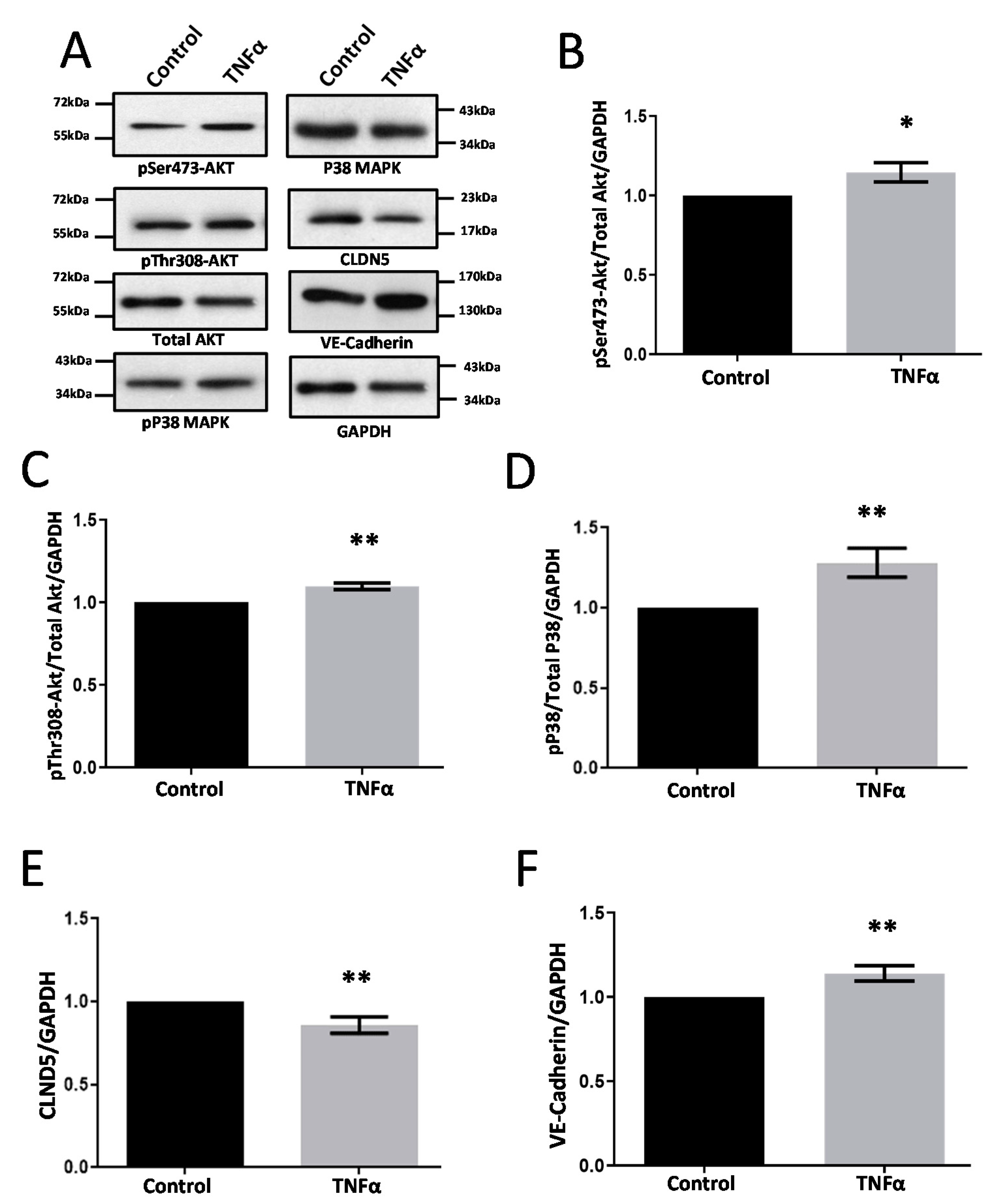
3.2. TNFα Induces Modest Changes in Cell-Junction Protein Expression in HRECs
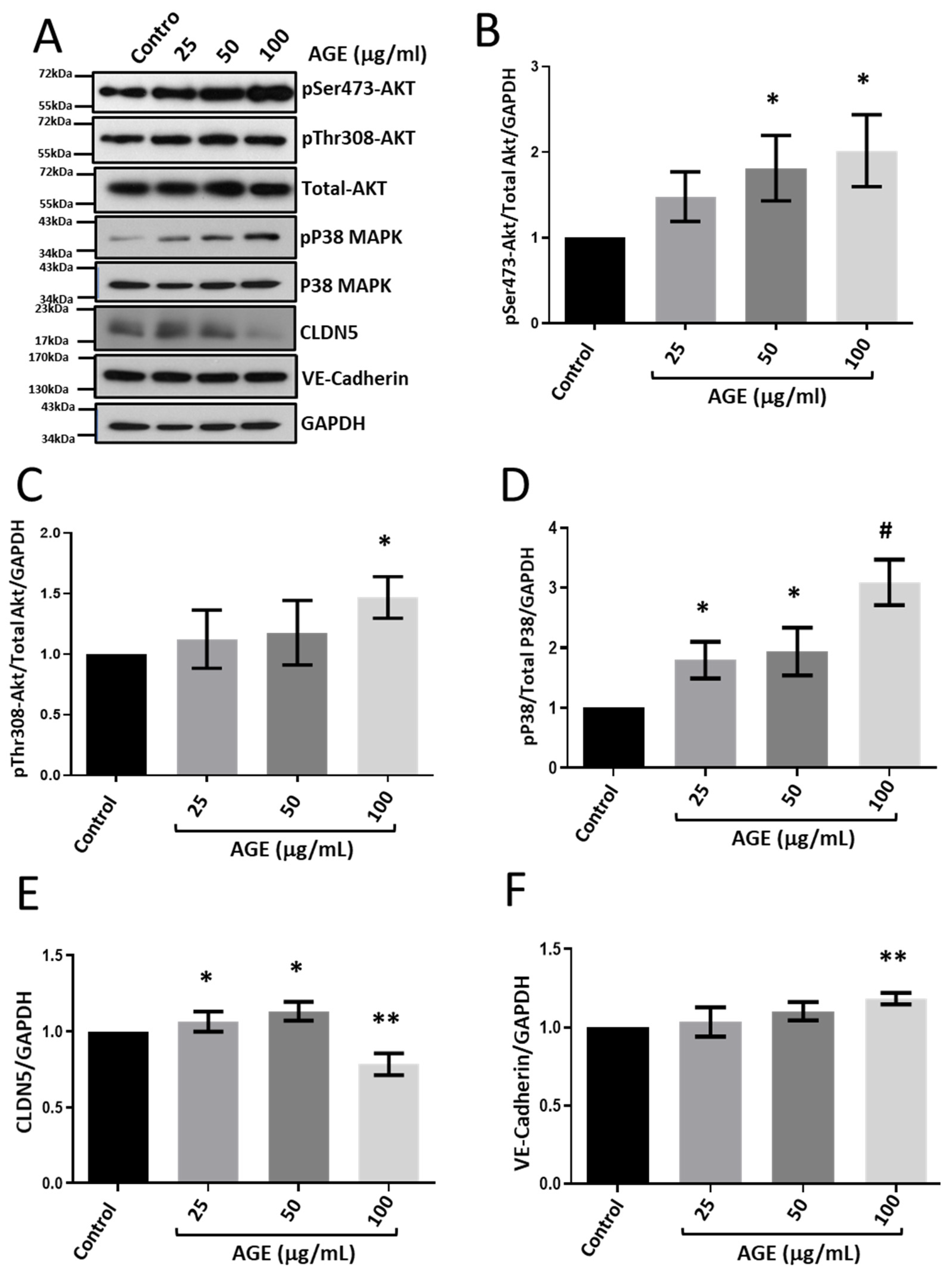
3.3. Treatment with BSA-Bound AGE Modulates CLDN5 Expression in HRECs
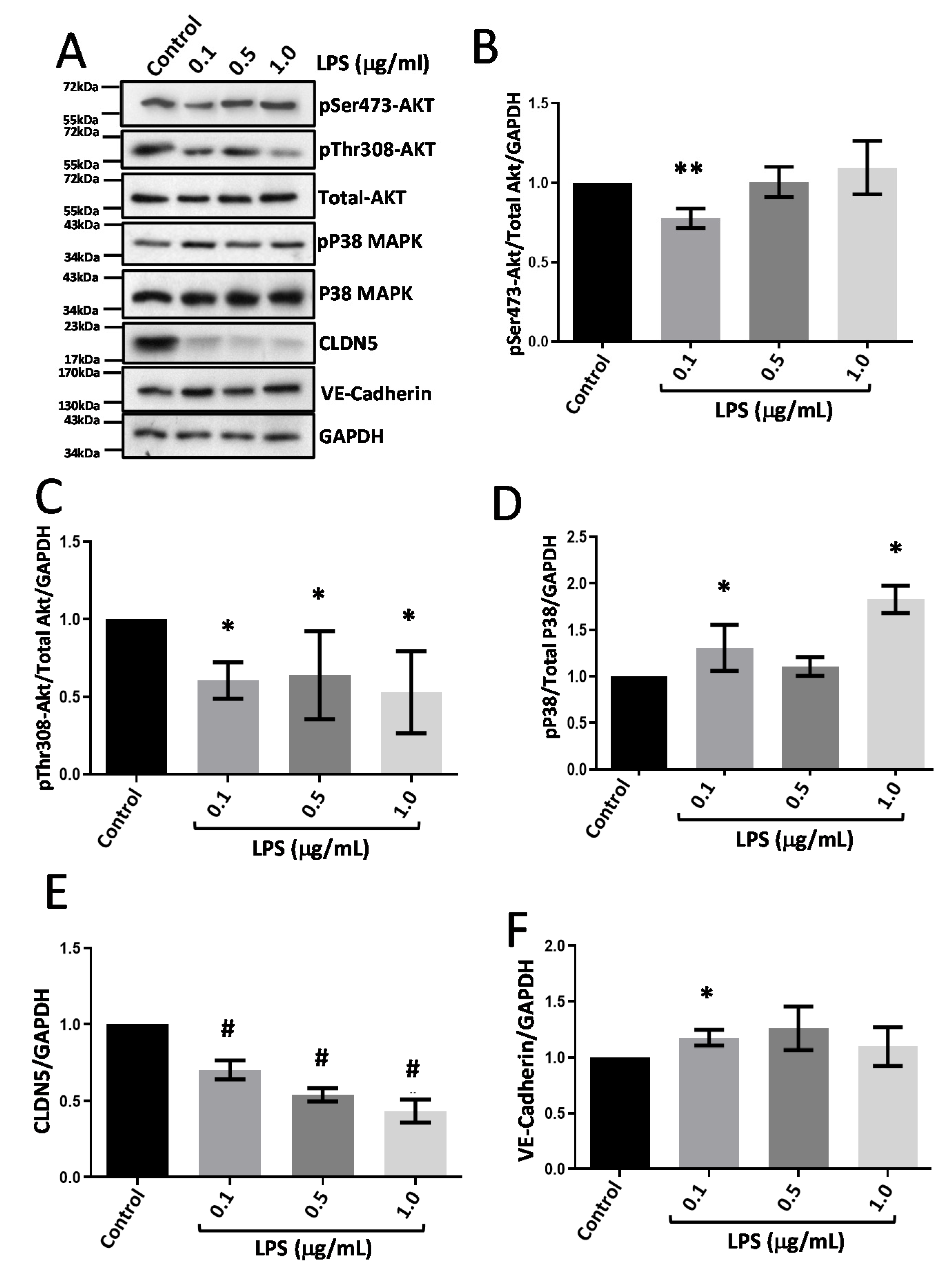
3.4. LPS Treatment Suppresses CLDN5 Expression with No Changes in VE-Cadherin
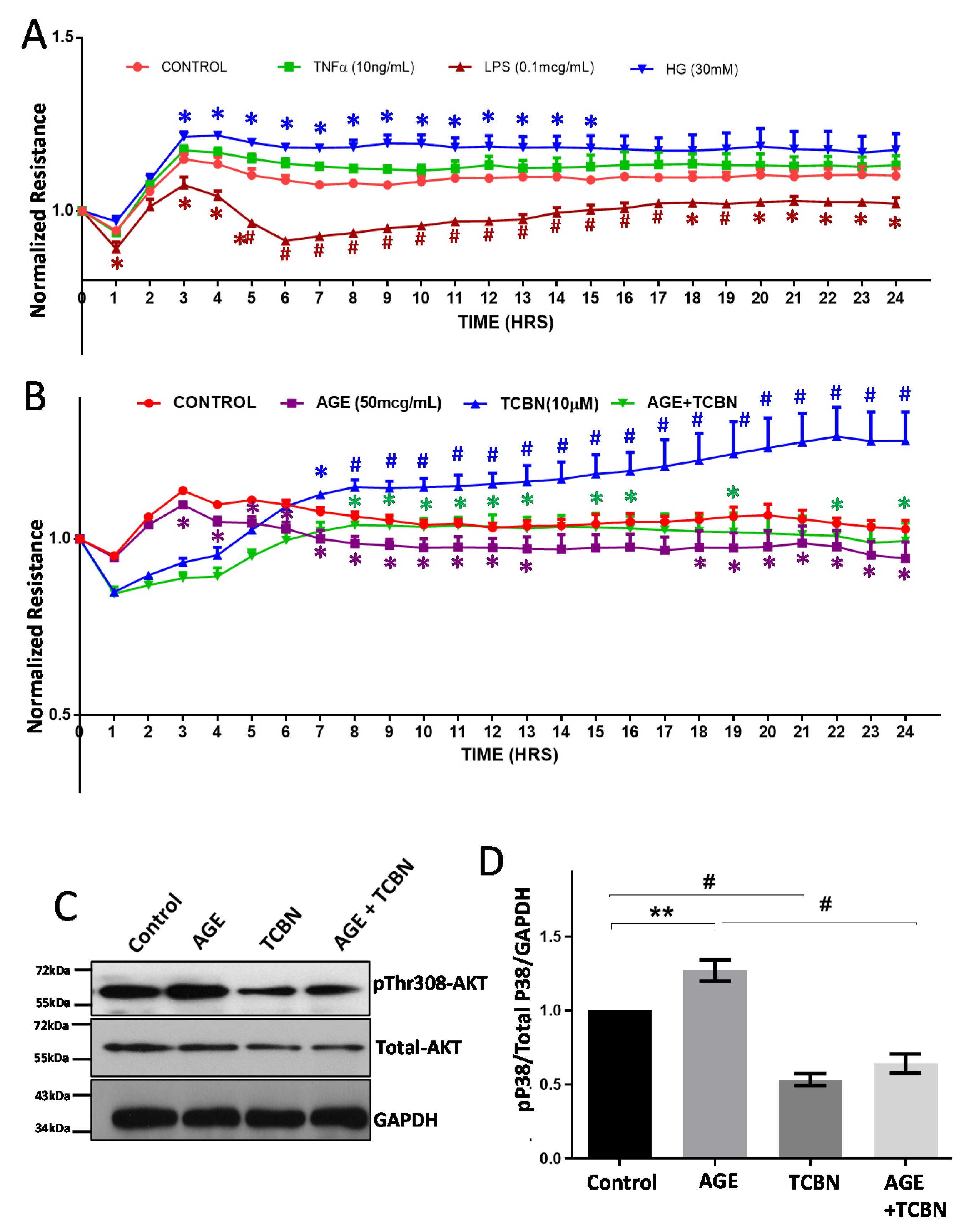
3.5. Modulators of Diabetes and Infection Associated Inflammation Have Distinct Effects and Mechanisms to Modulate the HREC Barrier
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Working Dilution (Primary) | Working Dilution (Secondary) |
|---|---|---|
| pSer 473 Akt | 1:1000 | 1:2000 |
| Pan Akt | 1:1000 | 1:2000 |
| pP38 MAPK | 1:1000 | 1:2000 |
| Total P38MAPK | 1:1000 | 1:2000 |
| CLDN5 | 1:2000 | 1:5000 |
| VE-Cadherin | 1:1000 | 1:2000 |
| GAPDH | 1:5000 | 1:10,000 |
References
- GBD 2019 Blindness; Vision Impairment Collaborators; the Vision Loss Expert Group of the Global Burden of Disease Study. Causes of blindness and vision impairment in 2020 and trends over 30 years, and prevalence of avoidable blindness in relation to VISION 2020: The Right to Sight: An analysis for the Global Burden of Disease Study. Lancet Glob. Health 2021, 9, e144–e160. [Google Scholar] [CrossRef]
- Cunha-Vaz, J.; Bernardes, R.; Lobo, C. Blood-Retinal Barrier. Eur. J. Ophthalmol. 2010, 21, 3–9. [Google Scholar] [CrossRef]
- Cunha-Vaz, J. The Blood-Retinal Barrier in the Management of Retinal Disease: EURETINA Award Lecture. Ophthalmologica 2017, 237, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.-Z.; Le, Y.-Z. Significance of Outer Blood–Retina Barrier Breakdown in Diabetes and Ischemia. Investig. Ophthalmol. Vis. Sci. 2011, 52, 2160–2164. [Google Scholar] [CrossRef] [PubMed]
- Rudraraju, M.; Narayanan, S.P.; Somanath, P.R. Regulation of blood-retinal barrier cell-junctions in diabetic retinopathy. Pharmacol. Res. 2020, 161, 105115. [Google Scholar] [CrossRef]
- Fresta, C.G.; Fidilio, A.; Caruso, G.; Caraci, F.; Giblin, F.J.; Leggio, G.M.; Salomone, S.; Drago, F.; Bucolo, C. A New Human Blood-Retinal Barrier Model Based on Endothelial Cells, Pericytes, and Astrocytes. Int. J. Mol. Sci. 2020, 21, 1636. [Google Scholar] [CrossRef] [Green Version]
- Campbell, M.; Humphries, P. The Blood-Retina Barrier. In Biology and Regulation of Blood-Tissue Barriers; Cheng, C.Y., Ed.; Springer: New York, NY, USA, 2013; pp. 70–84. [Google Scholar]
- Bauer, H.-C.; Krizbai, I.A.; Bauer, H.; Traweger, A. “You Shall Not Pass”—Tight junctions of the blood brain barrier. Front. Neurosci. 2014, 8, 392. [Google Scholar] [CrossRef]
- Hou, J. The kidney tight junction (Review). Int. J. Mol. Med. 2014, 34, 1451–1457. [Google Scholar] [CrossRef] [Green Version]
- Koval, M. Tight junctions, but not too tight: Fine control of lung permeability by claudins. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L217–L218. [Google Scholar] [CrossRef] [Green Version]
- Naylor, A.; Hopkins, A.; Hudson, N.; Campbell, M. Tight Junctions of the Outer Blood Retina Barrier. Int. J. Mol. Sci. 2019, 21, 211. [Google Scholar] [CrossRef] [Green Version]
- Adil, M.S.; Narayanan, S.P.; Somanath, P.R. Cell-cell junctions: Structure and regulation in physiology and pathology. Tissue Barriers 2021, 9, 1848212. [Google Scholar] [CrossRef] [PubMed]
- Gui, F.; You, Z.; Fu, S.; Wu, H.; Zhang, Y. Endothelial Dysfunction in Diabetic Retinopathy. Front. Endocrinol. 2020, 11, 591. [Google Scholar] [CrossRef]
- Kim, S.R.; Suh, W. Beneficial effects of the Src inhibitor, dasatinib, on breakdown of the blood-retinal barrier. Arch. Pharm. Res. 2017, 40, 197–203. [Google Scholar] [CrossRef]
- Alizadeh, E.; Mammadzada, P.; André, H. The Different Facades of Retinal and Choroidal Endothelial Cells in Response to Hypoxia. Int. J. Mol. Sci. 2018, 19, 3846. [Google Scholar] [CrossRef] [Green Version]
- Eelen, G.; de Zeeuw, P.; Treps, L.; Harjes, U.; Wong, B.W.; Carmeliet, P. Endothelial Cell Metabolism. Physiol. Rev. 2018, 98, 3–58. [Google Scholar] [CrossRef] [PubMed]
- Grammas, P.; Riden, M. Retinal endothelial cells are more susceptible to oxidative stress and increased permeability than brain-derived endothelial cells. Microvasc. Res. 2003, 65, 18–23. [Google Scholar] [CrossRef]
- Bharadwaj, A.S.; Appukuttan, B.; Wilmarth, P.A.; Pan, Y.; Stempel, A.J.; Chipps, T.J.; Benedetti, E.E.; Zamora, D.O.; Choi, D.; David, L.L.; et al. Role of the retinal vascular endothelial cell in ocular disease. Prog. Retin. Eye Res. 2013, 32, 102–180. [Google Scholar] [CrossRef] [Green Version]
- Curcio, F.; Ceriello, A. Decreased cultured endothelial cell proliferation in high glucose medium is reversed by antioxidants: New insights on the pathophysiological mechanisms of diabetic vascular complications. Vitr. Cell. Dev. Biol.-Anim. 1992, 28a, 787–790. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Schoenkerman, A.; Lowe, W.L. Activation of members of the mitogen-activated protein kinase family by glucose in endothelial cells. Am. J. Physiol.-Endocrinol. Metab. 2000, 279, E782–E790. [Google Scholar] [CrossRef] [PubMed]
- Kamal, K.; Du, W.; Mills, I.; Sumpio, B.E. Antiproliferative effect of elevated glucose in human microvascular endothelial cells. J. Cell Biochem. 1998, 71, 491–501. [Google Scholar] [CrossRef]
- Graier, W.F.; Grubenthal, I.; Dittrich, P.; Wascher, T.C.; Kostner, G.M. Intracellular mechanism of high D-glucose-induced modulation of vascular cell proliferation. Eur. J. Pharmacol. 1995, 294, 221–229. [Google Scholar] [CrossRef]
- La Selva, M.; Beltramo, E.; Pagnozzi, F.; Bena, E.; Molinatti, P.A.; Molinatti, G.M.; Porta, M. Thiamine corrects delayed replication and decreases production of lactate and advanced glycation end-products in bovine retinal and human umbilical vein endothelial cells cultured under high glucose conditions. Diabetologia 1996, 39, 1263–1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morisaki, N.; Watanabe, S.; Fukuda, K.; Saito, Y. Angiogenic interaction between retinal endothelial cells and pericytes from normal and diabetic rabbits, and phenotypic changes of diabetic cells. Cell. Mol. Biol. 1999, 45, 67–77. [Google Scholar] [PubMed]
- Rymaszewski, Z.; Szymanski, P.T.; Abplanalp, W.A.; Myatt, L.; Di Salvo, J.; Cohen, R.M. Human retinal vascular cells differ from umbilical cells in synthetic functions and their response to glucose. Proc. Soc. Exp. Biol. Med. 1992, 199, 183–191. [Google Scholar] [CrossRef]
- Huang, Q.; Sheibani, N. High glucose promotes retinal endothelial cell migration through activation of Src,PI3K/Akt1/eNOS, and ERKs. Am. J. Physiol. -Cell Physiol. 2008, 295, C1647–C1657. [Google Scholar] [CrossRef] [Green Version]
- Klaassen, I.; Van Noorden, C.J.; Schlingemann, R.O. Molecular basis of the inner blood-retinal barrier and its breakdown in diabetic macular edema and other pathological conditions. Prog. Retin. Eye Res. 2013, 34, 19–48. [Google Scholar] [CrossRef]
- Bhattacharjee, P.S.; Huq, T.S.; Potter, V.; Young, A.; Davenport, I.R.; Graves, R.; Mandal, T.K.; Clement, C.; McFerrin, H.E.; Muniruzzaman, S.; et al. High-glucose-induced endothelial cell injury is inhibited by a Peptide derived from human apolipoprotein E. PLoS ONE 2012, 7, e52152. [Google Scholar] [CrossRef]
- Gao, F.; Sabbineni, H.; Artham, S.; Somanath, P.R. Modulation of long-term endothelial-barrier integrity is conditional to the cross-talk between Akt and Srcsignaling. J. Cell. Physiol. 2017, 232, 2599–2609. [Google Scholar] [CrossRef]
- Gao, F.; Artham, S.; Sabbineni, H.; Al-Azayzih, A.; Peng, X.-D.; Hay, N.; Adams, R.H.; Byzova, T.V.; Somanath, P.R. Akt1 promotes stimuli-induced endothelial-barrier protection through FoxO-mediated tight-junction protein turnover. Cell. Mol. Life Sci. 2016, 73, 3917–3933. [Google Scholar] [CrossRef] [Green Version]
- McKenna, D.J.; Nelson, J.; Stitt, A.W. Advanced glycation alters expression of the 67kDa laminin receptor in retinal microvascular endothelial cells. Life Sci. 2001, 68, 2695–2703. [Google Scholar] [CrossRef]
- Aveleira, C.A.; Lin, C.M.; Abcouwer, S.F.; Ambrósio, A.F.; Antonetti, D.A. TNF-α signals through PKCζ/NF-κB to alter the tight junction complex and increase retinal endothelial cell permeability. Diabetes 2010, 59, 2872–2882. [Google Scholar] [CrossRef] [Green Version]
- Elner, S.G.; Petty, H.R.; Elner, V.M.; Yoshida, A.; Bian, Z.M.; Yang, D.; Kindezelskii, A.L. TLR4 mediates human retinal pigment epithelial endotoxin binding and cytokine expression. Trans. Am. Ophthalmol. Soc. 2005, 103, 126–135. [Google Scholar] [CrossRef]
- Halder, S.K.; Matsunaga, H.; Ishii, K.J.; Akira, S.; Miyake, K.; Ueda, H. Retinal cell type-specific prevention of ischemia-induced damages by LPS-TLR4 signaling through microglia. J. Neurochem. 2013, 126, 243–260. [Google Scholar] [CrossRef]
- Artham, S.; Verma, A.; Alwhaibi, A.; Adil, M.S.; Manicassamy, S.; Munn, D.H.; Somanath, P.R. Delayed Akt suppression in the lipopolysaccharide-induced acute lung injury promotes resolution that is associated with enhanced effector regulatory T cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 318, L750–L761. [Google Scholar] [CrossRef]
- Chen, J.; Somanath, P.R.; Razorenova, O.; Chen, W.S.; Hay, N.; Bornstein, P.; Byzova, T.V. Akt1 regulates pathological angiogenesis, vascular maturation and permeability in vivo. Nat. Med. 2005, 11, 1188–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Yu, X.W.; Zhang, D.D.; Fan, Z.G. Blood-retinal barrier as a converging pivot in understanding the initiation and development of retinal diseases. Chin. Med. J. 2020, 133, 2586–2594. [Google Scholar] [CrossRef]
- Lipski, D.A.; Foucart, V.; Dewispelaere, R.; Caspers, L.E.; Defrance, M.; Bruyns, C.; Willermain, F. Retinal endothelial cell phenotypic modifications during experimental autoimmune uveitis: A transcriptomic approach. BMC Ophthalmol. 2020, 20, 106. [Google Scholar] [CrossRef] [PubMed]
- Mirshahi, A.; Hoehn, R.; Lorenz, K.; Kramann, C.; Baatz, H. Anti-tumor necrosis factor alpha for retinal diseases: Current knowledge and future concepts. J. Ophthalmic. Vis. Res. 2012, 7, 39–44. [Google Scholar]
- Zhang, W.; Liu, H.; Rojas, M.; Caldwell, R.W.; Caldwell, R.B. Anti-inflammatory therapy for diabetic retinopathy. Immunotherapy 2011, 3, 609–628. [Google Scholar] [CrossRef] [Green Version]
- Lamers, M.L.; Almeida, M.E.; Vicente-Manzanares, M.; Horwitz, A.F.; Santos, M.F. High glucose-mediated oxidative stress impairs cell migration. PLoS ONE 2011, 6, e22865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kido, D.; Mizutani, K.; Takeda, K.; Mikami, R.; Matsuura, T.; Iwasaki, K.; Izumi, Y. Impact of diabetes on gingival wound healing via oxidative stress. PLoS ONE 2017, 12, e0189601. [Google Scholar] [CrossRef] [PubMed]
- Baumgartner-Parzer, S.M.; Wagner, L.; Pettermann, M.; Grillari, J.; Gessl, A.; Waldhäusl, W. High-glucose--triggered apoptosis in cultured endothelial cells. Diabetes 1995, 44, 1323–1327. [Google Scholar] [CrossRef] [PubMed]
- Deveci, M.; Gilmont, R.R.; Dunham, W.R.; Mudge, B.P.; Smith, D.J.; Marcelo, C.L. Glutathione enhances fibroblast collagen contraction and protects keratinocytes from apoptosis in hyperglycaemic culture. Br. J. Dermatol. 2005, 152, 217–224. [Google Scholar] [CrossRef]
- Liu, B.; Bhat, M.; Nagaraj, R.H. AlphaB-crystallin inhibits glucose-induced apoptosis in vascular endothelial cells. Biochem Biophys. Res. Commun. 2004, 321, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Dang, L.; Seale, J.P.; Qu, X. High glucose-induced human umbilical vein endothelial cell hyperpermeability is dependent on protein kinase C activation and independent of the Ca2+-nitric oxide signalling pathway. Clin. Exp. Pharmacol. Physiol. 2005, 32, 771–776. [Google Scholar] [CrossRef] [PubMed]
- Song, G.; Lin, D.; Bao, L.; Jiang, Q.; Zhang, Y.; Zheng, H.; Gao, Q. Effects of High Glucose on the Expression of LAMA1 and Biological Behavior of Choroid Retinal Endothelial Cells. J. Diabetes Res. 2018, 2018, 7504614. [Google Scholar] [CrossRef] [Green Version]
- Solomon, S.D.; Chew, E.; Duh, E.J.; Sobrin, L.; Sun, J.K.; VanderBeek, B.L.; Wykoff, C.C.; Gardner, T.W. Diabetic Retinopathy: A Position Statement by the American Diabetes Association. Diabetes Care 2017, 40, 412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGinn, S.; Saad, S.; Poronnik, P.; Pollock, C.A. High glucose-mediated effects on endothelial cell proliferation occur via p38 MAP kinase. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E708–E717. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Zhu, H.J.; Li, H.; Li, Q.X.; Li, F.M.; Cheng, L.; Liu, Y.G. p38-MAPK pathway is activated in retinopathy of microvascular disease of STZ-induced diabetic rat model. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 5789–5796. [Google Scholar] [CrossRef]
- Yang, J.; Caldwell, R.B.; Behzadian, M.A. Blockade of VEGF-induced GSK/beta-catenin signaling, uPAR expression and increased permeability by dominant negative p38alpha. Exp. Eye Res. 2012, 100, 101–108. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Duh, E.J.; Caldwell, R.B.; Behzadian, M.A. Antipermeability function of PEDF involves blockade of the MAP kinase/GSK/beta-catenin signaling pathway and uPAR expression. Investig. Ophthalmol. Vis. Sci. 2010, 51, 3273–3280. [Google Scholar] [CrossRef]
- Alwhaibi, A.; Verma, A.; Adil, M.S.; Somanath, P.R. The unconventional role of Akt1 in the advanced cancers and in diabetes-promoted carcinogenesis. Pharmacol. Res. 2019, 145, 104270. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.F.; Phung, T.; Shiojima, I.; Felske, T.; Upalakalin, J.N.; Feng, D.; Kornaga, T.; Dor, T.; Dvorak, A.M.; Walsh, K.; et al. Microvascular patterning is controlled by fine-tuning the Akt signal. Proc. Natl. Acad. Sci. USA 2005, 102, 128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, D.; Zhang, G.-M.; Xu, X.; Wang, L.-Y. The PI3K/Akt Signaling Pathway Mediates the High Glucose-Induced Expression of Extracellular Matrix Molecules in Human Retinal Pigment Epithelial Cells. J. Diabetes Res. 2015, 2015, 920280. [Google Scholar] [CrossRef] [PubMed]
- Das, F.; Ghosh-Choudhury, N.; Dey, N.; Bera, A.; Mariappan, M.M.; Kasinath, B.S.; Ghosh Choudhury, G. High Glucose Forces a Positive Feedback Loop Connecting Akt Kinase and FoxO1 Transcription Factor to Activate mTORC1 Kinase for Mesangial Cell Hypertrophy and Matrix Protein Expression. J. Biol. Chem. 2014, 289, 32703–32716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pushkarev, V. Activation of the PI3K/Akt/mTOR/p70S6K1 Signaling Cascade in the Mononuclear Cells of Peripheral Blood: Association with Insulin and Insulin-Like Growth Factor Levels in the Blood of Patients with Cancer and Diabetes. Cytol. Genet. 2019, 53, 489–493. [Google Scholar] [CrossRef]
- Huang, X.; Liu, G.; Guo, J.; Su, Z. The PI3K/AKT pathway in obesity and type 2 diabetes. Int. J. Biol. Sci. 2018, 14, 1483–1496. [Google Scholar] [CrossRef] [Green Version]
- Jacot, J.L.; Sherris, D. Potential Therapeutic Roles for Inhibition of the PI3K/Akt/mTOR Pathway in the Pathophysiology of Diabetic Retinopathy. J. Ophthalmol. 2011, 2011, 589813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, Y.; Saxena, S.; Mishra, A.; Saxena, A.; Natu, S.M. Advanced glycation end products and diabetic retinopathy. J. Ocul. Biol. Dis. Infor. 2013, 5, 63–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, X.; Li, Z.; Luo, D.; Huang, Y.; Zhu, J.; Wang, X.; Hu, H.; Patrick, C. Exogenous advanced glycosylation end products induce diabetes-like vascular dysfunction in normal rats: A factor in diabetic retinopathy. Graefe’s Arch. Clin. Exp. Ophthalmol. 2003, 241, 56–62. [Google Scholar] [CrossRef]
- Kandarakis, S.A.; Piperi, C.; Topouzis, F.; Papavassiliou, A.G. Emerging role of advanced glycation-end products (AGEs) in the pathobiology of eye diseases. Prog. Retin. Eye Res. 2014, 42, 85–102. [Google Scholar] [CrossRef]
- Lu, M.; Kuroki, M.; Amano, S.; Tolentino, M.; Keough, K.; Kim, I.; Bucala, R.; Adamis, A.P. Advanced glycation end products increase retinal vascular endothelial growth factor expression. J. Clin. Investig. 1998, 101, 1219–1224. [Google Scholar] [CrossRef]
- Yao, Y.; Li, R.; Du, J.; Li, X.; Zhao, L.; Long, L.; Li, D.; Lu, S. Tumor necrosis factor-alpha and diabetic retinopathy: Review and meta-analysis. Clin. Chim. Acta 2018, 485, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Cordero-Coma, M.; Sobrin, L. Anti-tumor necrosis factor-alpha therapy in uveitis. Surv. Ophthalmol. 2015, 60, 575–589. [Google Scholar] [CrossRef]
- Sainson, R.C.; Johnston, D.A.; Chu, H.C.; Holderfield, M.T.; Nakatsu, M.N.; Crampton, S.P.; Davis, J.; Conn, E.; Hughes, C.C. TNF primes endothelial cells for angiogenic sprouting by inducing a tip cell phenotype. Blood 2008, 111, 4997–5007. [Google Scholar] [CrossRef]
- Augustine, J.; Troendle, E.P.; Barabas, P.; McAleese, C.A.; Friedel, T.; Stitt, A.W.; Curtis, T.M. The Role of Lipoxidation in the Pathogenesis of Diabetic Retinopathy. Front. Endocrinol. 2020, 11, 621938. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Gandhi, J.K.; Zhong, X.; Wei, Y.; Gong, J.; Duh, E.J.; Vinores, S.A. TNFα Is Required for Late BRB Breakdown in Diabetic Retinopathy, and Its Inhibition Prevents Leukostasis and Protects Vessels and Neurons from Apoptosis. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1336–1344. [Google Scholar] [CrossRef] [Green Version]
- Park, B.S.; Lee, J.-O. Recognition of lipopolysaccharide pattern by TLR4 complexes. Exp. Mol. Med. 2013, 45, e66. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Jiang, Y.; Steinle, J.J. Toll-Like Receptor 4 Reduces Occludin and Zonula Occludens 1 to Increase Retinal Permeability Both in vitro and in vivo. J. Vasc. Res. 2017, 54, 367–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dvoriantchikova, G.; Barakat, D.J.; Hernandez, E.; Shestopalov, V.I.; Ivanov, D. Toll-like receptor 4 contributes to retinal ischemia/reperfusion injury. Mol. Vis. 2010, 16, 1907–1912. [Google Scholar]
- Rangasamy, S.; Monickaraj, F.; Legendre, C.; Cabrera, A.P.; Llaci, L.; Bilagody, C.; McGuire, P.; Das, A. Transcriptomics analysis of pericytes from retinas of diabetic animals reveals novel genes and molecular pathways relevant to blood-retinal barrier alterations in diabetic retinopathy. Exp. Eye Res. 2020, 195, 108043. [Google Scholar] [CrossRef] [PubMed]
- Van Hove, I.; De Groef, L.; Boeckx, B.; Modave, E.; Hu, T.T.; Beets, K.; Etienne, I.; Van Bergen, T.; Lambrechts, D.; Moons, L.; et al. Single-cell transcriptome analysis of the Akimba mouse retina reveals cell-type-specific insights into the pathobiology of diabetic retinopathy. Diabetologia 2020, 63, 2235–2248. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rudraraju, M.; Narayanan, S.P.; Somanath, P.R. Distinct Mechanisms of Human Retinal Endothelial Barrier Modulation In Vitro by Mediators of Diabetes and Uveitis. Life 2022, 12, 33. https://doi.org/10.3390/life12010033
Rudraraju M, Narayanan SP, Somanath PR. Distinct Mechanisms of Human Retinal Endothelial Barrier Modulation In Vitro by Mediators of Diabetes and Uveitis. Life. 2022; 12(1):33. https://doi.org/10.3390/life12010033
Chicago/Turabian StyleRudraraju, Madhuri, S. Priya Narayanan, and Payaningal R. Somanath. 2022. "Distinct Mechanisms of Human Retinal Endothelial Barrier Modulation In Vitro by Mediators of Diabetes and Uveitis" Life 12, no. 1: 33. https://doi.org/10.3390/life12010033