
Targeting Oxidative Phosphorylation-Proteasome Activity in Extracellular Detached Cells Promotes Anoikis and Inhibits Metastasis

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagent
2.2. Cell Line
2.3. Transfection
2.4. Apoptosis Detection
2.5. Reactive Oxygen Species (ROS) Quantification
2.6. RNA-Seq
2.7. RNA Isolation and qRT-PCR
2.8. Protein Extraction and Immunoblotting
2.9. Intracellular ATP Detection
2.10. Tumor Models
2.11. Statistical Analysis
3. Results
3.1. ECM Detachment Reprograms Metabolism in Tumor Cells
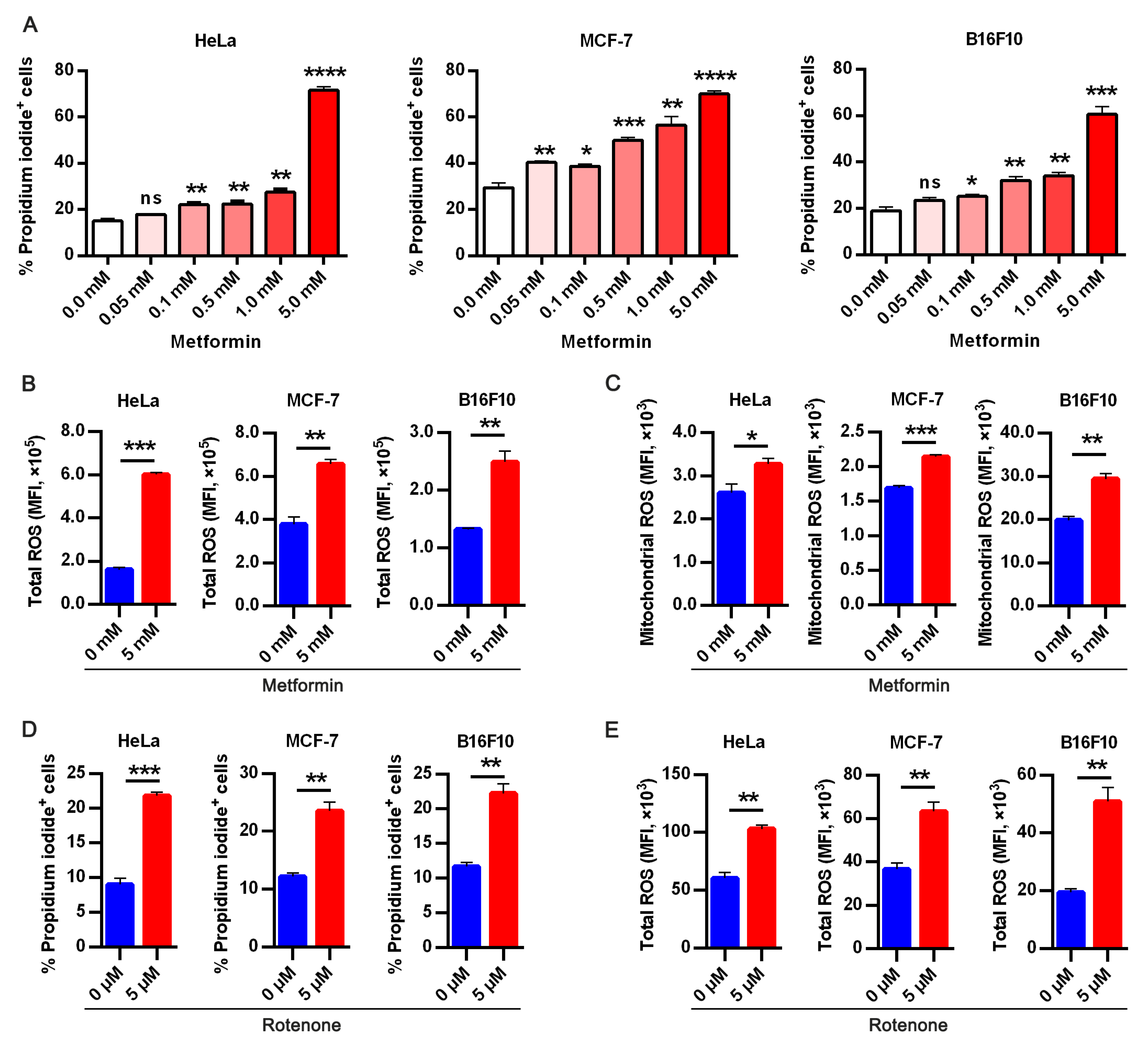
3.2. Targeting Oxidative Phosphorylation by Metformin Promotes Anoikis in Cancer Cells
3.3. Metformin Abrogates Upregulated Proteasome Activity in ECM Detached Tumor Cells
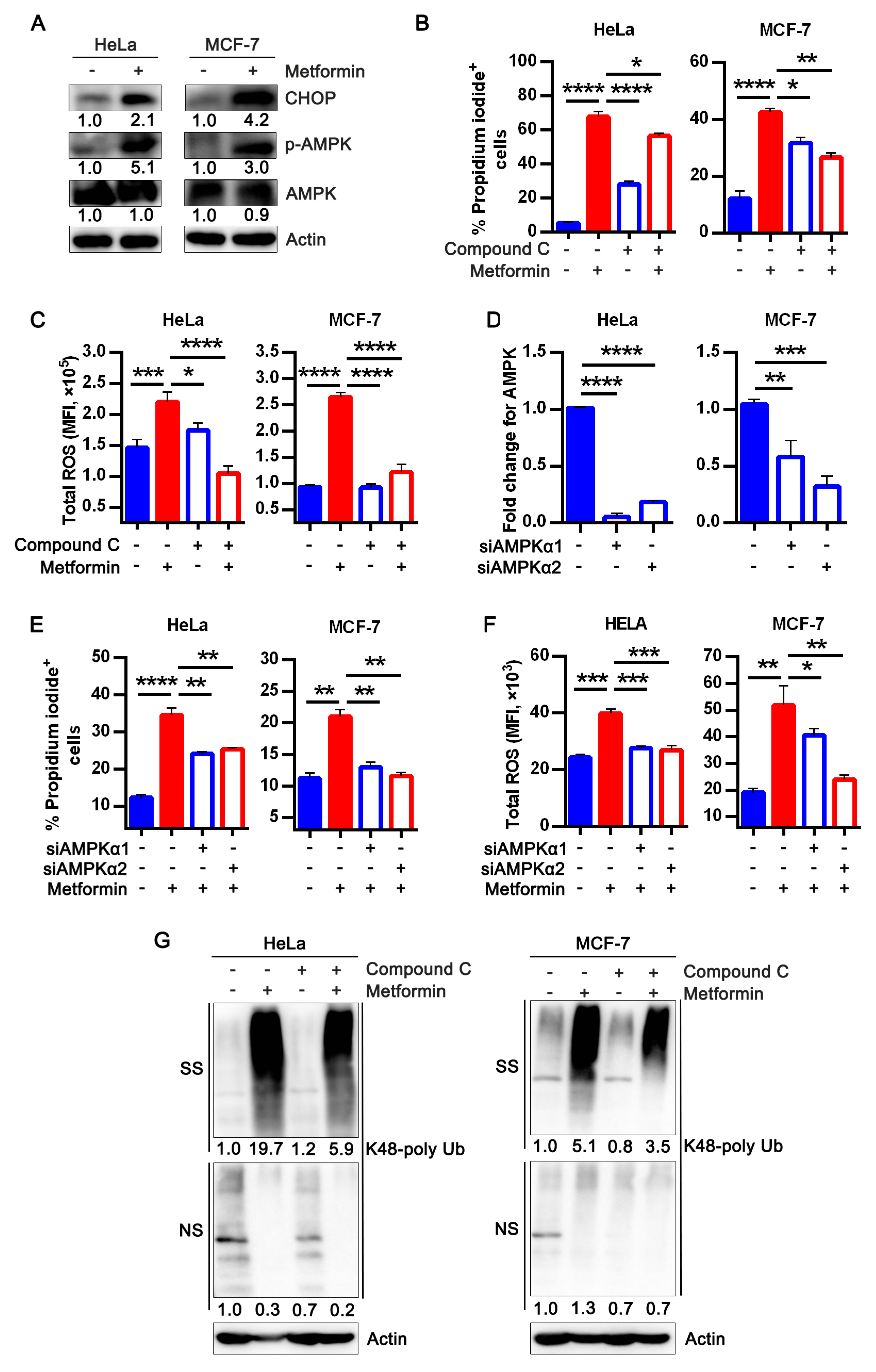
3.4. Metformin Treatment Activates AMPK and Apoptotic UPR Signaling Pathway
3.5. Metformin or PS341 Impedes Lung Metastasis In Vivo
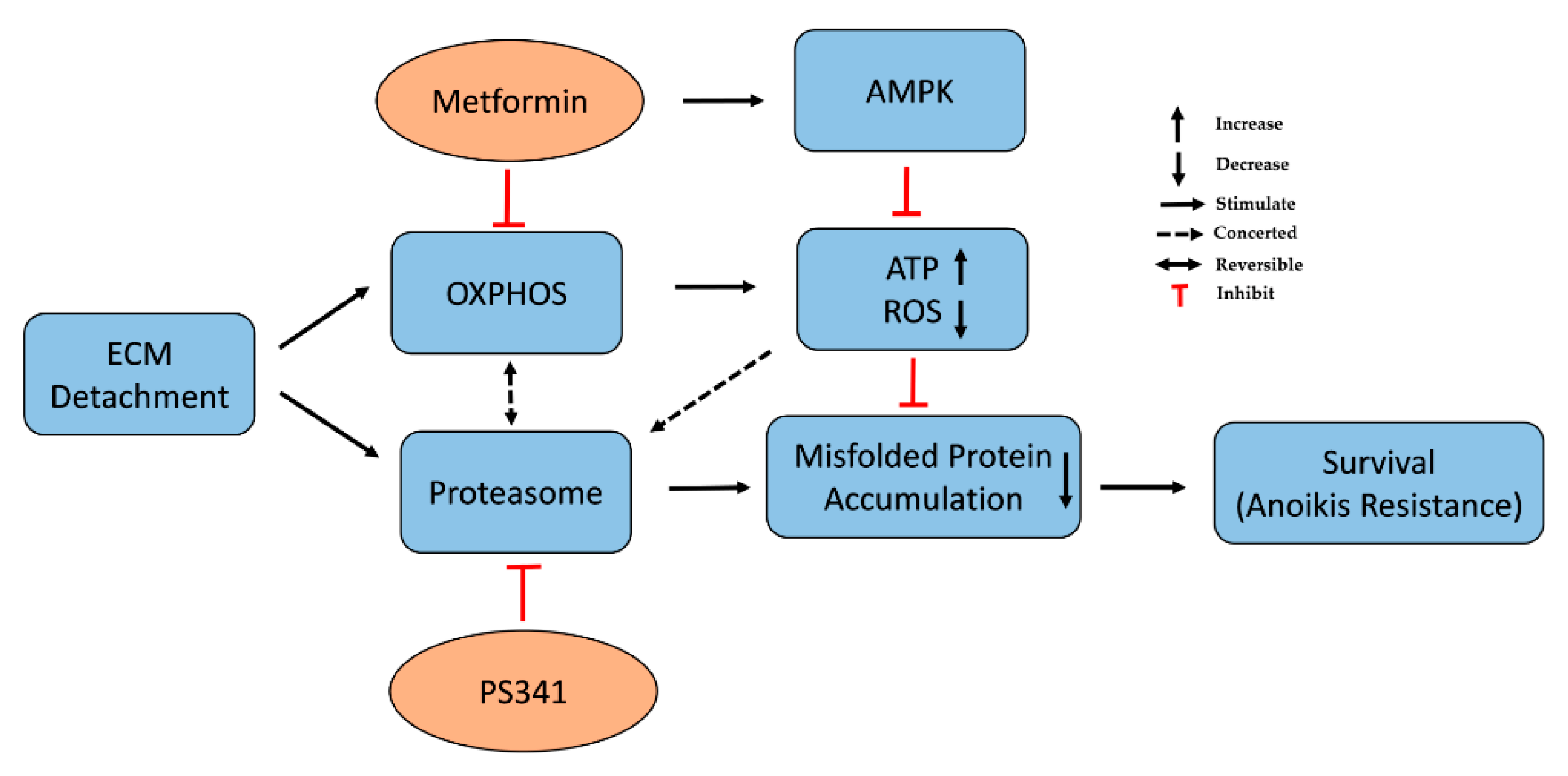
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- DeBerardinis, R.J.; Thompson, C.B. Cellular metabolism and disease: What do metabolic outliers teach us? Cell 2012, 148, 1132–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paoli, P.; Giannoni, E.; Chiarugi, P. Anoikis molecular pathways and its role in cancer progression. Biochim. Biophys. Acta 2013, 1833, 3481–3498. [Google Scholar] [CrossRef] [Green Version]
- Taddei, M.L.; Giannoni, E.; Fiaschi, T.; Chiarugi, P. Anoikis: An emerging hallmark in health and diseases. J. Pathol. 2012, 226, 380–393. [Google Scholar] [CrossRef] [PubMed]
- Adeshakin, F.O.; Adeshakin, A.O.; Afolabi, L.O.; Yan, D.; Zhang, G.; Wan, X. Mechanisms for Modulating Anoikis Resistance in Cancer and the Relevance of Metabolic Reprogramming. Front. Oncol. 2021, 11, 528. [Google Scholar] [CrossRef]
- Kim, Y.-N.; Koo, K.H.; Sung, J.Y.; Yun, U.-J.; Kim, H. Anoikis Resistance: An Essential Prerequisite for Tumor Metastasis. Int. J. Cell Biol. 2012, 2012, 306879. [Google Scholar] [CrossRef] [Green Version]
- Kamarajugadda, S.; Stemboroski, L.; Cai, Q.; Simpson, N.E.; Nayak, S.; Tan, M.; Lu, J. Glucose oxidation modulates anoikis and tumor metastasis. Mol. Cell. Biol. 2012, 32, 1893–1907. [Google Scholar] [CrossRef] [Green Version]
- Palorini, R.; Votta, G.; Pirola, Y.; De Vitto, H.; De Palma, S.; Airoldi, C.; Vasso, M.; Ricciardiello, F.; Lombardi, P.P.; Cirulli, C.; et al. Protein Kinase: A Activation Promotes Cancer Cell Resistance to Glucose Starvation and Anoikis. PLoS Genet. 2016, 12, e1005931. [Google Scholar] [CrossRef] [Green Version]
- Dillekås, H.; Rogers, M.S.; Straume, O. Are 90% of deaths from cancer caused by metastases? Cancer Med. 2019, 8, 5574–5576. [Google Scholar] [CrossRef] [Green Version]
- de Sousa Mesquita, A.P.; de Araújo Lopes, S.; Pernambuco Filho, P.C.A.; Nader, H.B.; Lopes, C.C. Acquisition of anoikis resistance promotes alterations in the Ras/ERK and PI3K/Akt signaling pathways and matrix remodeling in endothelial cells. Apoptosis Int. J. Program. Cell Death 2017, 22, 1116–1137. [Google Scholar] [CrossRef]
- Moulik, S.; Pal, S.; Biswas, J.; Chatterjee, A. Role of ERK in Modulating MMP 2 and MMP 9 with Respect to Tumour Invasiveness in Human Cancer Cell Line MCF-7 and MDA-MB-231. J. Tumor 2014, 2, 87–98. [Google Scholar]
- Yoshino, S.; Hara, T.; Nakaoka, H.J.; Kanamori, A.; Murakami, Y.; Seiki, M.; Sakamoto, T. The ERK signaling target RNF126 regulates anoikis resistance in cancer cells by changing the mitochondrial metabolic flux. Cell Discov. 2016, 2, 16019. [Google Scholar] [CrossRef] [Green Version]
- Adeshakin, F.O.; Adeshakin, A.O.; Liu, Z.; Lu, X.; Cheng, J.; Zhang, P.; Yan, D.; Zhang, G.; Wan, X. Upregulation of V-ATPase by STAT3 Activation Promotes Anoikis Resistance and Tumor Metastasis. J. Cancer 2021, 12, 4819–4829. [Google Scholar] [CrossRef]
- Hubrecht, R.C.; Carter, E. The 3Rs and Humane Experimental Technique: Implementing Change. Animals 2019, 9, 754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graham, M.L.; Prescott, M.J. The multifactorial role of the 3Rs in shifting the harm-benefit analysis in animal models of disease. Eur. J. Pharmacol. 2015, 759, 19–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.R.S.; et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife 2014, 3, e02242. [Google Scholar] [CrossRef]
- Saengboonmee, C.; Seubwal, W.; Cha’on, U.; Sawanyawisuth, K.; Wongkham, S.; Wongkham, C. Metformin Exerts Antiproliferative and Anti-metastatic Effects Against Cholangiocarcinoma Cells by Targeting STAT3 and NF-ĸB. Anticancer Res. 2017, 37, 115–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, G.; Lobanova, L.; Dawicki, W.; Groot, G.; Gordon, J.R.; Bowen, M.; Harkness, T.; Arnason, T. Metformin inhibits the development, and promotes the resensitization, of treatment-resistant breast cancer. PLoS ONE 2017, 12, e0187191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leclerc, G.M.; Leclerc, J.G.; Kuznetsov, J.N.; DeSalvo, J.; Barredo, J.C. Metformin induces apoptosis through AMPK-dependent inhibition of UPR signaling in ALL lymphoblasts. PLoS ONE 2013, 8, e74420. [Google Scholar] [CrossRef]
- De Santi, M.; Baldelli, G.; Diotallevi, A.; Galluzzi, L.; Schiavano, G.F.; Brandi, G. Metformin prevents cell tumorigenesis through autophagy-related cell death. Sci. Rep. 2019, 9, 66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, B.; Li, S.; Sheng, L.; Zhu, J.; Gu, L.; Shen, H.; La, D.; Hambly, B.D.; Bao, S.; Di, W. Metformin inhibits the development and metastasis of ovarian cancer. Oncol. Rep. 2012, 28, 903–908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, A.E.; Ito, H.; Rovira, I.I.; Kim, K.-S.; Takeda, K.; Yu, Z.-Y.; Ferrans, V.J.; Finkel, T. A Role for Reactive Oxygen Species in Endothelial Cell Anoikis. Circ. Res. 1999, 85, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Giannoni, E.; Buricchi, F.; Grimaldi, G.; Parri, M.; Cialdai, F.; Taddei, M.L.; Raugei, G.; Ramponi, G.; Chiarugi, P. Redox regulation of anoikis: Reactive oxygen species as essential mediators of cell survival. Cell Death Differ. 2008, 15, 867–878. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Darwish, T.; Larraufie, P.; Rimmington, D.; Cimino, I.; Goldspink, D.A.; Jenkins, B.; Koulman, A.; Brighton, C.A.; Ma, M.; et al. Inhibition of mitochondrial function by metformin increases glucose uptake, glycolysis and GDF-15 release from intestinal cells. Sci. Rep. 2021, 11, 2529. [Google Scholar] [CrossRef]
- Lipchick, B.C.; Fink, E.E.; Nikiforov, M.A. Oxidative stress and proteasome inhibitors in multiple myeloma. Pharmacol. Res. 2016, 105, 210–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maharjan, S.; Oku, M.; Tsuda, M.; Hoseki, J.; Sakai, Y. Mitochondrial impairment triggers cytosolic oxidative stress and cell death following proteasome inhibition. Sci. Rep. 2014, 4, 5896. [Google Scholar] [CrossRef] [Green Version]
- Kisselev, A.F.; van der Linden, W.A.; Overkleeft, H.S. Proteasome inhibitors: An expanding army attacking a unique target. Chem. Biol. 2012, 19, 99–115. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Frezza, M.; Schmitt, S.; Kanwar, J.; Dou, Q.P.L. Bortezomib as the first proteasome inhibitor anticancer drug: Current status and future perspectives. Curr. Cancer Drug Targets 2011, 11, 239–253. [Google Scholar] [CrossRef] [Green Version]
- Muñoz, C.; Francisco, J.S.; Gutiérrez, B.; González, J. Role of the Ubiquitin-Proteasome Systems in the Biology and Virulence of Protozoan Parasites. BioMed Res. Int. 2015, 2015, 141526. [Google Scholar] [CrossRef] [Green Version]
- Peth, A.; Uchiki, T.; Goldberg, A.L. ATP-dependent steps in the binding of ubiquitin conjugates to the 26S proteasome that commit to degradation. Mol. Cell 2010, 40, 671–681. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Mao, Y.; Zhou, T.; Luo, C.; Xie, J.; Qi, W.; Yang, Z.; Ma, J.X.; Gao, G.; Yang, X. Manganese superoxide dismutase mediates anoikis resistance and tumor metastasis in nasopharyngeal carcinoma. Oncotarget 2016, 7, 32408–32420. [Google Scholar] [CrossRef] [Green Version]
- Kamarajugadda, S.; Cai, Q.; Chen, H.; Nayak, S.; Zhu, J.; He, M.; Jin, Y.; Zhang, Y.; Ai, L.; Martin, S.S.; et al. Manganese superoxide dismutase promotes anoikis resistance and tumor metastasis. Cell Death Dis. 2013, 4, e504. [Google Scholar] [CrossRef]
- Sousa, B.; Pereira, J.; Marques, R.; Grilo, L.F.; Pereira, S.P.; Sardão, V.A.; Schmitt, F.; Oliveira, P.J.; Paredes, J. P-cadherin induces anoikis-resistance of matrix-detached breast cancer cells by promoting pentose phosphate pathway and decreasing oxidative stress. Biochim. Biophys. Acta Mol. Basis Dis. 2020, 1866, 165964. [Google Scholar] [CrossRef]
- Hardie, D.G. AMP-activated protein kinase: An energy sensor that regulates all aspects of cell function. Genes Dev. 2011, 25, 1895–1908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Hu, X.; Liu, Y.; Dong, S.; Wen, Z.; He, W.; Zhang, S.; Huang, Q.; Shi, M. ROS signaling under metabolic stress: Cross-talk between AMPK and AKT pathway. Mol. Cancer 2017, 16, 79. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Tian, M.; Ding, C.; Yu, S. The C/EBP Homologous Protein (CHOP) Transcription Factor Functions in Endoplasmic Reticulum Stress-Induced Apoptosis and Microbial Infection. Front. Immunol. 2019, 9, 3083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suganya, N.; Bhakkiyalakshmi, E.; Suriyanarayanan, S.; Paulmurugan, R.; Ramkumar, K.M. Quercetin ameliorates tunicamycin-induced endoplasmic reticulum stress in endothelial cells. Cell Prolif. 2014, 47, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Kapałczyńska, M.; Kolenda, T.; Przybyła, W.; Zajączkowska, M.; Teresiak, A.; Filas, V.; Ibbs, M.; Bliźniak, R.; Łuczewski, L.; Lamperska, K. 2D and 3D cell cultures—A comparison of different types of cancer cell cultures. Arch. Med. Sci. 2018, 14, 910–919. [Google Scholar] [CrossRef]
- Breslin, S.; O’Driscoll, L. The relevance of using 3D cell cultures, in addition to 2D monolayer cultures, when evaluating breast cancer drug sensitivity and resistance. Oncotarget 2016, 7, 45745–45756. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-C.; Lou, X.; Zhang, Z.; Ingram, P.; Yoon, E. High-Throughput Cancer Cell Sphere Formation for Characterizing the Efficacy of Photo Dynamic Therapy in 3D Cell Cultures. Sci. Rep. 2015, 5, 12175. [Google Scholar] [CrossRef] [Green Version]
- Lawrenson, K.; Grun, B.; Gayther, S.A. Heterotypic three-dimensional in vitro modeling of stromal-epithelial interactions during ovarian cancer initiation and progression. J. Vis. Exp. 2012, e4206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, W.-C.; Kim, H.; Kim, Y.-J.; Jeon, B.-M.; Kang, H.-B.; Ko, H. Catechol inhibits epidermal growth factor-induced epithelial-to-mesenchymal transition and stem cell-like properties in hepatocellular carcinoma cells. Sci. Rep. 2020, 10, 7620. [Google Scholar] [CrossRef]
- Chunhacha, P.; Sriuranpong, V.; Chanvorachote, P. Epithelial-mesenchymal transition mediates anoikis resistance and enhances invasion in pleural effusion-derived human lung cancer cells. Oncol. Lett. 2013, 5, 1043–1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Z.; Zhou, L.; Xie, N.; Nice, E.C.; Zhang, T.; Cui, Y.; Huang, C. Overcoming cancer therapeutic bottleneck by drug repurposing. Signal Transduct. Target. Ther. 2020, 5, 113. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.J. Metformin: Historical overview. Diabetologia 2017, 60, 1566–1576. [Google Scholar] [CrossRef] [Green Version]
- Ko, Y.; Choi, A.; Lee, M.; Lee, J.A. Metformin displays in vitro and in vivo antitumor effect against osteosarcoma. Korean J. Pediatr. 2016, 59, 374–380. [Google Scholar] [CrossRef] [Green Version]
- Amable, G.; Martínez-León, E.; Picco, M.E.; Di Siervi, N.; Davio, C.; Rozengurt, E.; Rey, O.L. Metformin inhibits β-catenin phosphorylation on Ser-552 through an AMPK/PI3K/Akt pathway in colorectal cancer cells. Int. J. Biochem. Cell Biol. 2019, 112, 88–94. [Google Scholar] [CrossRef]
- Saini, N.; Yang, X. Metformin as an anti-cancer agent: Actions and mechanisms targeting cancer stem cells. Acta Biochim. Biophys. Sin. 2017, 50, 133–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adeshakin, F.O.; Zhang, G.; Adeshakin, A.O.; Wan, X. Abstract 2869, Blockade of oxidative phosphorylation by metformin promotes anoikis. Cancer Res. 2021, 81 (Suppl. 13), 2869. [Google Scholar]
- Huang, Z.; Wu, Y.; Zhou, X.; Xu, J.; Zhu, W.; Shu, Y.; Liu, P. Efficacy of therapy with bortezomib in solid tumors: A review based on 32 clinical trials. Future Oncol. 2014, 10, 1795–1807. [Google Scholar] [CrossRef]
- Roeten, M.S.F.; Cloos, J.; Jansen, G. Positioning of proteasome inhibitors in therapy of solid malignancies. Cancer Chemother. Pharmacol. 2018, 81, 227–243. [Google Scholar] [CrossRef] [Green Version]
- Aljofan, M.; Riethmacher, D. Anticancer activity of metformin: A systematic review of the literature. Future Sci. OA 2019, 5, FSO410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y.; Kover, K.L.; Moore, W.V. New Insight into Metformin Mechanism of Action and Clinical Application; IntechOpen: London, UK, 2020. [Google Scholar]
- Amodio, G.; Moltedo, O.; Faraonio, R.; Remondelli, P. Targeting the Endoplasmic Reticulum Unfolded Protein Response to Counteract the Oxidative Stress-Induced Endothelial Dysfunction. Oxidative Med. Cell. Longev. 2018, 2018, 4946289. [Google Scholar] [CrossRef]
- Yang, L.; Sha, H.; Davisson, R.L.; Qi, L. Phenformin Activates the Unfolded Protein Response in an AMP-activated Protein Kinase (AMPK)-dependent Manner. J. Biol. Chem. 2013, 288, 13631–13638. [Google Scholar] [CrossRef] [Green Version]
- Meares, G.P.; Hughes, K.J.; Naatz, A.; Papa, F.R.; Urano, F.; Hansen, P.A.; Benveniste, E.N.; Corbett, J.A. IRE1-dependent activation of AMPK in response to nitric oxide. Mol. Cell. Biol. 2011, 31, 4286–4297. [Google Scholar] [CrossRef] [Green Version]
- Kimura, Y.; Irie, K.; Mizuno, T. Expression control of the AMPK regulatory subunit and its functional significance in yeast ER stress response. Sci. Rep. 2017, 7, 46713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zimmermann, K.; Baldinger, J.; Mayerhofer, B.; Atanasov, A.G.; Dirsch, V.M.; Heiss, E.H. Activated AMPK boosts the Nrf2/HO-1 signaling axis—A role for the unfolded protein response. Free. Radic. Biol. Med. 2015, 88, 417–426. [Google Scholar] [CrossRef] [Green Version]
- Conza, D.; Mirra, P.; Calì, G.; Insabato, L.; Fiory, F.; Beguinot, F.; Ulianich, L. Metformin Dysregulates the Unfolded Protein Response and the WNT/β-Catenin Pathway in Endometrial Cancer Cells through an AMPK-Independent Mechanism. Cells 2021, 10, 1067. [Google Scholar] [CrossRef] [PubMed]
- Del Prete, A.; Zaccagnino, P.; Di Paola, M.; Saltarella, M.; Celis, C.O.; Nico, B.; Santoro, G.; Lorusso, M. Role of mitochondria and reactive oxygen species in dendritic cell differentiation and functions. Free Radic. Biol. Med. 2008, 44, 1443–1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Ragheb, K.; Lawler, G.; Sturgis, J.; Rajwa, B.; Melendez, J.A.; Robinson, J.P. Mitochondrial Complex I Inhibitor Rotenone Induces Apoptosis through Enhancing Mitochondrial Reactive Oxygen Species Production. J. Biol. Chem. 2003, 278, 8516–8525. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Liu, S.; Zhu, M.; Zhang, H.; Wang, J.; Xu, Q.; Lin, K.; Zhou, X.; Tao, M.; Li, C.; et al. PS341 inhibits hepatocellular and colorectal cancer cells through the FOXO3/CTNNB1 signaling pathway. Sci. Rep. 2016, 6, 22090. [Google Scholar] [CrossRef]
- Yamashita, T.; Kato, K.; Fujihara, S.; Iwama, H.; Morishita, A.; Yamana, H.; Kobayashi, K.; Kamada, H.; Chiyo, T.; Kobara, H.; et al. Anti-diabetic drug metformin inhibits cell proliferation and tumor growth in gallbladder cancer via G0/G1 cell cycle arrest. Anti-Cancer Drugs 2020, 31, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Munoz, L.E.; Huang, H.L.; Guin, R.N.; Bommireddy, R.; Selvaraj, P. Metformin reduces PD-L1 expression in the tumor and enhances the efficacy of vaccine generated CD8 T cells in a murine model of triple negative breast cancer. J. Immunol. 2020, 204 (Suppl. S1), 239.33. [Google Scholar]
- Bahrambeigi, S.; Shafiei-Irannejad, V. Immune-mediated anti-tumor effects of metformin; targeting metabolic reprogramming of T cells as a new possible mechanism for anti-cancer effects of metformin. Biochem. Pharmacol. 2020, 174, 113787. [Google Scholar] [CrossRef]
- Schumacher, L.Y.; Vo, D.D.; Garban, H.J.; Comin-Anduix, B.; Owens, S.K.; Dissette, V.B.; Glaspy, J.A.; McBride, W.H.; Bonavida, B.; Economou, J.S.; et al. Immunosensitization of Tumor Cells to Dendritic Cell-Activated Immune Responses with the Proteasome Inhibitor Bortezomib (PS-341, Velcade). J. Immunol. 2006, 176, 4757–4765. [Google Scholar] [CrossRef] [Green Version]
- Song, C.W.; Lee, H.; Dings, R.P.M.; Williams, B.; Powers, J.; Santos, T.D.; Choi, B.-H.; Park, H.J. Metformin kills and radiosensitizes cancer cells and preferentially kills cancer stem cells. Sci. Rep. 2012, 2, 362. [Google Scholar] [CrossRef] [Green Version]
- Chuang, H.-C.; Chou, C.-C.; Kulp, S.K.; Chen, C.-S. AMPK as a potential anticancer target—Friend or foe? Curr. Pharm. Des. 2014, 20, 2607–2618. [Google Scholar] [CrossRef] [Green Version]
- Guo, P.; Qiu, Y.; Ma, X.; Li, T.; Ma, X.; Zhu, L.; Lin, Y.; Han, L. Tripartite motif 31 promotes resistance to anoikis of hepatocarcinoma cells through regulation of p53-AMPK axis. Exp. Cell Res. 2018, 368, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Krämer, L.; Groh, C.; Herrmann, J.M. The proteasome: Friend and foe of mitochondrial biogenesis. FEBS Lett. 2021, 595, 1223–1238. [Google Scholar] [CrossRef]
- Lavie, J.; De Belvalet, H.; Sonon, S.; Ion, A.M.; Dumon, E.; Melser, S.; Lacombe, D.; Dupuy, J.-W.; Lalou, C.; Bénard, G. Ubiquitin-Dependent Degradation of Mitochondrial Proteins Regulates Energy Metabolism. Cell Rep. 2018, 23, 2852–2863. [Google Scholar] [CrossRef]
- Meul, T.; Berschneider, K.; Schmitt, S.; Mayr, C.H.; Mattner, L.F.; Schiller, H.B.; Yazgili, A.S.; Wang, X.; Lukas, C.; Schlesser, C.; et al. Mitochondrial Regulation of the 26S Proteasome. Cell Rep. 2020, 32, 108059. [Google Scholar] [CrossRef] [PubMed]
- Schafer, Z.T.; Grassian, A.R.; Song, L.; Jiang, Z.; Gerhart-Hines, Z.; Irie, H.Y.; Gao, S.; Puigserver, P.; Brugge, J.S. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 2009, 461, 109–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jagannathan, S.; Abdel-Malek, M.A.Y.; Malek, E.; Vad, N.; Latif, T.; Anderson, K.C.; Driscoll, J.J. Pharmacologic screens reveal metformin that suppresses GRP78-dependent autophagy to enhance the anti-myeloma effect of bortezomib. Leukemia 2015, 29, 2184–2191. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adeshakin, F.O.; Adeshakin, A.O.; Liu, Z.; Cheng, J.; Zhang, P.; Yan, D.; Zhang, G.; Wan, X. Targeting Oxidative Phosphorylation-Proteasome Activity in Extracellular Detached Cells Promotes Anoikis and Inhibits Metastasis. Life 2022, 12, 42. https://doi.org/10.3390/life12010042
Adeshakin FO, Adeshakin AO, Liu Z, Cheng J, Zhang P, Yan D, Zhang G, Wan X. Targeting Oxidative Phosphorylation-Proteasome Activity in Extracellular Detached Cells Promotes Anoikis and Inhibits Metastasis. Life. 2022; 12(1):42. https://doi.org/10.3390/life12010042
Chicago/Turabian StyleAdeshakin, Funmilayo O., Adeleye O. Adeshakin, Zhao Liu, Jian Cheng, Pengchao Zhang, Dehong Yan, Guizhong Zhang, and Xiaochun Wan. 2022. "Targeting Oxidative Phosphorylation-Proteasome Activity in Extracellular Detached Cells Promotes Anoikis and Inhibits Metastasis" Life 12, no. 1: 42. https://doi.org/10.3390/life12010042
APA StyleAdeshakin, F. O., Adeshakin, A. O., Liu, Z., Cheng, J., Zhang, P., Yan, D., Zhang, G., & Wan, X. (2022). Targeting Oxidative Phosphorylation-Proteasome Activity in Extracellular Detached Cells Promotes Anoikis and Inhibits Metastasis. Life, 12(1), 42. https://doi.org/10.3390/life12010042