
Spinal Cord Involvement in Adult Mitochondrial Diseases: A Cohort Study

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Patients
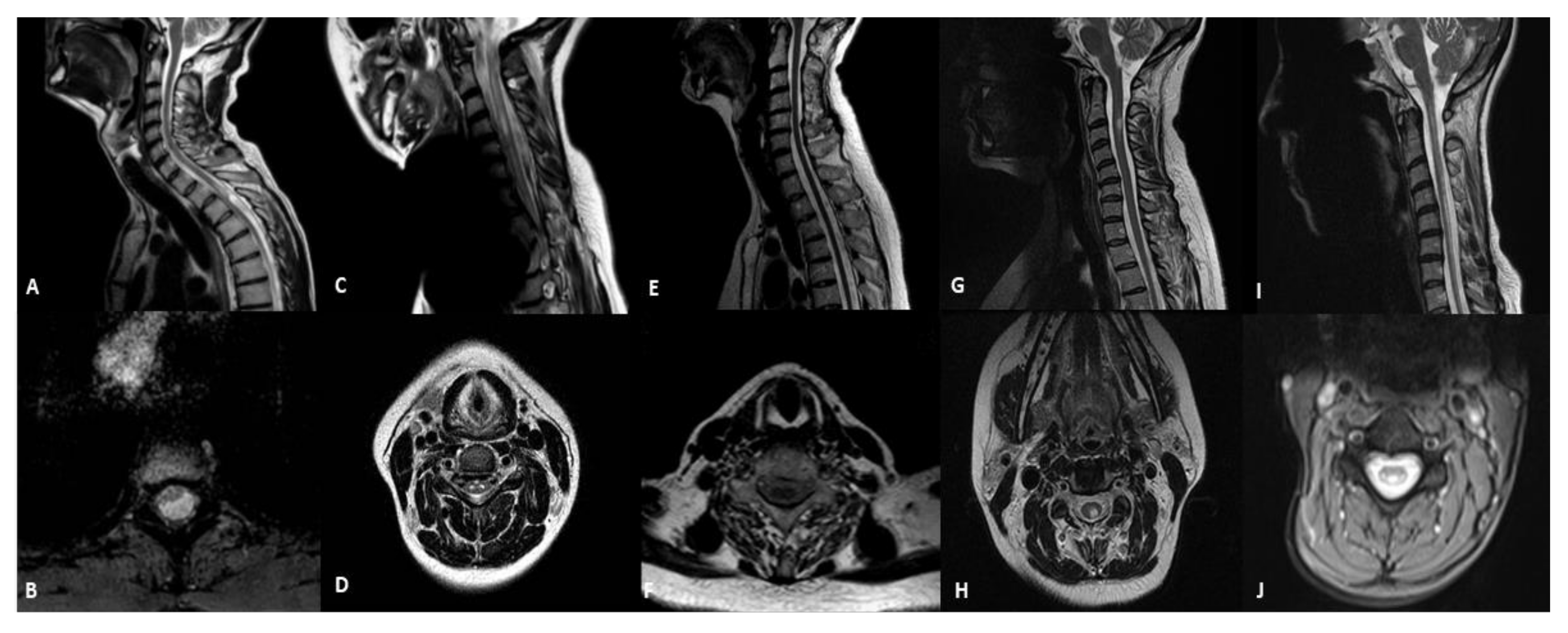
3.2. Spinal-Cord Imaging Findings
3.3. Brain Imaging Findings
3.4. Clinical and Laboratory Findings
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ng, Y.S.; Bindoff, L.A.; Gorman, G.S.; Klopstock, T.; Kornblum, C.; Mancuso, M.; McFarland, R.; Sue, C.M.; Suomalainen, A.; Taylor, R.W.; et al. Mitochondrial disease in adults: Recent advances and future promise. Lancet Neurol. 2021, 20, 573–584. [Google Scholar] [CrossRef]
- Lax, N.Z.; Gorman, G.S.; Turnbull, D.M. Review: Central nervous system involvement in mitochondrial disease. Neuropathol. Appl. Neurobiol. 2017, 43, 102–118. [Google Scholar] [CrossRef] [PubMed]
- Mascalchi, M.; Montomoli, M.; Guerrini, R. Neuroimaging in mitochondrial disorders. Essays Biochem. 2018, 62, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Finsterer, J. Cerebral imaging in adult mitochondrial disorders. J. Neurol. Sci. 2019, 404, 29–35. [Google Scholar] [CrossRef] [PubMed]
- Bindu, P.S.; Arvinda, H.; Taly, A.B.; Govindaraju, C.; Sonam, K.; Chiplunkar, S.; Kumar, R.; Gayathri, N.; Bharath, S.; Nagappa, M.; et al. Magnetic resonance imaging correlates of genetically characterized patients with mitochondrial disorders: A study from south India. Mitochondrion 2015, 25, 6–16. [Google Scholar] [CrossRef] [PubMed]
- Marelli, C.; Salsano, E.; Politi, L.S.; Labauge, P. Spinal cord involvement in adult-onset metabolic and genetic diseases. J. Neurol. Neurosurg. Psychiatry 2019, 90, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Luca, P.; Alessia, G.; Camilla, R.M.; Antonio, N.; Diego, M.; Federica, D.; Daria, D.; Rosalba, C.; Carlo, D.V.; Daniela, L. Spinal cord involvement in Kearns-Sayre syndrome: A neuroimaging study. Neuroradiology 2020, 62, 1315–1321. [Google Scholar] [CrossRef] [PubMed]
- Alves, C.A.P.F.; Teixeira, S.R.; Martin-Saavedra, J.S.; Guimarães Gonçalves, F.; Lo Russo, F.; Muraresku, C.; McCormick, E.M.; Falk, M.J.; Zolkipli-Cunningham, Z.; Ganetzky, R.; et al. Pediatric Leigh Syndrome: Neuroimaging Features and Genetic Correlations. Ann. Neurol. 2020, 88, 218–232. [Google Scholar] [CrossRef] [PubMed]
- Alves, C.A.P.F.; Goldstein, A.; Teixeira, S.R.; Martin-Saavedra, J.S.; de Barcelos, I.P.; Fadda, G.; Caschera, L.; Kidd, M.; Gonçalves, F.G.; McCormick, E.M.; et al. Involvement of the Spinal Cord in Primary Mitochondrial Disorders: A Neuroimaging Mimicker of Inflammation and Ischemia in Children. AJNR Am. J. Neuroradiol. 2021, 42, 389–396. [Google Scholar] [CrossRef] [PubMed]
- van Berge, L.; Hamilton, E.M.; Linnankivi, T.; Uziel, G.; Steenweg, M.E.; Isohanni, P.; Wolf, N.I.; Krägeloh-Mann, I.; Brautaset, N.J.; Andrews, P.I.; et al. Leukoencephalopathy with brainstem and spinal cord involvement and lactate elevation: Clinical and genetic characterization and target for therapy. Brain 2014, 137, 1019–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pfeffer, G.; Burke, A.; Yu-Wai-Man, P.; Compston, D.A.; Chinnery, P.F. Clinical features of MS associated with Leber hereditary optic neuropathy mtDNA mutations. Neurology 2013, 81, 2073–2081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mascalchi, M.; Bartolini, E.; Bianchi, A.; Gulino, P.; Procopio, E. Teaching NeuroImages: Spinal cord gray matter involvement in complex I deficiency mitochondriopathy. Neurology 2016, 87, 106–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souza, P.V.; Pinto, W.B.; Oliveira, A.S. Teaching NeuroImages: Longitudinally extensive transverse myelitis in MELAS. Neurology 2016, 86, e37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souza, P.V.S.; Bortholin, T.; Teixeira, C.A.C.; Seneor, D.D.; Marin, V.D.G.B.; Dias, R.B.; Farias, I.B.; Badia, B.M.L.; Silva, L.H.L.; Pinto, W.B.V.R.; et al. Leigh syndrome caused by mitochondrial DNA-maintenance defects revealed by whole exome sequencing. Mitochondrion 2019, 49, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, S.E.; Somoza, A.; Gilbert, D.L. Rare autosomal dominant POLG1 mutation in a family with metabolic strokes, posterior column spinal degeneration, and multi-endocrine disease. J. Child. Neurol. 2010, 2, 752–756. [Google Scholar] [CrossRef] [PubMed]
- Zsurka, G.; Kunz, W.S. Mitochondrial dysfunction and seizures: The neuronal energy crisis. Lancet Neurol. 2015, 14, 956–966. [Google Scholar] [CrossRef]
- Harris, J.J.; Attwell, D. The energetics of CNS white matter. J. Neurosci. 2012, 32, 356–371. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Phenotype | Number of Patients | Sex | Age (y Range) | Genotype |
|---|---|---|---|---|
| PEO | 21 | 7 M, 14 F | 18–78 | sDel, mDel, POLG, TWNK, SLC25A4, m.3243A > G |
| ANS | 3 | 1M, 2 F | 50–61 | POLG |
| MELAS | 3 | 2 M, 1 F | 32–62 | m.3243A > G |
| MIDD | 6 | 2 M, 4 F | 40–52 | m.3243A > G |
| MERRF | 7 | 1 M, 6 F | 18–58 | m.8344A > G, m.8356T > G, m.8356T > C |
| LHON | 3 | 1 M, 2 F | 19–45 | 14484T > C, m.11778G > A |
| ADOA | 2 | 1 M, 1 F | 42–45 | OPA1 |
| LS | 1 | F | 18 | m.13513G > A |
| LBSL | 1 | F | 41 | DARS2 |
| MNGIE | 2 | 2 M | 24–44 | TYMP |
| Others | 2 | 1 M, 1 F | 18–50 | m.3243A > G, m.13042G > A |
| Patient | Sex | Age at Onset | Age at MRI | Mutations | Phenotype | Spinal Cord Abnormalities |
|---|---|---|---|---|---|---|
| 1 | M | 28 | 45 years | m.3243A > G | MIDD | cystic-like lesion located in the white matter of dorsal and lateral columns |
| 2 | F | 11 | 18 years | m.13513G > A | LS | hyperintensity not involving a selective spinal tract with medullary atrophy |
| 3 | F | 50 | 63 years | POLG | ANS | medullary atrophy without signal alterations |
| 4 | F | 27 | 27 years | m.11778G > A | LHON | selective WMH of dorsalcolumns and lateral columns |
| 5 | F | 8 | 41 years | DARS2 | LBSL | hyperintensity of dorsalcolumns |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Primiano, G.; Mariotti, P.; Turrini, I.; Sancricca, C.; Sabino, A.; Torraco, A.; Carrozzo, R.; Servidei, S. Spinal Cord Involvement in Adult Mitochondrial Diseases: A Cohort Study. Life 2022, 12, 5. https://doi.org/10.3390/life12010005
Primiano G, Mariotti P, Turrini I, Sancricca C, Sabino A, Torraco A, Carrozzo R, Servidei S. Spinal Cord Involvement in Adult Mitochondrial Diseases: A Cohort Study. Life. 2022; 12(1):5. https://doi.org/10.3390/life12010005
Chicago/Turabian StylePrimiano, Guido, Paolo Mariotti, Ida Turrini, Cristina Sancricca, Andrea Sabino, Alessandra Torraco, Rosalba Carrozzo, and Serenella Servidei. 2022. "Spinal Cord Involvement in Adult Mitochondrial Diseases: A Cohort Study" Life 12, no. 1: 5. https://doi.org/10.3390/life12010005