
Ginsenoside F1 Protects the Brain against Amyloid Beta-Induced Toxicity by Regulating IDE and NEP

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Culture
2.3. Oligomerization of Aβ1–42
2.4. Measurement of Cell Viability and Cytotoxicity
2.5. Trichloroacetic Acid Precipitation
2.6. RNA Preparation and Quantitative Real-Time PCR
2.7. Western Blot Analysis
2.8. Transfection with Small Interfering RNA
2.9. ELISA
2.10. Thioflavin-T Assay
2.11. Animals
2.12. Liquid Chromatography with Tandem Mass Spectrometry (LC-MS/MS) Analysis
2.13. Histology and Staining
2.14. Statistical Analysis
3. Results
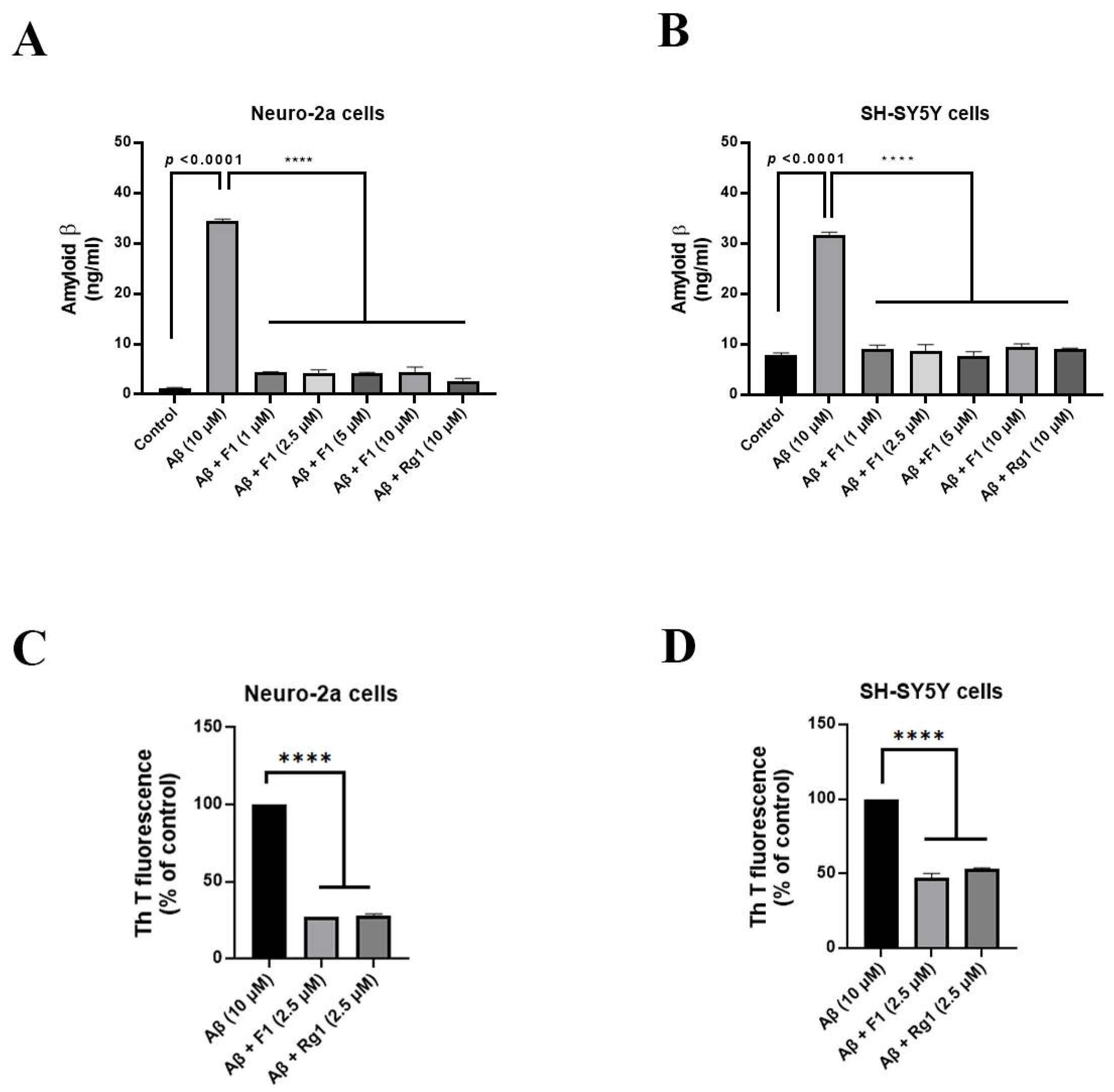
3.1. Ginsenoside F1 Reduces Aβ1–42-Induced Cytotoxicity in Neuronal Cells
3.2. Ginsenoside F1 Treatment Reduces the Secretion of Aβ1–42 in Neuronal Cells
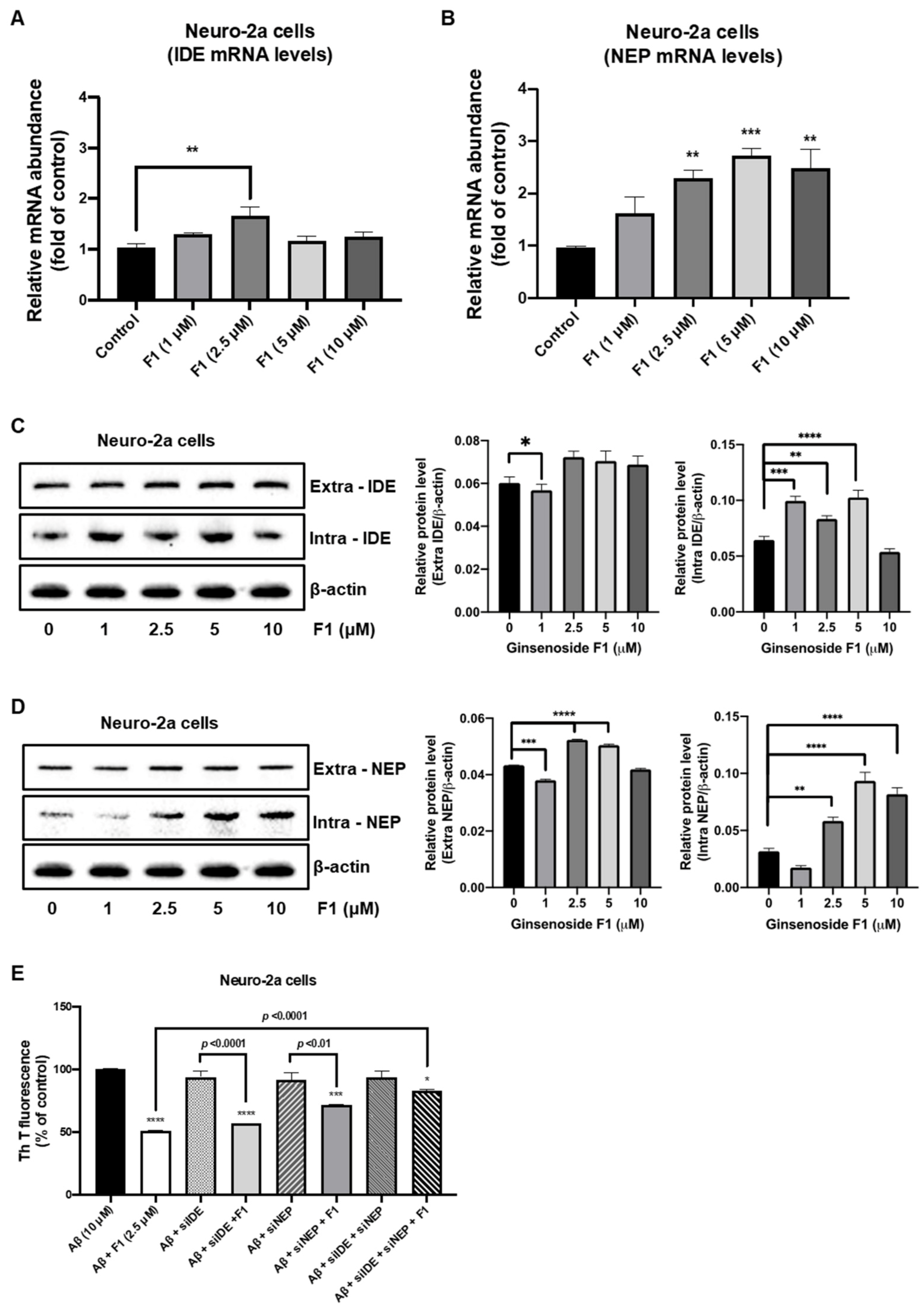
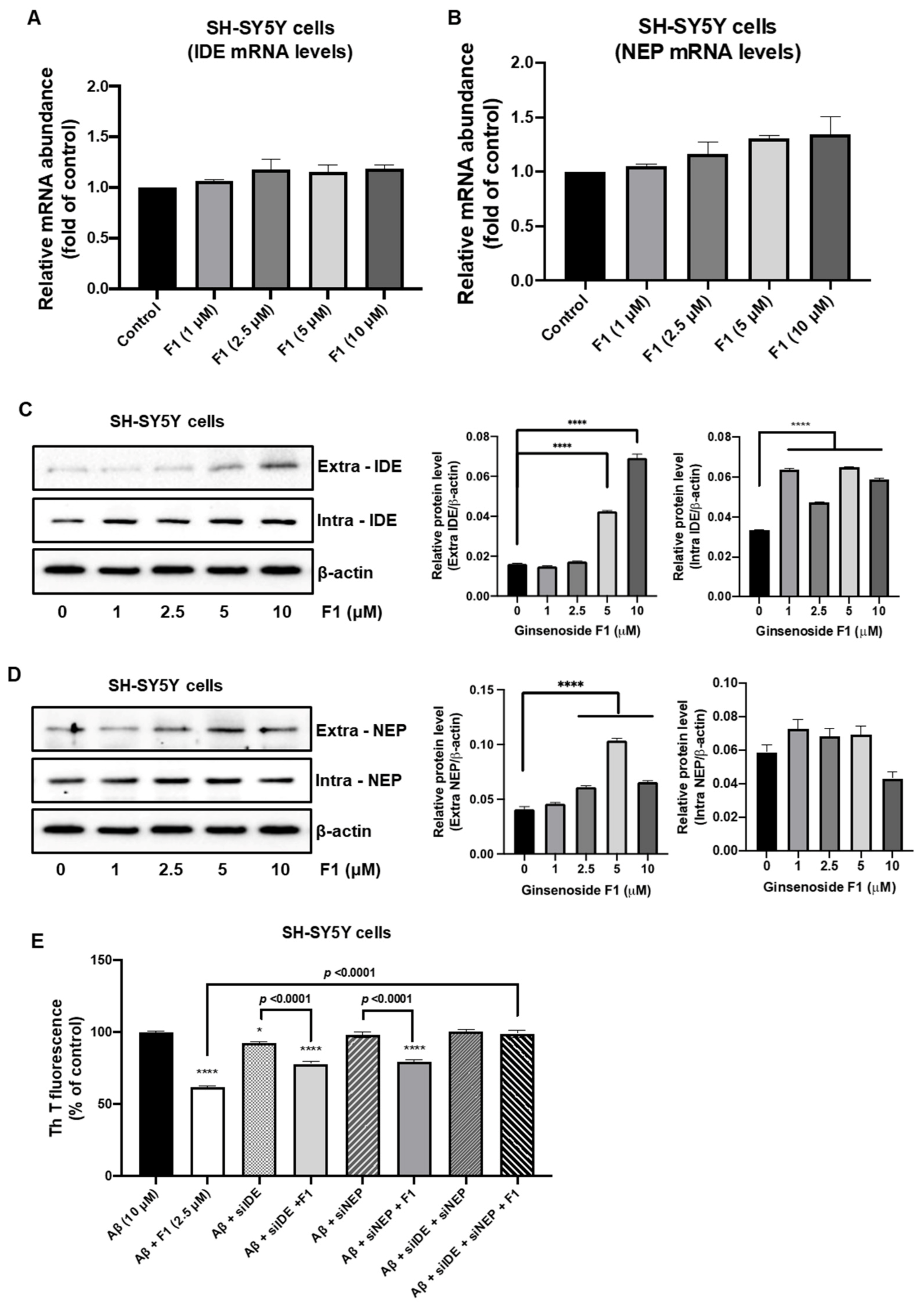
3.3. Ginsenoside F1 Upregulates the Expression of Aβ-Degrading Peptidases in Neuronal Cells
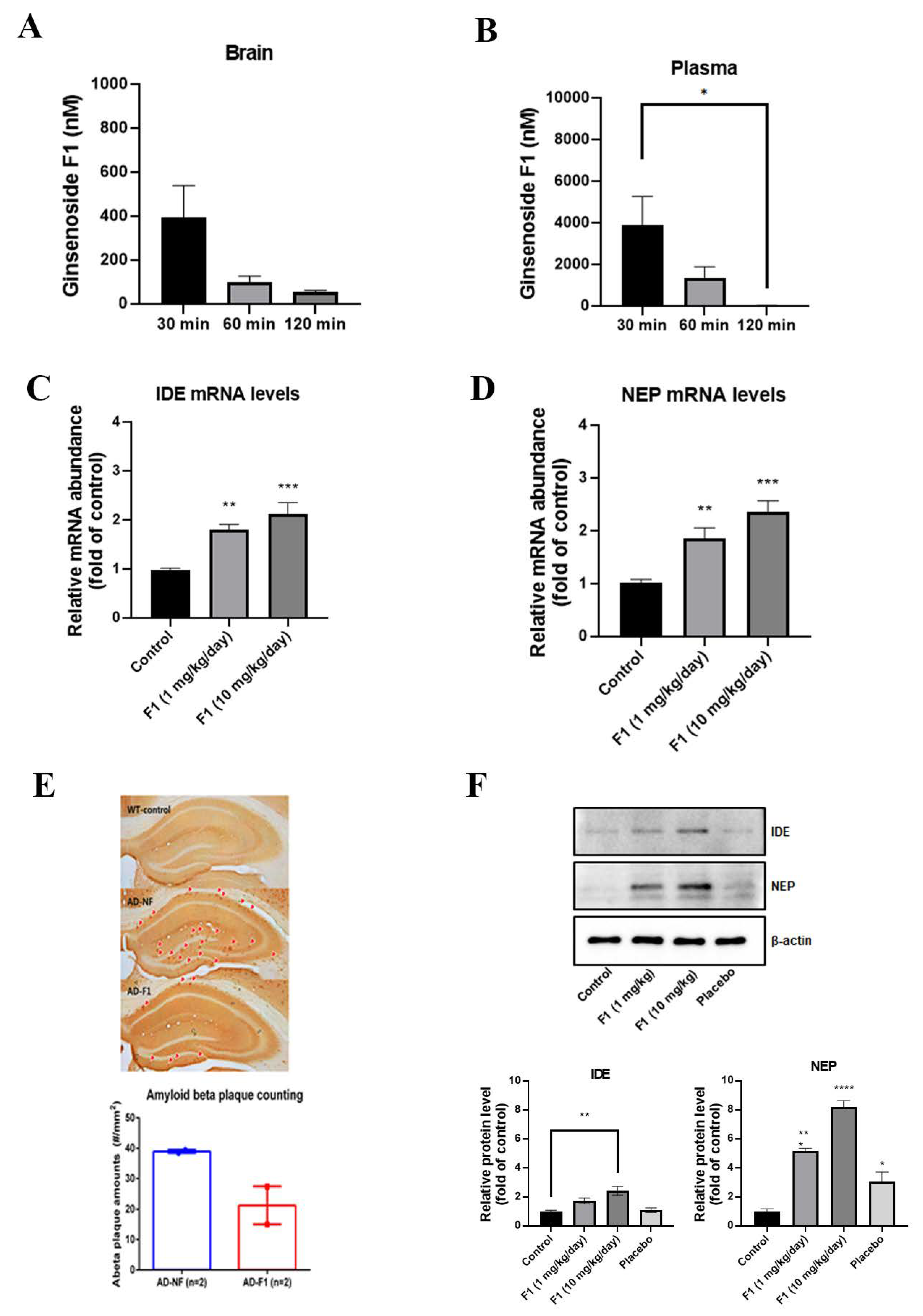
3.4. Ginsenoside F1 Can Pass BBB
3.5. Ginsenoside F1 Reduces the Aβ Plaque Formation through Aβ Peptidases in an AD Mouse Model
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Soto, C.; Kindy, M.S.; Baumann, M.; Frangione, B. Inhibition of Alzheimer’s amyloidosis by peptides that prevent beta-sheet conformation. Biochem. Biophys. Res. Commun. 1996, 226, 672–680. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Translating cell biology into therapeutic advances in Alzheimer’s disease. Nature 1999, 399, A23–A31. [Google Scholar] [CrossRef] [PubMed]
- Roher, A.E.; Chaney, M.O.; Kuo, Y.M.; Webster, S.D.; Stine, W.B.; Haverkamp, L.J.; Woods, A.S.; Cotter, R.J.; Tuohy, J.M.; Krafft, G.A.; et al. Morphology and toxicity of Abeta-(1–42) dimer derived from neuritic and vascular amyloid deposits of Alzheimer’s disease. J. Biol. Chem. 1996, 271, 20631–20635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid beta-peptide. Nat. Rev. Mol. Cell. Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Nalivaeva, N.N.; Belyaev, N.D.; Kerridge, C.; Turner, A.J. Amyloid-clearing proteins and their epigenetic regulation as a therapeutic target in Alzheimer’s disease. Front. Aging Neurosci. 2014, 6, 235. [Google Scholar] [CrossRef]
- Nalivaeva, N.N.; Beckett, C.; Belyaev, N.D.; Turner, A.J. Are amyloid-degrading enzymes viable therapeutic targets in Alzheimer’s disease? J. Neurochem. 2012, 120, 167–185. [Google Scholar] [CrossRef]
- Jha, N.K.; Jha, S.K.; Kumar, D.; Kejriwal, N.; Sharma, R.; Ambasta, R.K.; Kumar, P. Impact of insulin degrading enzyme and neprilysin in Alzheimer’s disease biology: Characterization of putative cognates for therapeutic applications. J. Alzheimers Dis. 2015, 48, 891–917. [Google Scholar] [CrossRef]
- Qi, L.-W.; Wang, C.-Z.; Yuan, C.-S. Ginsenosides from American ginseng: Chemical and pharmacological diversity. Phytochemistry 2011, 72, 689–699. [Google Scholar] [CrossRef] [Green Version]
- Chae, S.; Kang, K.A.; Chang, W.Y.; Kim, M.J.; Lee, S.J.; Lee, Y.S.; Kim, H.S.; Kim, D.H.; Hyun, J.W. Effect of Compound K, a metabolite of ginseng saponin, combined with γ-Ray radiation in human lung cancer cells in vitro and in vivo. J. Agric. Food Chem. 2009, 57, 5777–5782. [Google Scholar] [CrossRef]
- Mochizuki, M.; Yoo, Y.C.; Matsuzawa, K.; Sato, K.; Saiki, I.; Tonooka, S.; Samukawa, K.; Azuma, I. Inhibitory effect of tumor metastasis in mice by saponins, Ginsenoside-Rb2, 20 (R)-and 20 (S)-Ginsenoside-Rg3, of red ginseng. Biol. Pharm. Bull. 1995, 18, 1197–1202. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.-Q.; Luo, X.-Y.; Liu, G.-Z.; Chen, Y.-P.; Wang, Z.-C.; Sun, Y.-X. In vitro study of the relationship between the structure of ginsenoside and its antioxidative or prooxidative activity in free radical induced hemolysis of human erythrocytes. J. Agric. Food Chem. 2003, 51, 2555–2558. [Google Scholar] [CrossRef]
- Stavro, P.M.; Woo, M.; Heim, T.; Leiter, L.A.; Vuksan, V. North American ginseng exerts a neutral effect on blood pressure in individuals with hypertension. Hypertension 2005, 46, 406–411. [Google Scholar] [CrossRef] [Green Version]
- Jiang, B.; Xiong, Z.; Yang, J.; Wang, W.; Wang, Y.; Hu, Z.-L.; Wang, F.; Chen, J.-G. Antidepressant-like effects of ginsenoside Rg1 are due to activation of the BDNF signalling pathway and neurogenesis in the hippocampus. Br. J. Pharmacol. 2012, 166, 1872–1887. [Google Scholar] [CrossRef] [Green Version]
- Feng, L.; Wang, L.; Hu, C.; Jiang, X. Pharmacokinetics, Tissue Distribution, Metabolism, and Excretion of Ginsenoside Rg1 in Rats. Arch. Pharm. Res. 2010, 33, 1975–1984. [Google Scholar] [CrossRef]
- Bae, E.-A.; Shin, J.-E.; Kim, D.-H. Metabolism of ginsenoside Re by human intestinal microflora and its estrogenic effect. Biol. Pharm. Bull. 2005, 28, 1903–1908. [Google Scholar] [CrossRef] [Green Version]
- Cui, C.H.; Jeon, B.M.; Fu, Y.; Im, W.T.; Kim, S.C. High-density immobilization of a ginsenoside-transforming β-glucosidase for enhanced food-grade production of minor ginsenosides. Appl. Microbiol. Biotechnol. 2019, 103, 7003–7015. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L. Voluntary oral administration of drugs in mice. Protoc. Exch 2011. [Google Scholar] [CrossRef]
- Townsend, K.P.; Pratico, D. Novel therapeutic opportunities for Alzheimer’s disease: Focus on nonsteroidal anti-inflammatory drugs. FASEB J. 2005, 19, 1592–1601. [Google Scholar] [CrossRef]
- Dorfman, V.B.; Pasquini, L.; Riudavets, M.; López-Costa, J.J.; Villegas, A.; Troncoso, J.C.; Lopera, F.; Castaño, E.M.; Morelli, L. Differential cerebral deposition of IDE and NEP in sporadic and familial Alzheimer’s disease. Neurobiol. Aging 2010, 31, 1743–1757. [Google Scholar] [CrossRef] [Green Version]
- Iwata, N.; Tsubuki, S.; Takaki, Y.; Shirotani, K.; Lu, B.; Gerard, N.P.; Gerard, C.; Hama, E.; Lee, H.J.; Saido, T.C. Metabolic regulation of brain Abeta by neprilysin. Science 2001, 292, 1550–1552. [Google Scholar] [CrossRef]
- Farris, W.; Mansourian, S.; Chang, Y.; Lindsley, L.; Eckman, E.A.; Frosch, M.P.; Eckman, C.B.; Tanzi, R.E.; Selkoe, D.J.; Guenette, S. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc. Natl. Acad. Sci. USA 2003, 100, 4162–4167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; He, P.; Cui, J.; Staufenbiel, M.; Harada, N.; Shen, Y. Brain endogenous estrogen levels determine responses to estrogen replacement therapy via regulation of BACE1 and NEP in female Alzheimer’s transgenic mice. Mol. Neurobiol. 2013, 47, 857–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leissring, M.A.; Farris, W.; Chang, A.Y.; Walsh, D.M.; Wu, X.; Sun, X.; Frosch, M.P.; Selkoe, D.J. Enhanced proteolysis of beta-amyloid in APP transgenic mice prevents plaque formation, secondary pathology, and premature death. Neuron 2003, 40, 1087–1093. [Google Scholar] [CrossRef] [Green Version]
- Ertekin-Taner, N.; Allen, M.; Fadale, D.; Scanlin, L.; Younkin, L.; Petersen, R.C.; Graff-Radford, N.; Younkin, S.G. Genetic variants in a haplotype block spanning IDE are significantly associated with plasma Abeta42 levels and risk for Alzheimer disease. Hum. Mutat. 2004, 23, 334–342. [Google Scholar] [CrossRef]
- Vekrellis, K.; Ye, Z.; Qiu, W.Q.; Walsh, D.; Hartley, D.; Chesneau, V.; Rosner, M.R.; Selkoe, D.J. Neurons regulate extracellular levels of amyloid beta-protein via proteolysis by insulin-degrading enzyme. J. Neurosci. 2000, 20, 1657–1665. [Google Scholar] [CrossRef] [Green Version]
- Kurochkin, I.V.; Goto, S. Alzheimer’s beta-amyloid peptide specifically interacts with and is degraded by insulin degrading enzyme. FEBS Lett. 1994, 345, 33–37. [Google Scholar] [CrossRef] [Green Version]
- Pérez, A.; Morelli, L.; Cresto, J.C.; Castaño, E.M. Degradation of soluble amyloid beta-peptides 1–40, 1–42, and the Dutch variant 1–40Q by insulin degrading enzyme from Alzheimer disease and control brains. Neurochem. Res. 2000, 25, 247–255. [Google Scholar] [CrossRef]
- Reilly, C.E. Neprilysin content is reduced in Alzheimer brain areas. J. Neurol. 2001, 248, 159–160. [Google Scholar] [CrossRef]
- Apelt, J.; Ach, K.; Schliebs, R. Aging-related down-regulation of neprilysin, a putative beta-amyloid-degrading enzyme, in transgenic Tg2576 Alzheimer-like mouse brain is accompanied by an astroglial upregulation in the vicinity of beta-amyloid plaques. Neurosci. Lett. 2003, 339, 183–186. [Google Scholar] [CrossRef]
- Caccamo, A.; Oddo, S.; Sugarman, M.C.; Akbari, Y.; LaFerla, F.M. Age- and region-dependent alterations in Abeta-degrading enzymes: Implications for Abeta-induced disorders. Neurobiol. Aging 2005, 26, 645–654. [Google Scholar] [CrossRef]
- Kubo, T.; Nishimura, S.; Kumagae, Y.; Kaneko, I. In vivo conversion of racemized beta-amyloid ([D-Ser 26]A beta 1–40) to truncated and toxic fragments ([D-Ser 26]A beta 25–35/40) and fragment presence in the brains of Alzheimer’s patients. J. Neurosci. Res. 2002, 70, 474–483. [Google Scholar] [CrossRef]
- Gowing, E.; Roher, A.E.; Woods, A.S.; Cotter, R.J.; Chaney, M.; Little, S.P.; Ball, M.J. Chemical characterization of A beta 17–42 peptide, a component of diffuse amyloid deposits of Alzheimer disease. J. Biol. Chem. 1994, 269, 10987–10990. [Google Scholar] [CrossRef]
- Tarozzi, A.; Morroni, F.; Merlicco, A.; Bolondi, C.; Teti, G.; Falconi, M.; Cantelli-Forti, G.; Hrelia, P. Neuroprotective effects of cyanidin 3-O-glucopyranoside on amyloid beta (25–35) oligomer-induced toxicity. Neurosci. Lett. 2010, 473, 72–76. [Google Scholar] [CrossRef]
- Selkoe, D.J. Clearing the brain’s amyloid cobwebs. Neuron 2001, 32, 177–180. [Google Scholar] [CrossRef] [Green Version]
- Pardridge, W.M. Biopharmaceutical drug targeting to the brain. J. Drug Target. 2010, 18, 157–167. [Google Scholar] [CrossRef]
- Kelsey, N.A.; Wilkins, H.M.; Linseman, D.A. Nutraceutical antioxidants as novel neuroprotective agents. Molecules 2010, 15, 7792–7814. [Google Scholar] [CrossRef] [Green Version]
- Nabavi, S.F.; Braidy, N.; Habtemariam, S.; Orhan, I.E.; Daglia, M.; Manayi, A.; Gortzi, O.; Nabavi, S.M. Neuroprotective effects of chrysin: From chemistry to medicine. Neurochem. Int. 2015, 90, 224–231. [Google Scholar] [CrossRef]
- Brown, R.C.; Lockwood, A.H.; Sonawane, B.R. Neurodegenerative diseases: An overview of environmental risk factors. Environ. Health Perspect. 2005, 113, 1250–1256. [Google Scholar] [CrossRef] [Green Version]
- de Andrade Teles, R.B.; Diniz, T.C.; Costa Pinto, T.C.; de Oliveira Júnior, R.G.; Gama E Silva, M.; de Lavor, É.M.; Fernandes, A.W.C.; de Oliveira, A.P.; de Almeida Ribeiro, F.P.R.; da Silva, A.A.M.; et al. Flavonoids as Therapeutic Agents in Alzheimer’s and Parkinson’s Diseases: A Systematic Review of Preclinical Evidences. Oxid. Med. Cell. Longev. 2018, 7043213. [Google Scholar] [CrossRef]
- Marković, Z.; Đorović, J.; Petrović, Z.D.; Petrović, V.P.; Simijonović, D. Investigation of the antioxidant and radical scavenging activities of some phenolic Schiff bases with different free radicals. J. Mol. Model. 2015, 21, 293. [Google Scholar] [CrossRef]
- Nday, C.M.; Halevas, E.; Jackson, G.E.; Salifoglou, A. Quercetin encapsulation in modified silica nanoparticles: Potential use against Cu(II)-induced oxidative stress in neurodegeneration. J. Inorg. Biochem. 2015, 145, 51–64. [Google Scholar] [CrossRef]
- Elbaz, A.; Carcaillon, L.; Kab, S.; Moisan, F. Epidemiology of Parkinson’s disease. Rev. Neurol. 2016, 172, 14–26. [Google Scholar] [CrossRef]
- Han, J.; Oh, J.-P.; Yoo, M.; Cui, C.-H.; Jeon, B.-M.; Kim, S.-C.; Han, J.-H. Minor ginsenoside F1 improves memory in APP/PS1 mice. Mol. Brain. 2019, 12, 77. [Google Scholar] [CrossRef] [Green Version]
- Hou, J.; Cui, C.; Kim, S.; Sung, C.; Choi, C. Ginsenoside F1 suppresses astrocytic senescence-associated secretory phenotype. Chem. Biol. Interact. 2018, 283, 75–83. [Google Scholar] [CrossRef]
- Bastola, T.; Pariyar, R.; Jeon, B.; Baek, J.; Chang, B.; Kim, S.-C.; Kim, S.; Seo, J. Protective effects of SGB121, ginsenoside F1-enriched ginseng extract, on scopolamine-induced cytotoxicity and memory impairments. J. Funct. Foods 2020, 74, 104165. [Google Scholar] [CrossRef]
- Yu, D.; Liu, C.; Zhang, H.; Ren, J.; Qu, X. Glycoengineering artificial receptors for microglia to phagocytose Aβ aggregates. Chem. Sci. 2021, 12, 4963–4969. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yun, Y.-J.; Park, B.-H.; Hou, J.; Oh, J.-P.; Han, J.-H.; Kim, S.-C. Ginsenoside F1 Protects the Brain against Amyloid Beta-Induced Toxicity by Regulating IDE and NEP. Life 2022, 12, 58. https://doi.org/10.3390/life12010058
Yun Y-J, Park B-H, Hou J, Oh J-P, Han J-H, Kim S-C. Ginsenoside F1 Protects the Brain against Amyloid Beta-Induced Toxicity by Regulating IDE and NEP. Life. 2022; 12(1):58. https://doi.org/10.3390/life12010058
Chicago/Turabian StyleYun, Yee-Jin, Bong-Hwan Park, Jingang Hou, Jung-Pyo Oh, Jin-Hee Han, and Sun-Chang Kim. 2022. "Ginsenoside F1 Protects the Brain against Amyloid Beta-Induced Toxicity by Regulating IDE and NEP" Life 12, no. 1: 58. https://doi.org/10.3390/life12010058
APA StyleYun, Y.-J., Park, B.-H., Hou, J., Oh, J.-P., Han, J.-H., & Kim, S.-C. (2022). Ginsenoside F1 Protects the Brain against Amyloid Beta-Induced Toxicity by Regulating IDE and NEP. Life, 12(1), 58. https://doi.org/10.3390/life12010058