
Two Novel Functional Mutations in Promoter Region of SCN3B Gene Associated with Atrial Fibrillation

Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Mutation Analysis of SCN3B
2.3. Bioinformatics-Based Prediction
2.4. Clones and Site-Directed Mutagenesis
2.5. Dual Luciferase Reporter Assays
2.6. Western Blotting
2.7. RNA Isolation and Quantitative RT-PCR
2.8. Statistical Analysis
3. Results
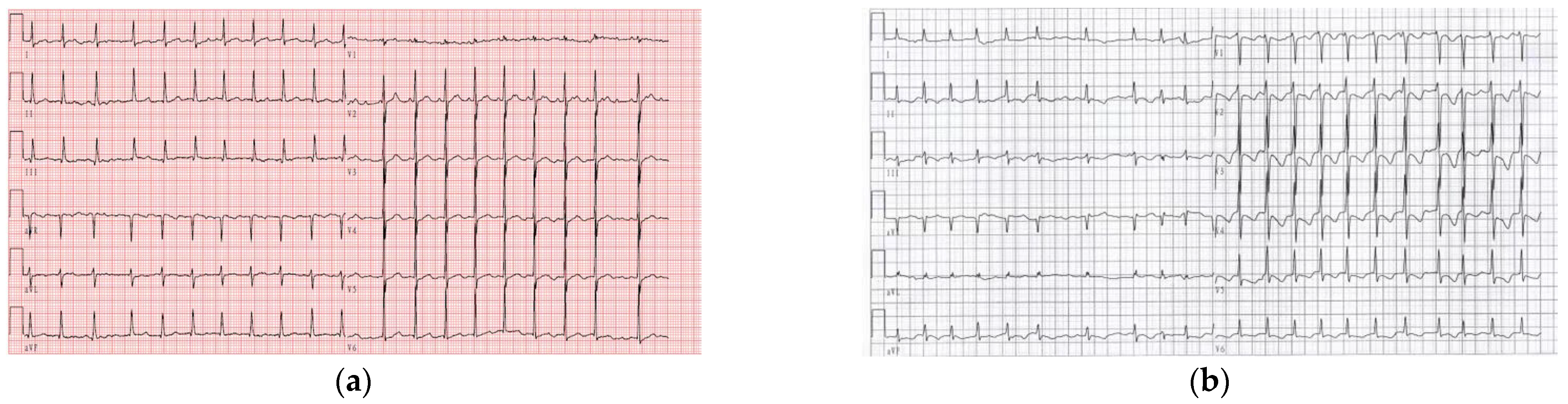
3.1. Identification of Two Novel Susceptibility Mutations in SCN3B
3.2. Transcriptional Activity and Expression Level Enhanced in the Alternative a Allele of Rare Variant c.-324C>A (rs976125894)
3.3. c.-324C>A Variant (rs976125894) Regulates the Expression of SCN3B through Interacting with Transcription Factor GATA4
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zimetbaum, P. Atrial Fibrillation. Ann. Intern. Med. 2017, 166, ITC33–ITC48. [Google Scholar] [CrossRef] [PubMed]
- Piccini, J.P.; Hammill, B.G.; Sinner, M.F.; Jensen, P.N.; Hernandez, A.F.; Heckbert, S.R.; Benjamin, E.J.; Curtis, L.H. Incidence and prevalence of atrial fibrillation and associated mortality among Medicare beneficiaries, 1993–2007. Circ. Cardiovasc. Qual. Outcomes 2012, 5, 85–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuster, V.; Rydén, L.; Cannom, D.S.; Crijns, H.J.; Curtis, A.B.; Ellenbogen, K.A.; Halperin, J.L.; Le, H.J.; Kay, G.N.; Lowe, J.E. ACC/AHA/ESC 2006 Guidelines for the Management of Patients with Atrial Fibrillation: A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Revise the 2001 Guidelines for the Management of Patients With Atrial Fibrillation): Developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Eur. Heart J. 2006, 38, 1231–1265. [Google Scholar] [CrossRef] [Green Version]
- Tsao, C.W.; Vasan, R.S. Cohort Profile: The Framingham Heart Study (FHS): Overview of milestones in cardiovascular epidemiology. Int. J. Epidemiol. 2015, 44, 1800–1813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, J.D.; Gollob, M.H. Impact of genetic discoveries on the classification of lone atrial fibrillation. J. Am. Coll. Cardiol. 2010, 55, 705–712. [Google Scholar] [CrossRef] [Green Version]
- Christophersen, I.E.; Ravn, L.S.; Budtz-Joergensen, E.; Skytthe, A.; Haunsoe, S.; Svendsen, J.H.; Christensen, K. Familial aggregation of atrial fibrillation: A study in Danish twins. Circ. Arrhythm. Electrophysiol. 2009, 2, 378–383. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.H.; Xu, S.J.; Bendahhou, S.; Wang, X.L.; Wang, Y.; Xu, W.Y.; Jin, H.W.; Sun, H.; Su, X.Y.; Zhuang, Q.N.; et al. KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science 2003, 299, 251–254. [Google Scholar] [CrossRef]
- Xiang, R.; Xu, C.; Zhan, C.; Yang, Y.; Shi, L.; Fan, W.; Wang, C.; Xia, Y.; Yang, B.; Gang, W. Identification of NPPA variants associated with atrial fibrillation in a Chinese GeneID population. Clin. Chim. Acta 2010, 411, 481–485. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, S.; Yoo, S.; Chakrabarti, S.; Zhang, T.; Ke, T.; Oberti, C.; Yong, S.L.; Fang, F.; Li, L. Mutation in Nuclear Pore Component NUP155 Leads to Atrial Fibrillation and Early Sudden Cardiac Death. Cell 2008, 135, 1017–1027. [Google Scholar] [CrossRef] [Green Version]
- Olesen, M.S.; Nielsen, M.W.; Haunso, S.; Svendsen, J.H. Atrial fibrillation: The role of common and rare genetic variants. Eur. J. Hum. Genet. 2014, 22, 297–306. [Google Scholar] [CrossRef]
- Chengqi, X.; Rongfeng, Z.; Yunlong, X.; Liang, X.; Wei, Y.; Pengyun, W. Annotation of susceptibility SNPs associated with atrial fibrillation. Aging 2020, 12, 16981–16998. [Google Scholar] [CrossRef]
- Choi, S.H.; Weng, L.C.; Roselli, C.; Lin, H.; Haggerty, C.M.; Shoemaker, M.B.; Barnard, J.; Arking, D.E.; Chasman, D.I.; Albert, C.M.; et al. Association Between Titin Loss-of-Function Variants and Early-Onset Atrial Fibrillation. JAMA 2018, 320, 2354–2364. [Google Scholar] [CrossRef] [PubMed]
- Olson, T.M.; Michels, V.V.; Ballew, J.D.; Reyna, S.P.; Karst, M.L.; Herron, K.J.; Horton, S.C.; Rodeheffer, R.J.; Anderson, J.L. Sodium Channel Mutations and Susceptibility to Heart Failure and Atrial Fibrillation. JAMA 2005, 293, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Darbar, D.; Kaiser, D.W.; Jiramongkolchai, K.; Chopra, S.; Donahue, B.S.; Kannankeril, P.J.; Roden, D.M. Mutations in sodium channel β1- and β2-subunits associated with atrial fibrillation. Circ. Arrhythmia Electrophysiol. 2009, 2, 268–275. [Google Scholar] [CrossRef] [Green Version]
- Hu, D.; Barajas-Martinez, H.; Burashnikov, E.; Springer, M.; Wu, Y.; Varro, A.; Pfeiffer, R.; Koopmann, T.T.; Cordeiro, J.M.; Guerchicoff, A. A Mutation in the β3 Subunit of the Cardiac Sodium Channel Associated With Brugada ECG Phenotype. Circ. Cardiovasc. Genet. 2009, 2, 270. [Google Scholar] [CrossRef]
- Medeiros-Domingo, A.; Kaku, T.; Tester, D.J.; Iturralde-Torres, P.; Itty, A.; Ye, B.; Valdivia, C.; Ueda, K.; Canizales-Quinteros, S.; Tusié-Luna, M.T. SCN4B-Encoded Sodium Channel β4 Subunit in Congenital Long-QT Syndrome. Circ. Cardiovasc. Genet. 2007, 116, 134. [Google Scholar] [CrossRef] [Green Version]
- Li, R.G.; Wang, Q.; Xu, Y.J.; Zhang, M.; Qu, X.K.; Liu, X.; Fang, W.Y.; Yang, Y.Q. Mutations of the SCN4B-encoded sodium channel beta4 subunit in familial atrial fibrillation. Int. J. Mol. Med. 2013, 32, 144–150. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, H.; Koopmann, T.T.; Scouarnec, S.L.; Tao, Y.; Bezzina, C.R. Sodium channel β1 subunit mutations associated with Brugada syndrome and cardiac conduction disease in humans. J. Clin. Investig. 2008, 118, 2260–2268. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Yang, Q.; Wu, X.; Yang, Y.; Shi, L.; Wang, C.; Wu, G.; Xia, Y.; Yang, B.; Zhang, R.; et al. Functional dominant-negative mutation of sodium channel subunit gene SCN3B associated with atrial fibrillation in a Chinese GeneID population. Biochem. Biophys. Res. Commun. 2010, 398, 98–104. [Google Scholar] [CrossRef] [Green Version]
- Olesen, M.S.; Jespersen, T.; Nielsen, J.B.; Bo, L.; Møller, D.V.; Hedley, P.; Christiansen, M.; Varró, A.; Olesen, S.P.; Haunsø, S. Mutations in sodium channel β-subunit SCN3B are associated with early-onset lone atrial fibrillation. Cardiovasc. Res. 2011, 89, 786–793. [Google Scholar] [CrossRef]
- Hakim, P.; Brice, N.; Thresher, R.; Lawrence, J.; Zhang, Y.; Jackson, A.P.; Grace, A.A.; Huang, C.L. Scn3b knockout mice exhibit abnormal sino-atrial and cardiac conduction properties. Acta Physiol. 2010, 198, 47–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdivia, C.R.; Medeiros-Domingo, A.; Ye, B.; Shen, W.K.; Algiers, T.J.; Ackerman, M.J.; Makielski, J.C. Loss-of-function mutation of the SCN3B-encoded sodium channel β3 subunit associated with a case of idiopathic ventricular fibrillation. Cardiovasc. Res. 2010, 86, 392–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellinor, P.T.; Nam, E.G.; Shea, M.A.; Milan, D.J.; Ruskin, J.N.; MacRae, C.A. Cardiac sodium channel mutation in atrial fibrillation. Heart Rhythm. 2008, 5, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Huang, H.; Liu, G.; Lam, K.; Rutberg, J.; Green, M.S.; Birnie, D.H.; Lemery, R.; Chahine, M.; Gollob, M.H. Gain-of-function mutation of Nav1.5 in atrial fibrillation enhances cellular excitability and lowers the threshold for action potential firing. Biochem. Biophys. Res. Commun. 2009, 380, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Lieve, K.V.; Verkerk, A.O.; Podliesna, S.; van der Werf, C.; Tanck, M.W.; Hofman, N.; van Bergen, P.F.; Beekman, L.; Bezzina, C.R.; Wilde, A.A.M.; et al. Gain-of-function mutation in SCN5A causes ventricular arrhythmias and early onset atrial fibrillation. Int. J. Cardiol. 2017, 236, 187–193. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, K.; Konno, T.; Tada, H.; Tani, S.; Liu, L.; Fujino, N.; Nohara, A.; Hodatsu, A.; Tsuda, T.; Tanaka, Y.; et al. Functional Characterization of Rare Variants Implicated in Susceptibility to Lone Atrial Fibrillation. Circ. Arrhythm. Electrophysiol. 2015, 8, 1095–1104. [Google Scholar] [CrossRef]
- Zhu, Y.; Tazearslan, C.; Suh, Y. Challenges and progress in interpretation of non-coding genetic variants associated with human disease. Exp. Biol. Med. 2017, 242, 1325–1334. [Google Scholar] [CrossRef] [Green Version]
- Kraft, P.; Wacholder, S.; Cornelis, M.C.; Hu, F.B.; Hayes, R.B.; Thomas, G.; Hoover, R.; Hunter, D.J.; Chanock, S. Beyond odds ratios—Communicating disease risk based on genetic profiles. Nat. Rev. Genet. 2009, 10, 264–269. [Google Scholar] [CrossRef]
- Sampsa, P.; Heikki, T.; Risto, K.; Heikki, R.J. GATA transcription factors in the developing and adult heart. Cardiovasc. Res. 2004, 2, 196–207. [Google Scholar] [CrossRef]
- Bruneau, B.G. The developmental genetics of congenital heart disease. Nature 2008, 451, 943–948. [Google Scholar] [CrossRef]
- Ang, Y.S.; Rivas, R.N.; Ribeiro, A.J.S.; Srivas, R.; Rivera, J.; Stone, N.R.; Pratt, K.; Mohamed, T.M.A.; Fu, J.D.; Spencer, C.I.; et al. Disease Model of GATA4 Mutation Reveals Transcription Factor Cooperativity in Human Cardiogenesis. Cell 2016, 167, 1734–1749.e22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eric, N.O. Gene regulatory networks in the evolution and development of the heart. Science 2006, 313, 1922–1927. [Google Scholar] [CrossRef] [Green Version]
- Song, K.; Nam, Y.J.; Luo, X.; Qi, X.; Tan, W.; Huang, G.N.; Acharya, A.; Smith, C.L.; Tallquist, M.D.; Neilson, E.G.; et al. Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature 2012, 485, 599–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, J.Q.; Shen, F.F.; Fang, W.Y.; Liu, X.; Yang, Y.Q. Novel GATA4 mutations in lone atrial fibrillation. Int. J. Mol. Med. 2011, 28, 1025–1032. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.Q.; Wang, M.Y.; Zhang, X.L.; Tan, H.W.; Shi, H.F.; Jiang, W.F.; Wang, X.H.; Fang, W.Y.; Liu, X. GATA4 loss-of-function mutations in familial atrial fibrillation. Clin. Chim. Acta 2011, 412, 1825–1830. [Google Scholar] [CrossRef]
- Posch, M.G.; Boldt, L.H.; Polotzki, M.; Richter, S.; Rolf, S.; Perrot, A.; Dietz, R.; Ozcelik, C.; Haverkamp, W. Mutations in the cardiac transcription factor GATA4 in patients with lone atrial fibrillation. Eur. J. Med. Genet. 2010, 53, 201–203. [Google Scholar] [CrossRef]
- Nielsen, J.B.; Thorolfsdottir, R.B.; Fritsche, L.G.; Zhou, W.; Skov, M.W.; Graham, S.E.; Herron, T.J.; McCarthy, S.; Schmidt, E.M.; Sveinbjornsson, G.; et al. Biobank-driven genomic discovery yields new insight into atrial fibrillation biology. Nat. Genet. 2018, 50, 1234–1239. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence | |
|---|---|---|
| rs976125894 (Sanger) | F | CCCCACTGGACCTCCCCAGT |
| R | GATTCCAGTCGGAACGCAAC | |
| rs976125894 (HRM) | F | CCTCCCCAGTTCGAGGGAGC |
| R | CCACAGCCTGGCTGCTAGGC | |
| rs1284768362 (Sanger) | F | CCCAGGGGGCGACTTTCTGA |
| R | GAACGCAACCGATCCTGGGGA | |
| rs1284768362 (HRM) | F | CCGATCAGCCGCTCCGCGCC |
| R | AGCAGTGCGACTCCCTTCCGA |
| Target | Sequence | |
|---|---|---|
| 18S rRNA | F | CTCACTGAGGATGAGGTGG |
| R | GTTCAAGAACCAGTCTGGGA | |
| β-actin | F | GAAGATCAAGATCATTGCTCCTC |
| R | ATCCACATCTGCTGGAAGG | |
| SCN3B | F | TCTACTGGGTCAGTGTCTG |
| R | CTTCATGCAGGAGATGCAG | |
| GATA4 | F | AGATGCGTCCCATCAAGAC |
| R | CAGAGGCATTCAGGATGTG | |
| WT1 | F | CGTTTCTCACTGGTCTCAG |
| R | ATCCTCCGAACCCAAAAGCC | |
| IKZF1 | F | AAGTTTCAGGGAAGGAAAGC |
| R | CTCTTGGAGCTTTGCTGTC |
| Demographic and Clinical Feature | AF Population |
|---|---|
| Number of study subjects | 355 |
| Gender (number of females and %) | 145 (40.85%) |
| Age (mean ± SD years) | 44.05 ± 12.85 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, L.; Li, K.; Tian, B.; Jia, M.; Wang, Q.; Xu, C.; Xiong, L.; Wang, Q.; Zeng, Y.; Wang, P. Two Novel Functional Mutations in Promoter Region of SCN3B Gene Associated with Atrial Fibrillation. Life 2022, 12, 1794. https://doi.org/10.3390/life12111794
Lin L, Li K, Tian B, Jia M, Wang Q, Xu C, Xiong L, Wang Q, Zeng Y, Wang P. Two Novel Functional Mutations in Promoter Region of SCN3B Gene Associated with Atrial Fibrillation. Life. 2022; 12(11):1794. https://doi.org/10.3390/life12111794
Chicago/Turabian StyleLin, Liyan, Ke Li, Beijia Tian, Mengru Jia, Qianyan Wang, Chengqi Xu, Liang Xiong, Qing Wang, Yali Zeng, and Pengyun Wang. 2022. "Two Novel Functional Mutations in Promoter Region of SCN3B Gene Associated with Atrial Fibrillation" Life 12, no. 11: 1794. https://doi.org/10.3390/life12111794
APA StyleLin, L., Li, K., Tian, B., Jia, M., Wang, Q., Xu, C., Xiong, L., Wang, Q., Zeng, Y., & Wang, P. (2022). Two Novel Functional Mutations in Promoter Region of SCN3B Gene Associated with Atrial Fibrillation. Life, 12(11), 1794. https://doi.org/10.3390/life12111794