
Abdominal Aortic Aneurysm Formation with a Focus on Vascular Smooth Muscle Cells


{kind=link}
{kind=link}
Abstract
:1. Introduction
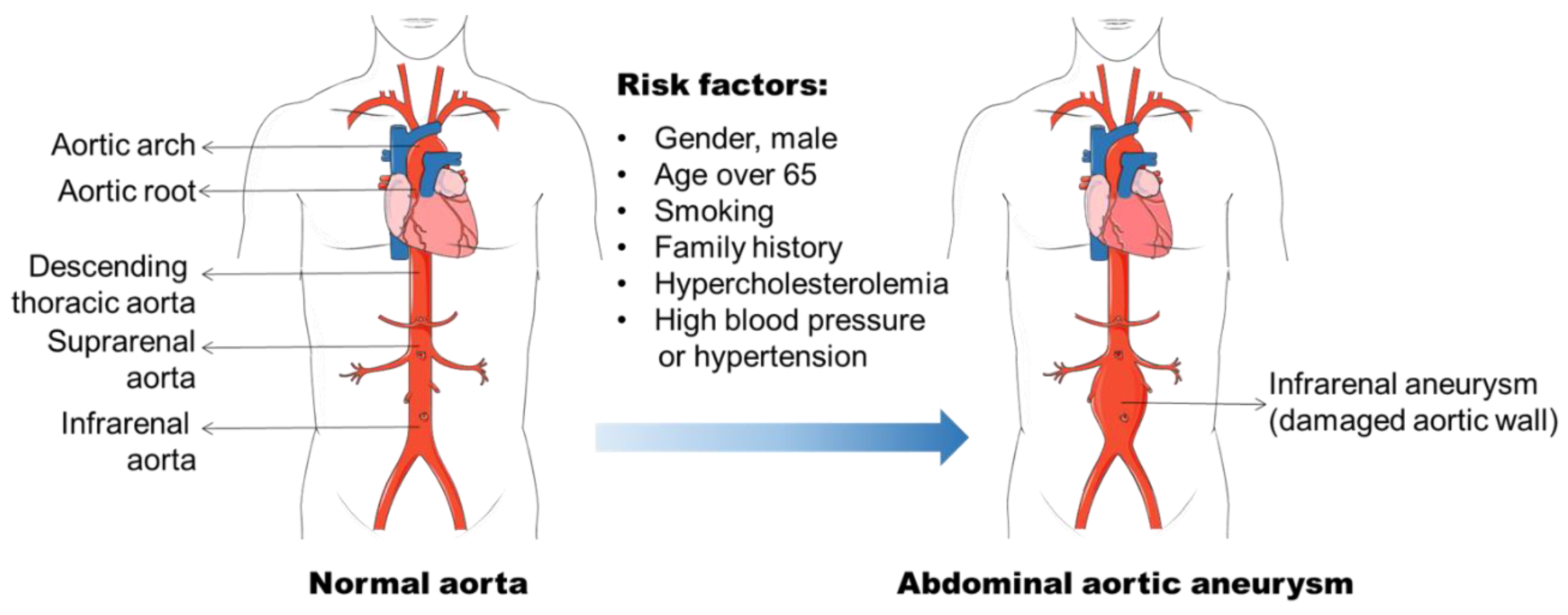
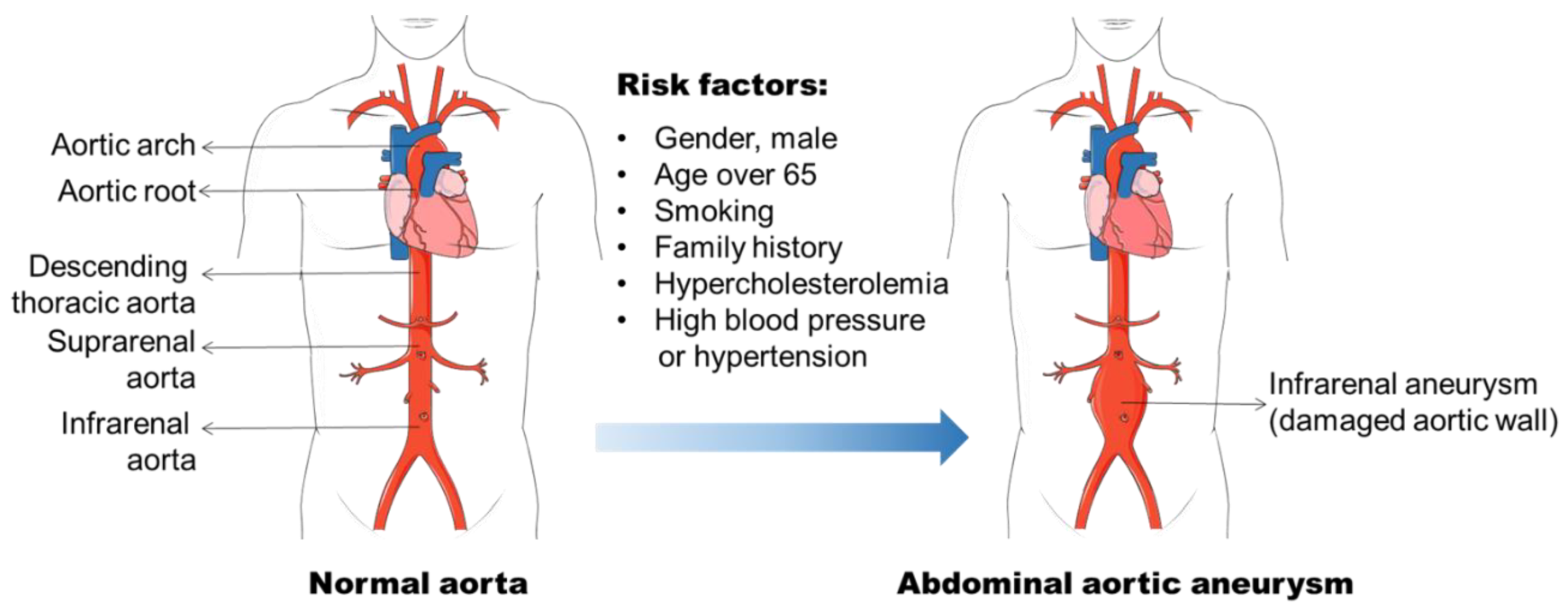
2. AAA Formation
2.1. Risk Factors for AAA
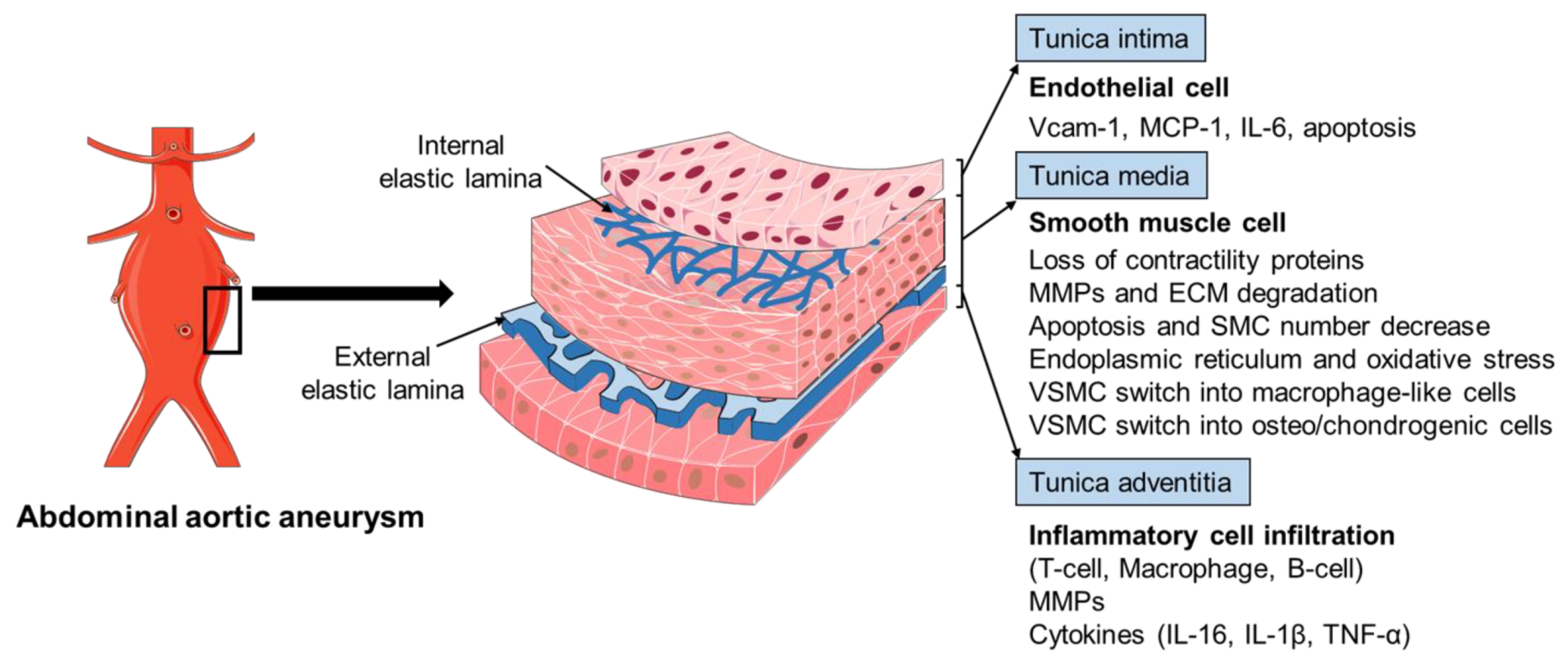
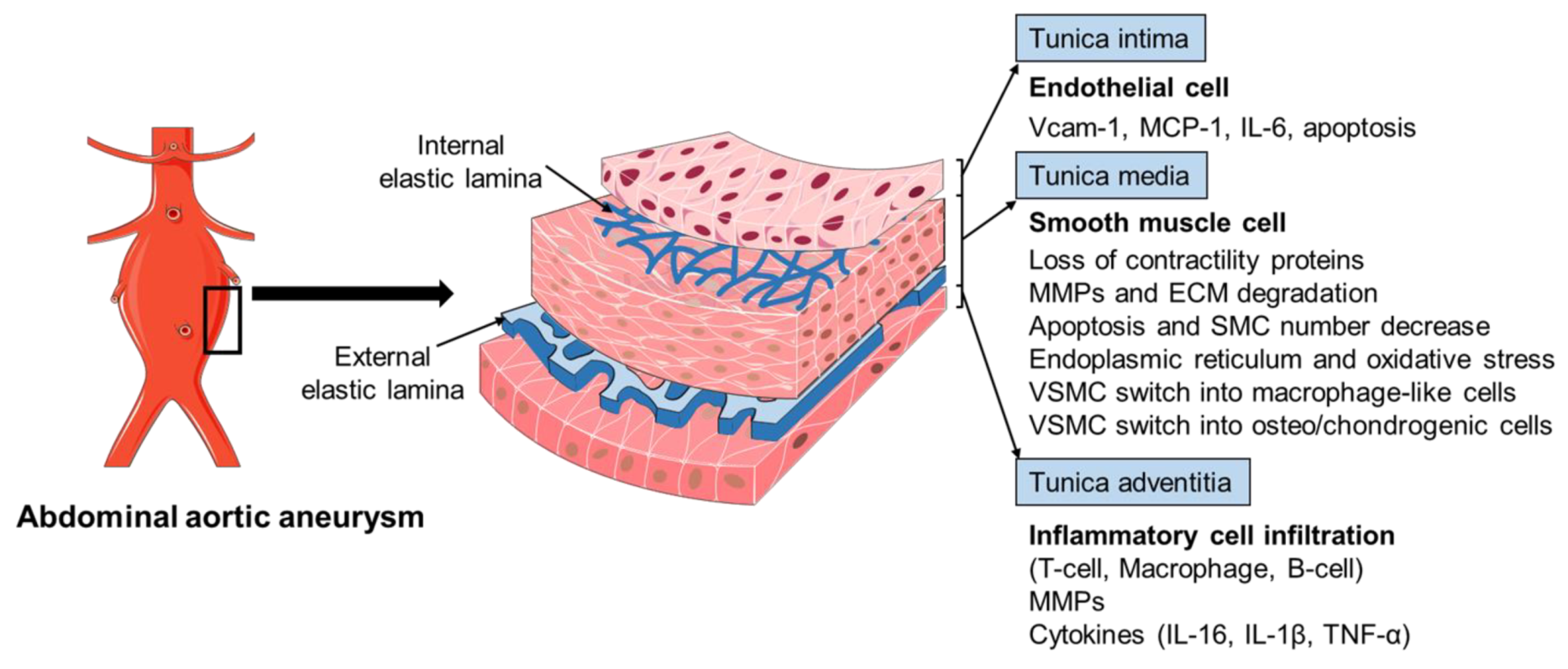
2.2. Histopathology of AAA
3. Vascular Smooth Muscle Cells in AAA Formation
3.1. VSMC Phenotypic Plasticity
3.2. VSMC Phenotypic Modulation in AAA
3.2.1. VSMC Contractility and TGF-β
3.2.2. SMCs Express Proteolytic Enzymes to Induce ECM Disorganization
3.2.3. Endoplasmic Reticulum Stress and Oxidative Stress
3.2.4. Apoptosis and SMC Loss
3.2.5. VSMCs Inflammatory Phenotypic Change and Transdifferentiation into Macrophage-like Cells
3.2.6. VSMC Phenotypic Switch into Osteo/Chondrogenic VSMCs
4. Animal Models Used to Investigate SMC Phenotypic Change and AAA
5. Application of Single-Cell RNA-Sequencing (scRNA Seq) in AAA Studies
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AAA | abdominal aortic aneurysm |
| Ang II | angiotensin II |
| α-SMA | α-smooth muscle actin |
| CNN | SM-calponin |
| EC | endothelial cell |
| ECM | extracellular matrix |
| HDACs | histone deacetylases |
| IL-1β | interleukin-1β |
| MCP-1 | monocyte chemoattractant protein 1 |
| MMP | matrix metalloproteinase |
| NOX | nicotinamide adenine dinucleotide phosphate oxidase |
| PDGF-BB | platelet-derived growth factor-BB |
| PPE | porcine pancreatic elastase |
| RIPK1/3 | receptor-interacting protein kinase 1/3 |
| ROS | reactive oxygen species |
| SMMHC | SM myosin heavy chain |
| SM22α | smooth muscle 22α |
| TAA | thoracic aortic aneurysm |
| TFEB | transcription factor EB |
| TGF-β | transforming growth factor-β |
| TGFBR1/2 | TGF-β receptor 1/2 |
| TIMPs | tissue inhibitor of metalloproteinases |
| TNF-α | tumor necrosis factor-α |
| Vcam-1 | vascular cell adhesion molecule 1 |
| VSMC | vascular smooth muscle cell |
References
- Johnston, K.W.; Rutherford, R.B.; Tilson, M.D.; Shah, D.M.; Hollier, L.; Stanley, J.C. Suggested standards for reporting on arterial aneurysms. Subcommittee on Reporting Standards for Arterial Aneurysms, Ad Hoc Committee on Reporting Standards, Society for Vascular Surgery and North American Chapter, International Society for Cardiovascular Surgery. J. Vasc. Surg. 1991, 13, 452–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jersey, A.M.; Foster, D.M. Cerebral Aneurysm; StatPearls Publishing LLC: Treasure Island, FL, USA, 2021. [Google Scholar]
- Quintana, R.A.; Taylor, W.R. Cellular Mechanisms of Aortic Aneurysm Formation. Circ. Res. 2019, 124, 607–618. [Google Scholar] [CrossRef] [PubMed]
- Kainth, A.; Smeds, M.R. Popliteal Aneurysm Repair; StatPearls Publishing LLC: Treasure Island, FL, USA, 2021. [Google Scholar]
- Anderson, P.L.; Arons, R.R.; Moskowitz, A.J.; Gelijns, A.; Magnell, C.; Faries, P.L.; Clair, D.; Nowygrod, R.; Kent, K.C. A statewide experience with endovascular abdominal aortic aneurysm repair: Rapid diffusion with excellent early results. J. Vasc. Surg. 2004, 39, 10–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nordon, I.M.; Hinchliffe, R.J.; Loftus, I.M.; Thompson, M.M. Pathophysiology and epidemiology of abdominal aortic aneurysms. Nat. Rev. Cardiol. 2011, 8, 92–102. [Google Scholar] [CrossRef]
- Eickhoff, J. Incidence of diagnosis, operation and death from abdominal aortic aneurysms in Danish hospitals: Results from a nation-wide survey, 1977–1990. Eur. J. Surg. Acta Chir. 1993, 159, 619–623. [Google Scholar]
- Moll, F.L.; Powell, J.T.; Fraedrich, G.; Verzini, F.; Haulon, S.; Waltham, M.; van Herwaarden, J.A.; Holt, P.J.; van Keulen, J.W.; Rantner, B. Management of abdominal aortic aneurysms clinical practice guidelines of the European society for vascular surgery. Eur. J. Vasc. Endovasc. Surg. 2011, 41, S1–S58. [Google Scholar] [CrossRef] [Green Version]
- Kantonen, I.; Lepantalo, M.; Brommels, M.; Luther, M.; Salenius, J.P.; Ylonen, K. Mortality in ruptured abdominal aortic aneurysms. The Finnvasc Study Group. Eur. J. Vasc. Endovasc. Surg. 1999, 17, 208–212. [Google Scholar] [CrossRef] [Green Version]
- Earnshaw, J.J.; Lees, T. Update on Screening for Abdominal Aortic Aneurysm. Eur. J. Vasc. Endovasc. Surg. 2017, 54, 1–2. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, T.F.X.; Landon, B.E.; Schermerhorn, M.L. AAA Screening Should Be Expanded. Circulation 2019, 140, 889–890. [Google Scholar] [CrossRef]
- Hallin, A.; Bergqvist, D.; Holmberg, L. Literature review of surgical management of abdominal aortic aneurysm. Eur. J. Vasc. Endovasc. Surg. 2001, 22, 197–204. [Google Scholar] [CrossRef] [Green Version]
- Kuivaniemi, H.; Ryer, E.J.; Elmore, J.R.; Tromp, G. Understanding the pathogenesis of abdominal aortic aneurysms. Expert Rev. Cardiovasc. 2015, 13, 975–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altobelli, E.; Rapacchietta, L.; Profeta, V.F.; Fagnano, R. Risk factors for abdominal aortic aneurysm in population-based studies: A systematic review and meta-analysis. Int. J. Environ. Res. Public Health 2018, 15, 2805. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilson, M. Aortic aneurysms and atherosclerosis. Circulation 1992, 85, 378–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonamigo, T.P.; Siqueira, I. Screening for abdominal aortic aneurysms. Rev. Hosp. Clin. Fac. Med. Sao Paulo 2003, 58, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Kim, L.G.; Thompson, S.G.; Marteau, T.M.; Scott, R.A.; Multicentre Aneurysm Screening Study, G. Screening for abdominal aortic aneurysms: The effects of age and social deprivation on screening uptake, prevalence and attendance at follow-up in the MASS trial. J. Med. Screen. 2004, 11, 50–53. [Google Scholar] [CrossRef] [Green Version]
- Saratzis, A.; Dattani, N.; Brown, A.; Shalhoub, J.; Bosanquet, D.; Sidloff, D.; Stather, P.; Vascular, T.; Endovascular Research, N. Multi-Centre Study on Cardiovascular Risk Management on Patients Undergoing AAA Surveillance. Eur. J. Vasc. Endovasc. Surg. 2017, 54, 116–122. [Google Scholar] [CrossRef] [Green Version]
- Svensjo, S.; Bjorck, M.; Gurtelschmid, M.; Djavani Gidlund, K.; Hellberg, A.; Wanhainen, A. Low prevalence of abdominal aortic aneurysm among 65-year-old Swedish men indicates a change in the epidemiology of the disease. Circulation 2011, 124, 1118–1123. [Google Scholar] [CrossRef] [Green Version]
- Norman, P.E.; Curci, J.A. Understanding the effects of tobacco smoke on the pathogenesis of aortic aneurysm. Arter. Thromb. Vasc. Biol. 2013, 33, 1473–1477. [Google Scholar] [CrossRef] [Green Version]
- Stolle, K.; Berges, A.; Lietz, M.; Lebrun, S.; Wallerath, T. Cigarette smoke enhances abdominal aortic aneurysm formation in angiotensin II-treated apolipoprotein E-deficient mice. Toxicol. Lett. 2010, 199, 403–409. [Google Scholar] [CrossRef]
- Maegdefessel, L.; Azuma, J.; Toh, R.; Deng, A.; Merk, D.R.; Raiesdana, A.; Leeper, N.J.; Raaz, U.; Schoelmerich, A.M.; McConnell, M.V.; et al. MicroRNA-21 blocks abdominal aortic aneurysm development and nicotine-augmented expansion. Sci. Transl. Med. 2012, 4, 122ra122. [Google Scholar] [CrossRef] [Green Version]
- Daugherty, A.; Manning, M.W.; Cassis, L.A. Angiotensin II promotes atherosclerotic lesions and aneurysms in apolipoprotein E-deficient mice. J. Clin. Investig. 2000, 105, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuivaniemi, H.; Platsoucas, C.D.; Tilson III, M.D. Aortic aneurysms: An immune disease with a strong genetic component. Circulation 2008, 117, 242–252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahlgren, C.M.; Larsson, E.; Magnusson, P.K.; Hultgren, R.; Swedenborg, J. Genetic and environmental contributions to abdominal aortic aneurysm development in a twin population. J. Vasc. Surg. 2010, 51, 3–7; discussion 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Vlijmen-van Keulen, C.J.; Pals, G.; Rauwerda, J.A. Familial abdominal aortic aneurysm: A systematic review of a genetic background. Eur. J. Vasc. Endovasc. Surg. 2002, 24, 105–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, C.W.; Barber, G.G.; Bouchard, A.G.; McPhail, N.V.; Roberge, C.; Waddell, W.G.; Wellington, J.L. Abdominal aortic aneurysm: Consequences of a positive family history. Can. J. Surg. 1989, 32, 117–120. [Google Scholar] [PubMed]
- Larsson, E.; Granath, F.; Swedenborg, J.; Hultgren, R. A population-based case-control study of the familial risk of abdominal aortic aneurysm. J. Vasc. Surg. 2009, 49, 47–50; discussion 51. [Google Scholar] [CrossRef] [Green Version]
- Vaughan, C.J.; Casey, M.; He, J.; Veugelers, M.; Henderson, K.; Guo, D.; Campagna, R.; Roman, M.J.; Milewicz, D.M.; Devereux, R.B. Identification of a chromosome 11q23. 2-q24 locus for familial aortic aneurysm disease, a genetically heterogeneous disorder. Circulation 2001, 103, 2469–2475. [Google Scholar] [CrossRef] [Green Version]
- Shibamura, H.; Olson, J.M.; van Vlijmen-Van Keulen, C.; Buxbaum, S.G.; Dudek, D.M.; Tromp, G.; Ogata, T.; Skunca, M.; Sakalihasan, N.; Pals, G.; et al. Genome scan for familial abdominal aortic aneurysm using sex and family history as covariates suggests genetic heterogeneity and identifies linkage to chromosome 19q13. Circulation 2004, 109, 2103–2108. [Google Scholar] [CrossRef] [Green Version]
- Jones, G.T.; Tromp, G.; Kuivaniemi, H.; Gretarsdottir, S.; Baas, A.F.; Giusti, B.; Strauss, E.; Van’t Hof, F.N.; Webb, T.R.; Erdman, R.; et al. Meta-Analysis of Genome-Wide Association Studies for Abdominal Aortic Aneurysm Identifies Four New Disease-Specific Risk Loci. Circ. Res. 2017, 120, 341–353. [Google Scholar] [CrossRef]
- Singh, T.P.; Field, M.A.; Bown, M.J.; Jones, G.T.; Golledge, J. Systematic review of genome-wide association studies of abdominal aortic aneurysm. Atherosclerosis 2021, 327, 39–48. [Google Scholar] [CrossRef]
- Vats, S.; Sundquist, K.; Wang, X.; Zarrouk, M.; Agren-Witteschus, S.; Sundquist, J.; Gottsater, A.; Memon, A.A. Associations of global DNA methylation and homocysteine levels with abdominal aortic aneurysm: A cohort study from a population-based screening program in Sweden. Int. J. Cardiol. 2020, 321, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Toghill, B.J.; Saratzis, A.; Harrison, S.C.; Verissimo, A.R.; Mallon, E.B.; Bown, M.J. The potential role of DNA methylation in the pathogenesis of abdominal aortic aneurysm. Atherosclerosis 2015, 241, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Takagi, H.; Umemoto, T.; Group, A. Association of Hypertension with Abdominal Aortic Aneurysm Expansion. Ann. Vasc. Surg. 2017, 39, 74–89. [Google Scholar] [CrossRef] [PubMed]
- Nyberg, A.; Skagius, E.; Englund, E.; Nilsson, I.; Ljungh, A.; Henriksson, A.E. Abdominal aortic aneurysm and the impact of infectious burden. Eur. J. Vasc. Endovasc. Surg. 2008, 36, 292–296. [Google Scholar] [CrossRef] [Green Version]
- Jablonska, A.; Zagrapan, B.; Paradowska, E.; Neumayer, C.; Eilenberg, W.; Brostjan, C.; Klinger, M.; Nanobachvili, J.; Huk, I. Abdominal aortic aneurysm and virus infection: A potential causative role for cytomegalovirus infection? J. Med. Virol. 2021, 93, 5017–5024. [Google Scholar] [CrossRef]
- Wolinsky, H.; Glagov, S. A lamellar unit of aortic medial structure and function in mammals. Circ. Res. 1967, 20, 99–111. [Google Scholar] [CrossRef] [Green Version]
- Crawford, E.S.; Cohen, E.S. Aortic aneurysm: A multifocal disease: Presidential address. Arch. Surg. 1982, 117, 1393–1400. [Google Scholar] [CrossRef]
- Koch, A.E.; Haines, G.K.; Rizzo, R.J.; Radosevich, J.A.; Pope, R.M.; Robinson, P.G.; Pearce, W.H. Human abdominal aortic aneurysms. Immunophenotypic analysis suggesting an immune-mediated response. Am. J. Pathol. 1990, 137, 1199. [Google Scholar]
- Gregory, A.K.; Yin, N.X.; Capella, J.; Xia, S.; Newman, K.M.; Tilson, M.D. Features of autoimmunity in the abdominal aortic aneurysm. Arch. Surg. 1996, 131, 85–88. [Google Scholar] [CrossRef]
- Yuan, Z.; Lu, Y.; Wei, J.; Wu, J.; Yang, J.; Cai, Z. Abdominal Aortic Aneurysm: Roles of Inflammatory Cells. Front. Immunol. 2020, 11, 609161. [Google Scholar] [CrossRef]
- Marculescu, R.; Sodeck, G.; Domanovits, H.; Hobusch, G.; Exner, M.; Heinzl, H.; Huber, K.; Mannhalter, C.; Minar, E.; Wagner, O. Interleukin-1 gene cluster variants and abdominal aortic aneurysms. Thromb. Haemost. 2005, 94, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Bown, M.J.; Burton, P.R.; Horsburgh, T.; Nicholson, M.L.; Bell, P.R.; Sayers, R.D. The role of cytokine gene polymorphisms in the pathogenesis of abdominal aortic aneurysms: A case-control study. J. Vasc. Surg. 2003, 37, 999–1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, K.G.; Brull, D.J.; Brown, L.C.; Sian, M.; Greenhalgh, R.M.; Humphries, S.E.; Powell, J.T. Interleukin-6 (IL-6) and the prognosis of abdominal aortic aneurysms. Circulation 2001, 103, 2260–2265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anidjar, S.; Dobrin, P.B.; Eichorst, M.; Graham, G.P.; Chejfec, G. Correlation of inflammatory infiltrate with the enlargement of experimental aortic aneurysms. J. Vasc. Surg. 1992, 16, 139–147. [Google Scholar] [CrossRef] [Green Version]
- Juvonen, J.; Surcel, H.-M.; Satta, J.; Teppo, A.-M.; Bloigu, A.; Syrjälä, H.; Airaksinen, J.; Leinonen, M.; Saikku, P.; Juvonen, T. Elevated circulating levels of inflammatory cytokines in patients with abdominal aortic aneurysm. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2843–2847. [Google Scholar] [CrossRef]
- Colonnello, J.S.; Hance, K.A.; Shames, M.L.; Wyble, C.W.; Ziporin, S.J.; Leidenfrost, J.E.; Ennis, T.L.; Upchurch Jr, G.R.; Thompson, R.W. Transient exposure to elastase induces mouse aortic wall smooth muscle cell production of MCP-1 and RANTES during development of experimental aortic aneurysm. J. Vasc. Surg. 2003, 38, 138–146. [Google Scholar] [CrossRef] [Green Version]
- Hance, K.A.; Tataria, M.; Ziporin, S.J.; Lee, J.K.; Thompson, R.W. Monocyte chemotactic activity in human abdominal aortic aneurysms: Role of elastin degradation peptides and the 67–kD cell surface elastin receptor. J. Vasc. Surg. 2002, 35, 254–261. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Deng, H.; Zhou, Z.; Xiong, X.; Gao, L. Endothelium as a Potential Target for Treatment of Abdominal Aortic Aneurysm. Oxid. Med. Cell. Longev. 2018, 2018, 6306542. [Google Scholar] [CrossRef] [Green Version]
- Spartalis, E.; Spartalis, M.; Athanasiou, A.; Paschou, S.A.; Patelis, N.; Voudris, V.; Iliopoulos, D.C. Endothelium in Aortic Aneurysm Disease: New Insights. Curr. Med. Chem. 2020, 27, 1081–1088. [Google Scholar] [CrossRef]
- Lopez-Candales, A.; Holmes, D.R.; Liao, S.; Scott, M.J.; Wickline, S.A.; Thompson, R.W. Decreased vascular smooth muscle cell density in medial degeneration of human abdominal aortic aneurysms. Am. J. Pathol. 1997, 150, 993–1007. [Google Scholar]
- Miller, F.J., Jr.; Sharp, W.J.; Fang, X.; Oberley, L.W.; Oberley, T.D.; Weintraub, N.L. Oxidative stress in human abdominal aortic aneurysms: A potential mediator of aneurysmal remodeling. Arter. Thromb. Vasc. Biol. 2002, 22, 560–565. [Google Scholar] [CrossRef] [Green Version]
- Airhart, N.; Arif, B.; Curci, J. Vascular smooth muscle cells from abdominal aortic aneurysms have uniquely high elastolytic potential associated with activation of MMP-2. J. Surg. Res. 2013, 179, 282. [Google Scholar] [CrossRef]
- Thompson, R.W.; Liao, S.; Curci, J.A. Vascular smooth muscle cell apoptosis in abdominal aortic aneurysms. Coron. Artery Dis. 1997, 8, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Mazurek, R.; Dave, J.M.; Chandran, R.R.; Misra, A.; Sheikh, A.Q.; Greif, D.M. Vascular Cells in Blood Vessel Wall Development and Disease. Adv. Pharm. 2017, 78, 323–350. [Google Scholar] [CrossRef] [Green Version]
- Xie, C.; Huang, H.; Sun, X.; Guo, Y.; Hamblin, M.; Ritchie, R.P.; Garcia-Barrio, M.T.; Zhang, J.; Chen, Y.E. MicroRNA-1 regulates smooth muscle cell differentiation by repressing Kruppel-like factor 4. Stem Cells Dev. 2011, 20, 205–210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.; Jacquet, L.; Karamariti, E.; Xu, Q. Origin and differentiation of vascular smooth muscle cells. J. Physiol. 2015, 593, 3013–3030. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Chen, S.Y. Transforming growth factor-beta and smooth muscle differentiation. World J. Biol. Chem. 2012, 3, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Owens, G.K.; Kumar, M.S.; Wamhoff, B.R. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 2004, 84, 767–801. [Google Scholar] [CrossRef]
- Wasteson, P.; Johansson, B.R.; Jukkola, T.; Breuer, S.; Akyürek, L.M.; Partanen, J.; Lindahl, P. Developmental origin of smooth muscle cells in the descending aorta in mice. Development 2008, 135, 1823–1832. [Google Scholar] [CrossRef] [Green Version]
- Alexander, M.R.; Owens, G.K. Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annu. Rev. Physiol. 2012, 74, 13–40. [Google Scholar] [CrossRef]
- Coll-Bonfill, N.; De La Cruz-Thea, B.; Pisano, M.; Musri, M. Noncoding RNAs in smooth muscle cell homeostasis: Implications in phenotypic switch and vascular disorders. Pflügers Arch.-Eur. J. Physiol. 2016, 468, 1071–1087. [Google Scholar] [CrossRef] [PubMed]
- Ailawadi, G.; Moehle, C.W.; Pei, H.; Walton, S.P.; Yang, Z.; Kron, I.L.; Lau, C.L.; Owens, G.K. Smooth muscle phenotypic modulation is an early event in aortic aneurysms. J. Thorac. Cardiovasc. Surg. 2009, 138, 1392–1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, T.; Kaestner, K.H.; Owens, G.K. Conditional deletion of Kruppel-like factor 4 delays downregulation of smooth muscle cell differentiation markers but accelerates neointimal formation following vascular injury. Circ. Res. 2008, 102, 1548–1557. [Google Scholar] [CrossRef]
- Chen, P.Y.; Qin, L.; Li, G.; Tellides, G.; Simons, M. Smooth muscle FGF/TGF β cross talk regulates atherosclerosis progression. EMBO Mol. Med. 2016, 8, 712–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, X.; Shi, N.; Cui, X.B.; Wang, J.N.; Fukui, Y.; Chen, S.Y. Dedicator of cytokinesis 2, a novel regulator for smooth muscle phenotypic modulation and vascular remodeling. Circ. Res. 2015, 116, e71–e80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crosas-Molist, E.; Meirelles, T.; López-Luque, J.; Serra-Peinado, C.; Selva, J.; Caja, L.; Gorbenko del Blanco, D.; Uriarte, J.J.; Bertran, E.; Mendizábal, Y. Vascular smooth muscle cell phenotypic changes in patients with Marfan syndrome. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 960–972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Vranckx, R.; Van Kien, P.K.; Lalande, A.; Boisset, N.; Mathieu, F.; Wegman, M.; Glancy, L.; Gasc, J.-M.; Brunotte, F. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat. Genet. 2006, 38, 343–349. [Google Scholar] [CrossRef]
- Li, W.; Li, Q.; Jiao, Y.; Qin, L.; Ali, R.; Zhou, J.; Ferruzzi, J.; Kim, R.W.; Geirsson, A.; Dietz, H.C.; et al. Tgfbr2 disruption in postnatal smooth muscle impairs aortic wall homeostasis. J. Clin. Investig. 2014, 124, 755–767. [Google Scholar] [CrossRef] [Green Version]
- Dai, X.; Shen, J.; Annam, N.P.; Jiang, H.; Levi, E.; Schworer, C.M.; Tromp, G.; Arora, A.; Higgins, M.; Wang, X.F.; et al. SMAD3 deficiency promotes vessel wall remodeling, collagen fiber reorganization and leukocyte infiltration in an inflammatory abdominal aortic aneurysm mouse model. Sci. Rep. 2015, 5, 10180. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Ait-Oufella, H.; Herbin, O.; Bonnin, P.; Ramkhelawon, B.; Taleb, S.; Huang, J.; Offenstadt, G.; Combadiere, C.; Renia, L.; et al. TGF-beta activity protects against inflammatory aortic aneurysm progression and complications in angiotensin II-infused mice. J. Clin. Investig. 2010, 120, 422–432. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Rateri, D.L.; Howatt, D.A.; Balakrishnan, A.; Moorleghen, J.J.; Cassis, L.A.; Daugherty, A. TGF-beta Neutralization Enhances AngII-Induced Aortic Rupture and Aneurysm in Both Thoracic and Abdominal Regions. PLoS ONE 2016, 11, e0153811. [Google Scholar] [CrossRef]
- Biros, E.; Walker, P.J.; Nataatmadja, M.; West, M.; Golledge, J. Downregulation of transforming growth factor, beta receptor 2 and Notch signaling pathway in human abdominal aortic aneurysm. Atherosclerosis 2012, 221, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Baas, A.F.; Medic, J.; van’t Slot, R.; de Kovel, C.G.; Zhernakova, A.; Geelkerken, R.H.; Kranendonk, S.E.; van Sterkenburg, S.M.; Grobbee, D.E.; Boll, A.P.; et al. Association of the TGF-beta receptor genes with abdominal aortic aneurysm. Eur. J. Hum. Genet. 2010, 18, 240–244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, P.Y.; Qin, L.; Li, G.; Malagon-Lopez, J.; Wang, Z.; Bergaya, S.; Gujja, S.; Caulk, A.W.; Murtada, S.I.; Zhang, X.; et al. Smooth Muscle Cell Reprogramming in Aortic Aneurysms. Cell Stem Cell 2020, 26, 542–557.e511. [Google Scholar] [CrossRef]
- Rangrez, A.Y.; Massy, Z.A.; Metzinger-Le Meuth, V.; Metzinger, L. miR-143 and miR-145: Molecular keys to switch the phenotype of vascular smooth muscle cells. Circ. Cardiovasc. Genet. 2011, 4, 197–205. [Google Scholar] [CrossRef]
- Xin, M.; Small, E.M.; Sutherland, L.B.; Qi, X.; McAnally, J.; Plato, C.F.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. MicroRNAs miR-143 and miR-145 modulate cytoskeletal dynamics and responsiveness of smooth muscle cells to injury. Genes Dev. 2009, 23, 2166–2178. [Google Scholar] [CrossRef] [Green Version]
- Elia, L.; Quintavalle, M.; Zhang, J.; Contu, R.; Cossu, L.; Latronico, M.V.; Peterson, K.L.; Indolfi, C.; Catalucci, D.; Chen, J.; et al. The knockout of miR-143 and -145 alters smooth muscle cell maintenance and vascular homeostasis in mice: Correlates with human disease. Cell Death Differ. 2009, 16, 1590–1598. [Google Scholar] [CrossRef]
- Van Varik, B.; Rennenberg, R.; Reutelingsperger, C.; Kroon, A.; de Leeuw, P.; Schurgers, L.J. Mechanisms of arterial remodeling: Lessons from genetic diseases. Front. Genet. 2012, 3, 290. [Google Scholar] [CrossRef] [Green Version]
- Bendeck, M.P.; Irvin, C.; Reidy, M.A. Inhibition of matrix metalloproteinase activity inhibits smooth muscle cell migration but not neointimal thickening after arterial injury. Circ. Res. 1996, 78, 38–43. [Google Scholar] [CrossRef]
- Sandford, R.; Bown, M.; London, N.; Sayers, R. The genetic basis of abdominal aortic aneurysms: A review. Eur. J. Vasc. Endovasc. Surg. 2007, 33, 381–390. [Google Scholar] [CrossRef] [Green Version]
- Tilson, M.D.; Reilly, J.M.; Brophy, C.M.; Webster, E.L.; Barnett, T.R. Expression and sequence of the gene for tissue inhibitor of metalloproteinases in patients with abdominal aortic aneurysms. J. Vasc. Surg. 1993, 18, 266–270. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Khalil, R.A. Matrix Metalloproteinases, Vascular Remodeling, and Vascular Disease. Adv. Pharm. 2018, 81, 241–330. [Google Scholar] [CrossRef]
- Raffetto, J.D.; Khalil, R.A. Matrix metalloproteinases and their inhibitors in vascular remodeling and vascular disease. Biochem. Pharm. 2008, 75, 346–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, G.T.; Phillips, V.L.; Harris, E.L.; Rossaak, J.I.; van Rij, A.M. Functional matrix metalloproteinase-9 polymorphism (C-1562T) associated with abdominal aortic aneurysm. J. Vasc. Surg. 2003, 38, 1363–1367. [Google Scholar] [CrossRef] [Green Version]
- Lamblin, N.; Bauters, C.; Hermant, X.; Lablanche, J.-M.; Helbecque, N.; Amouyel, P. Polymorphisms in the promoter regions of MMP-2, MMP-3, MMP-9 and MMP-12 genes as determinants of aneurysmal coronary artery disease. J. Am. Coll. Cardiol. 2002, 40, 43–48. [Google Scholar] [CrossRef] [Green Version]
- Eriksson, P.; Jormsjö-Pettersson, S.; Brady, A.; Deguchi, H.; Hamsten, A.; Powell, J. Genotype–phenotype relationships in an investigation of the role of proteases in abdominal aortic aneurysm expansion. J. Br. Surg. 2005, 92, 1372–1376. [Google Scholar] [CrossRef]
- Longo, G.M.; Buda, S.J.; Fiotta, N.; Xiong, W.; Griener, T.; Shapiro, S.; Baxter, B.T. MMP-12 has a role in abdominal aortic aneurysms in mice. Surgery 2005, 137, 457–462. [Google Scholar] [CrossRef]
- Brophy, C.M.; Marks, W.H.; Reilly, J.M.; Tilson, M.D. Decreased tissue inhibitor of metalloproteinases (TIMP) in abdominal aortic aneurysm tissue: A preliminary report. J. Surg. Res. 1991, 50, 653–657. [Google Scholar] [CrossRef]
- Eskandari, M.K.; Vijungco, J.D.; Flores, A.; Borensztajn, J.; Shively, V.; Pearce, W.H. Enhanced abdominal aortic aneurysm in TIMP-1-deficient mice. J. Surg. Res. 2005, 123, 289–293. [Google Scholar] [CrossRef]
- Rossaak, J.I.; van Rij, A.M.; Jones, G.T.; Harris, E.L. Association of the 4G/5G polymorphism in the promoter region of plasminogen activator inhibitor-1 with abdominal aortic aneurysms. J. Vasc. Surg. 2000, 31, 1026–1032. [Google Scholar] [CrossRef]
- Gurung, R.; Choong, A.M.; Woo, C.C.; Foo, R.; Sorokin, V. Genetic and Epigenetic Mechanisms Underlying Vascular Smooth Muscle Cell Phenotypic Modulation in Abdominal Aortic Aneurysm. Int. J. Mol. Sci. 2020, 21, 6334. [Google Scholar] [CrossRef] [PubMed]
- Boon, R.A.; Dimmeler, S. MicroRNAs and aneurysm formation. Trends Cardiovasc. Med. 2011, 21, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.W.; Kumar, S.; Son, D.J.; Jang, I.H.; Griendling, K.K.; Jo, H. Prevention of abdominal aortic aneurysm by anti-microRNA-712 or anti-microRNA-205 in angiotensin II-infused mice. Arter. Thromb. Vasc. Biol. 2014, 34, 1412–1421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galan, M.; Varona, S.; Orriols, M.; Rodriguez, J.A.; Aguilo, S.; Dilme, J.; Camacho, M.; Martinez-Gonzalez, J.; Rodriguez, C. Induction of histone deacetylases (HDACs) in human abdominal aortic aneurysm: Therapeutic potential of HDAC inhibitors. Dis. Model. Mech. 2016, 9, 541–552. [Google Scholar] [CrossRef] [Green Version]
- Vinh, A.; Gaspari, T.A.; Liu, H.B.; Dousha, L.F.; Widdop, R.E.; Dear, A.E. A novel histone deacetylase inhibitor reduces abdominal aortic aneurysm formation in angiotensin II-infused apolipoprotein E-deficient mice. J. Vasc. Res. 2008, 45, 143–152. [Google Scholar] [CrossRef]
- Zhang, S.; Liu, Z.; Xie, N.; Huang, C.; Li, Z.; Yu, F.; Fu, Y.; Cui, Q.; Kong, W. Pan-HDAC (Histone Deacetylase) Inhibitors Increase Susceptibility of Thoracic Aortic Aneurysm and Dissection in Mice. Arter. Thromb. Vasc. Biol. 2021, 41, 2848–2850. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, T.T.; Zhang, R.; Fu, W.Y.; Wang, X.; Wang, F.; Gao, P.; Ding, Y.N.; Xie, Y.; Hao, D.L.; et al. Calorie restriction protects against experimental abdominal aortic aneurysms in mice. J. Exp. Med. 2016, 213, 2473–2488. [Google Scholar] [CrossRef]
- Furmanik, M.; M Shanahan, C. Endoplasmic reticulum stress in arterial smooth muscle cells: A novel regulator of vascular disease. Curr. Cardiol. Rev. 2017, 13, 94–105. [Google Scholar]
- Navas-Madronal, M.; Rodriguez, C.; Kassan, M.; Fite, J.; Escudero, J.R.; Canes, L.; Martinez-Gonzalez, J.; Camacho, M.; Galan, M. Enhanced endoplasmic reticulum and mitochondrial stress in abdominal aortic aneurysm. Clin. Sci. 2019, 133, 1421–1438. [Google Scholar] [CrossRef]
- Qin, Y.; Wang, Y.; Liu, O.; Jia, L.; Fang, W.; Du, J.; Wei, Y. Tauroursodeoxycholic Acid Attenuates Angiotensin II Induced Abdominal Aortic Aneurysm Formation in Apolipoprotein E-deficient Mice by Inhibiting Endoplasmic Reticulum Stress. Eur. J. Vasc. Endovasc. Surg. 2017, 53, 337–345. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.; Fu, Y.; Cai, Z.; Yu, F.; Gong, Z.; Dai, R.; Hu, Y.; Zeng, L.; Xu, Q.; Kong, W. Unspliced XBP1 confers VSMC homeostasis and prevents aortic aneurysm formation via FoxO4 interaction. Circ. Res. 2017, 121, 1331–1345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, L.X.; Zhang, W.M.; Zhang, H.J.; Li, T.T.; Wang, Y.L.; Qin, Y.W.; Gu, H.; Du, J. Mechanical stretch-induced endoplasmic reticulum stress, apoptosis and inflammation contribute to thoracic aortic aneurysm and dissection. J. Pathol. 2015, 236, 373–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gavrila, D.; Li, W.G.; McCormick, M.L.; Thomas, M.; Daugherty, A.; Cassis, L.A.; Miller, F.J., Jr.; Oberley, L.W.; Dellsperger, K.C.; Weintraub, N.L. Vitamin E inhibits abdominal aortic aneurysm formation in angiotensin II-infused apolipoprotein E-deficient mice. Arter. Thromb. Vasc. Biol. 2005, 25, 1671–1677. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.-W.; Jia, L.-X.; Ni, X.-Q.; Zhao, L.; Chang, J.-R.; Zhang, J.-S.; Hou, Y.-L.; Zhu, Y.; Guan, Y.-F.; Yu, Y.-R. Intermedin1− 53 Attenuates Abdominal Aortic Aneurysm by Inhibiting Oxidative Stress. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 2176–2190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Branchetti, E.; Poggio, P.; Sainger, R.; Shang, E.; Grau, J.B.; Jackson, B.M.; Lai, E.K.; Parmacek, M.S.; Gorman, R.C.; Gorman, J.H. Oxidative stress modulates vascular smooth muscle cell phenotype via CTGF in thoracic aortic aneurysm. Cardiovasc. Res. 2013, 100, 316–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siu, K.L.; Li, Q.; Zhang, Y.; Guo, J.; Youn, J.Y.; Du, J.; Cai, H. NOX isoforms in the development of abdominal aortic aneurysm. Redox Biol. 2017, 11, 118–125. [Google Scholar] [CrossRef] [Green Version]
- McCormick, M.L.; Gavrila, D.; Weintraub, N.L. Role of oxidative stress in the pathogenesis of abdominal aortic aneurysms. Arter. Thromb. Vasc. Biol. 2007, 27, 461–469. [Google Scholar] [CrossRef] [Green Version]
- Xiong, W.; Mactaggart, J.; Knispel, R.; Worth, J.; Zhu, Z.; Li, Y.; Sun, Y.; Baxter, B.T.; Johanning, J. Inhibition of reactive oxygen species attenuates aneurysm formation in a murine model. Atherosclerosis 2009, 202, 128–134. [Google Scholar] [CrossRef] [Green Version]
- Konior, A.; Schramm, A.; Czesnikiewicz-Guzik, M.; Guzik, T.J. NADPH oxidases in vascular pathology. Antioxid. Redox Signal. 2014, 20, 2794–2814. [Google Scholar] [CrossRef] [Green Version]
- Soe, N.N.; Sowden, M.; Baskaran, P.; Kim, Y.; Nigro, P.; Smolock, E.M.; Berk, B.C. Acetylation of cyclophilin A is required for its secretion and vascular cell activation. Cardiovasc. Res. 2014, 101, 444–453. [Google Scholar] [CrossRef]
- Thomas, M.; Gavrila, D.; McCormick, M.L.; Miller, F.J., Jr.; Daugherty, A.; Cassis, L.A.; Dellsperger, K.C.; Weintraub, N.L. Deletion of p47phox attenuates angiotensin II-induced abdominal aortic aneurysm formation in apolipoprotein E-deficient mice. Circulation 2006, 114, 404–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Starke, R.M.; Thompson, J.W.; Ali, M.S.; Pascale, C.L.; Martinez Lege, A.; Ding, D.; Chalouhi, N.; Hasan, D.M.; Jabbour, P.; Owens, G.K. Cigarette smoke initiates oxidative stress-induced cellular phenotypic modulation leading to cerebral aneurysm pathogenesis. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 610–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowe, V.L.; Stevens, S.L.; Reddick, T.T.; Freeman, M.B.; Donnell, R.; Carroll, R.C.; Goldman, M.H. Vascular smooth muscle cell apoptosis in aneurysmal, occlusive, and normal human aortas. J. Vasc. Surg. 2000, 31, 567–576. [Google Scholar] [CrossRef]
- Yamanouchi, D.; Morgan, S.; Stair, C.; Seedial, S.; Lengfeld, J.; Kent, K.C.; Liu, B. Accelerated aneurysmal dilation associated with apoptosis and inflammation in a newly developed calcium phosphate rodent abdominal aortic aneurysm model. J. Vasc. Surg. 2012, 56, 455–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajagopalan, S.; Meng, X.P.; Ramasamy, S.; Harrison, D.G.; Galis, Z.S. Reactive oxygen species produced by macrophage-derived foam cells regulate the activity of vascular matrix metalloproteinases in vitro. Implications for atherosclerotic plaque stability. J. Clin. Investig. 1996, 98, 2572–2579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, L.-X.; Zhang, W.-M.; Li, T.-T.; Liu, Y.; Piao, C.-M.; Ma, Y.-C.; Lu, Y.; Wang, Y.; Liu, T.-T.; Qi, Y.-F. ER stress dependent microparticles derived from smooth muscle cells promote endothelial dysfunction during thoracic aortic aneurysm and dissection. Clin. Sci. 2017, 131, 1287–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagata, S. Fas ligand-induced apoptosis. Annu. Rev. Genet. 1999, 33, 29–55. [Google Scholar] [CrossRef]
- Waring, P.; Mullbacher, A. Cell death induced by the Fas/Fas ligand pathway and its role in pathology. Immunol. Cell. Biol. 1999, 77, 312–317. [Google Scholar] [CrossRef]
- Henderson, E.L.; Geng, Y.J.; Sukhova, G.K.; Whittemore, A.D.; Knox, J.; Libby, P. Death of smooth muscle cells and expression of mediators of apoptosis by T lymphocytes in human abdominal aortic aneurysms. Circulation 1999, 99, 96–104. [Google Scholar] [CrossRef] [Green Version]
- Lee, T.; Chau, L. Fas/Fas ligand-mediated death pathway is involved in oxLDL-induced apoptosis in vascular smooth muscle cells. Am J. Physiol. Cell Physiol. 2001, 280, C709–C718. [Google Scholar] [CrossRef]
- Kuiper, J.; Quax, P.H.; Bot, I. Anti-apoptotic serpins as therapeutics in cardiovascular diseases. Cardiovasc. Hematol. Disord. Drug Targets 2013, 13, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Ang, L.S.; Boivin, W.A.; Williams, S.J.; Zhao, H.; Abraham, T.; Carmine-Simmen, K.; McManus, B.M.; Bleackley, R.C.; Granville, D.J. Serpina3n attenuates granzyme B-mediated decorin cleavage and rupture in a murine model of aortic aneurysm. Cell Death Dis. 2011, 2, e209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, H.; Sun, J.; Liang, W.; Chang, Z.; Rom, O.; Zhao, Y.; Zhao, G.; Xiong, W.; Wang, H.; Zhu, T.; et al. Cyclodextrin Prevents Abdominal Aortic Aneurysm via Activation of Vascular Smooth Muscle Cell Transcription Factor EB. Circulation 2020, 142, 483–498. [Google Scholar] [CrossRef] [PubMed]
- Leeper, N.J.; Raiesdana, A.; Kojima, Y.; Chun, H.J.; Azuma, J.; Maegdefessel, L.; Kundu, R.K.; Quertermous, T.; Tsao, P.S.; Spin, J.M. MicroRNA-26a is a novel regulator of vascular smooth muscle cell function. J. Cell. Physiol. 2011, 226, 1035–1043. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.Y.; Trenner, M.; Boon, R.A.; Spin, J.M.; Maegdefessel, L. Long noncoding RNAs in key cellular processes involved in aortic aneurysms. Atherosclerosis 2020, 292, 112–118. [Google Scholar] [CrossRef] [Green Version]
- Kalayinia, S.; Arjmand, F.; Maleki, M.; Malakootian, M.; Singh, C.P. MicroRNAs: Roles in cardiovascular development and disease. Cardiovasc. Pathol. 2021, 50, 107296. [Google Scholar] [CrossRef]
- Li, D.Y.; Busch, A.; Jin, H.; Chernogubova, E.; Pelisek, J.; Karlsson, J.; Sennblad, B.; Liu, S.; Lao, S.; Hofmann, P.; et al. H19 Induces Abdominal Aortic Aneurysm Development and Progression. Circulation 2018, 138, 1551–1568. [Google Scholar] [CrossRef]
- Zhang, Z.; Zou, G.; Chen, X.; Lu, W.; Liu, J.; Zhai, S.; Qiao, G. Knockdown of lncRNA PVT1 Inhibits Vascular Smooth Muscle Cell Apoptosis and Extracellular Matrix Disruption in a Murine Abdominal Aortic Aneurysm Model. Mol. Cells 2019, 42, 218–227. [Google Scholar] [CrossRef]
- Wang, Q.; Liu, Z.; Ren, J.; Morgan, S.; Assa, C.; Liu, B. Receptor-interacting protein kinase 3 contributes to abdominal aortic aneurysms via smooth muscle cell necrosis and inflammation. Circ. Res. 2015, 116, 600–611. [Google Scholar] [CrossRef] [Green Version]
- Zhou, T.; Wang, Q.; Phan, N.; Ren, J.; Yang, H.; Feldman, C.C.; Feltenberger, J.B.; Ye, Z.; Wildman, S.A.; Tang, W.; et al. Identification of a novel class of RIP1/RIP3 dual inhibitors that impede cell death and inflammation in mouse abdominal aortic aneurysm models. Cell Death Dis. 2019, 10, 226. [Google Scholar] [CrossRef]
- Khoury, M.K.; Zhou, T.; Yang, H.; Prince, S.R.; Gupta, K.; Stranz, A.R.; Wang, Q.; Liu, B. GSK2593074A blocks progression of existing abdominal aortic dilation. JVS Vasc. Sci. 2020, 1, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; DeRoo, E.; Yang, H.; Stranz, A.; Wang, Q.; Ginnan, R.; Singer, H.A.; Liu, B. MLKL and CaMKII Are Involved in RIPK3-Mediated Smooth Muscle Cell Necroptosis. Cells 2021, 10, 2397. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; He, Y.; Wei, X.; Jiang, D.S. Targeting regulated cell death in aortic aneurysm and dissection therapy. Pharm. Res 2021, 176, 106048. [Google Scholar] [CrossRef]
- Abdul-Hussien, H.; Hanemaaijer, R.; Kleemann, R.; Verhaaren, B.F.; van Bockel, J.H.; Lindeman, J.H. The pathophysiology of abdominal aortic aneurysm growth: Corresponding and discordant inflammatory and proteolytic processes in abdominal aortic and popliteal artery aneurysms. J. Vasc. Surg. 2010, 51, 1479–1487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizas, K.D.; Ippagunta, N.; Tilson, M.D., 3rd. Immune cells and molecular mediators in the pathogenesis of the abdominal aortic aneurysm. Cardiol. Rev. 2009, 17, 201–210. [Google Scholar] [CrossRef] [PubMed]
- Shankman, L.S.; Gomez, D.; Cherepanova, O.A.; Salmon, M.; Alencar, G.F.; Haskins, R.M.; Swiatlowska, P.; Newman, A.A.; Greene, E.S.; Straub, A.C.; et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat. Med. 2015, 21, 628–637. [Google Scholar] [CrossRef] [Green Version]
- Toghill, B.J.; Saratzis, A.; Freeman, P.J.; Sylvius, N.; Collaborators, U.; Bown, M.J. SMYD2 promoter DNA methylation is associated with abdominal aortic aneurysm (AAA) and SMYD2 expression in vascular smooth muscle cells. Clin. Epigenetics 2018, 10, 29. [Google Scholar] [CrossRef]
- Xu, G.; Liu, G.; Xiong, S.; Liu, H.; Chen, X.; Zheng, B. The histone methyltransferase Smyd2 is a negative regulator of macrophage activation by suppressing interleukin 6 (IL-6) and tumor necrosis factor alpha (TNF-alpha) production. J. Biol. Chem. 2015, 290, 5414–5423. [Google Scholar] [CrossRef] [Green Version]
- Maegdefessel, L.; Spin, J.M.; Raaz, U.; Eken, S.M.; Toh, R.; Azuma, J.; Adam, M.; Nakagami, F.; Heymann, H.M.; Chernogubova, E.; et al. miR-24 limits aortic vascular inflammation and murine abdominal aneurysm development. Nat. Commun. 2014, 5, 5214. [Google Scholar] [CrossRef] [Green Version]
- Villa-Bellosta, R.; Millan, A.; Sorribas, V. Role of calcium-phosphate deposition in vascular smooth muscle cell calcification. Am. J. Physiol. Cell. Physiol. 2011, 300, C210–C220. [Google Scholar] [CrossRef] [Green Version]
- Duer, M.J.; Friscic, T.; Proudfoot, D.; Reid, D.G.; Schoppet, M.; Shanahan, C.M.; Skepper, J.N.; Wise, E.R. Mineral surface in calcified plaque is like that of bone: Further evidence for regulated mineralization. Arter. Thromb. Vasc. Biol. 2008, 28, 2030–2034. [Google Scholar] [CrossRef] [PubMed]
- Strauss, H.W.; Nakahara, T.; Narula, N.; Narula, J. Vascular Calcification: The Evolving Relationship of Vascular Calcification to Major Acute Coronary Events. J. Nucl. Med. 2019, 60, 1207–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buijs, R.V.; Willems, T.P.; Tio, R.A.; Boersma, H.H.; Tielliu, I.F.; Slart, R.H.; Zeebregts, C.J. Calcification as a risk factor for rupture of abdominal aortic aneurysm. Eur. J. Vasc. Endovasc. Surg. 2013, 46, 542–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petsophonsakul, P.; Furmanik, M.; Forsythe, R.; Dweck, M.; Schurink, G.W.; Natour, E.; Reutelingsperger, C.; Jacobs, M.; Mees, B.; Schurgers, L. Role of Vascular Smooth Muscle Cell Phenotypic Switching and Calcification in Aortic Aneurysm Formation. Arter. Thromb. Vasc. Biol. 2019, 39, 1351–1368. [Google Scholar] [CrossRef]
- Forsythe, R.O.; Newby, D.E.; Robson, J.M. Monitoring the biological activity of abdominal aortic aneurysms Beyond Ultrasound. Heart 2016, 102, 817–824. [Google Scholar] [CrossRef] [Green Version]
- Lindholt, J.S. Aneurysmal wall calcification predicts natural history of small abdominal aortic aneurysms. Atherosclerosis 2008, 197, 673–678. [Google Scholar] [CrossRef]
- Forsythe, R.O.; Dweck, M.R.; McBride, O.M.B.; Vesey, A.T.; Semple, S.I.; Shah, A.S.V.; Adamson, P.D.; Wallace, W.A.; Kaczynski, J.; Ho, W.; et al. (18)F-Sodium Fluoride Uptake in Abdominal Aortic Aneurysms: The SoFIA(3) Study. J. Am. Coll. Cardiol. 2018, 71, 513–523. [Google Scholar] [CrossRef]
- Reeps, C.; Essler, M.; Pelisek, J.; Seidl, S.; Eckstein, H.H.; Krause, B.J. Increased 18F-fluorodeoxyglucose uptake in abdominal aortic aneurysms in positron emission/computed tomography is associated with inflammation, aortic wall instability, and acute symptoms. J. Vasc. Surg. 2008, 48, 417–423; discussion 424. [Google Scholar] [CrossRef] [Green Version]
- Iyemere, V.P.; Proudfoot, D.; Weissberg, P.L.; Shanahan, C.M. Vascular smooth muscle cell phenotypic plasticity and the regulation of vascular calcification. J. Intern. Med. 2006, 260, 192–210. [Google Scholar] [CrossRef]
- Shanahan, C.M.; Crouthamel, M.H.; Kapustin, A.; Giachelli, C.M. Arterial calcification in chronic kidney disease: Key roles for calcium and phosphate. Circ. Res. 2011, 109, 697–711. [Google Scholar] [CrossRef] [Green Version]
- Shioi, A.; Nishizawa, Y.; Jono, S.; Koyama, H.; Hosoi, M.; Morii, H. Beta-glycerophosphate accelerates calcification in cultured bovine vascular smooth muscle cells. Arter. Thromb. Vasc. Biol. 1995, 15, 2003–2009. [Google Scholar] [CrossRef] [PubMed]
- Basalyga, D.M.; Simionescu, D.T.; Xiong, W.; Baxter, B.T.; Starcher, B.C.; Vyavahare, N.R. Elastin degradation and calcification in an abdominal aorta injury model: Role of matrix metalloproteinases. Circulation 2004, 110, 3480–3487. [Google Scholar] [CrossRef] [PubMed]
- Patelis, N.; Moris, D.; Schizas, D.; Damaskos, C.; Perrea, D.; Bakoyiannis, C.; Liakakos, T.; Georgopoulos, S. Animal models in the research of abdominal aortic aneurysms development. Physiol. Res. 2017, 66, 899–915. [Google Scholar] [CrossRef] [PubMed]
- Busch, A.; Holm, A.; Wagner, N.; Ergun, S.; Rosenfeld, M.; Otto, C.; Baur, J.; Kellersmann, R.; Lorenz, U. Extra- and Intraluminal Elastase Induce Morphologically Distinct Abdominal Aortic Aneurysms in Mice and Thus Represent Specific Subtypes of Human Disease. J. Vasc. Res. 2016, 53, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Bhamidipati, C.M.; Mehta, G.S.; Lu, G.; Moehle, C.W.; Barbery, C.; DiMusto, P.D.; Laser, A.; Kron, I.L.; Upchurch, G.R., Jr.; Ailawadi, G. Development of a novel murine model of aortic aneurysms using peri-adventitial elastase. Surgery 2012, 152, 238–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senemaud, J.; Caligiuri, G.; Etienne, H.; Delbosc, S.; Michel, J.B.; Coscas, R. Translational Relevance and Recent Advances of Animal Models of Abdominal Aortic Aneurysm. Arter. Thromb. Vasc. Biol. 2017, 37, 401–410. [Google Scholar] [CrossRef] [Green Version]
- Chiou, A.C.; Chiu, B.; Pearce, W.H. Murine aortic aneurysm produced by periarterial application of calcium chloride. J. Surg. Res. 2001, 99, 371–376. [Google Scholar] [CrossRef]
- Lu, G.; Su, G.; Davis, J.P.; Schaheen, B.; Downs, E.; Roy, R.J.; Ailawadi, G.; Upchurch, G.R., Jr. A novel chronic advanced stage abdominal aortic aneurysm murine model. J. Vasc. Surg. 2017, 66, 232–242.e234. [Google Scholar] [CrossRef] [Green Version]
- Fashandi, A.Z.; Hawkins, R.B.; Salmon, M.D.; Spinosa, M.D.; Montgomery, W.G.; Cullen, J.M.; Lu, G.; Su, G.; Ailawadi, G.; Upchurch, G.R., Jr. A novel reproducible model of aortic aneurysm rupture. Surgery 2018, 163, 397–403. [Google Scholar] [CrossRef]
- Gopal, K.; Kumar, K.; Nandini, R.; Jahan, P.; Kumar, M.J. High fat diet containing cholesterol induce aortic aneurysm through recruitment and proliferation of circulating agranulocytes in apoE knock out mice model. J. Thromb. Thrombolysis 2010, 30, 154–163. [Google Scholar] [CrossRef]
- Zhao, G.; Lu, H.; Chang, Z.; Zhao, Y.; Zhu, T.; Chang, L.; Guo, Y.; Garcia-Barrio, M.T.; Chen, Y.E.; Zhang, J. Single-cell RNA sequencing reveals the cellular heterogeneity of aneurysmal infrarenal abdominal aorta. Cardiovasc. Res. 2021, 117, 1402–1416. [Google Scholar] [CrossRef] [PubMed]
- Hadi, T.; Boytard, L.; Silvestro, M.; Alebrahim, D.; Jacob, S.; Feinstein, J.; Barone, K.; Spiro, W.; Hutchison, S.; Simon, R.; et al. Macrophage-derived netrin-1 promotes abdominal aortic aneurysm formation by activating MMP3 in vascular smooth muscle cells. Nat. Commun. 2018, 9, 5022. [Google Scholar] [CrossRef] [PubMed]
- Dawson, A.; Li, Y.; Li, Y.; Ren, P.; Vasquez, H.G.; Zhang, C.; Rebello, K.R.; Ageedi, W.; Azares, A.R.; Mattar, A.B.; et al. Single-Cell Analysis of Aneurysmal Aortic Tissue in Patients with Marfan Syndrome Reveals Dysfunctional TGF-β Signaling. Genes 2021, 13, 95. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qian, G.; Adeyanju, O.; Olajuyin, A.; Guo, X. Abdominal Aortic Aneurysm Formation with a Focus on Vascular Smooth Muscle Cells. Life 2022, 12, 191. https://doi.org/10.3390/life12020191
Qian G, Adeyanju O, Olajuyin A, Guo X. Abdominal Aortic Aneurysm Formation with a Focus on Vascular Smooth Muscle Cells. Life. 2022; 12(2):191. https://doi.org/10.3390/life12020191
Chicago/Turabian StyleQian, Guoqing, Oluwaseun Adeyanju, Ayobami Olajuyin, and Xia Guo. 2022. "Abdominal Aortic Aneurysm Formation with a Focus on Vascular Smooth Muscle Cells" Life 12, no. 2: 191. https://doi.org/10.3390/life12020191
APA StyleQian, G., Adeyanju, O., Olajuyin, A., & Guo, X. (2022). Abdominal Aortic Aneurysm Formation with a Focus on Vascular Smooth Muscle Cells. Life, 12(2), 191. https://doi.org/10.3390/life12020191