
In Silico Molecular Characterization of Human TMPRSS2 Protease Polymorphic Variants and Associated SARS-CoV-2 Susceptibility



Abstract
:1. Introduction
2. Methodology
2.1. Structure Retrieval, Molecular Modelling and Docking
2.2. Mutagenesis, Binding Affinity and Interfacial Contact Predictions
2.3. TMPRSS2 and Protease Inhibitors Interaction Analysis
3. Results
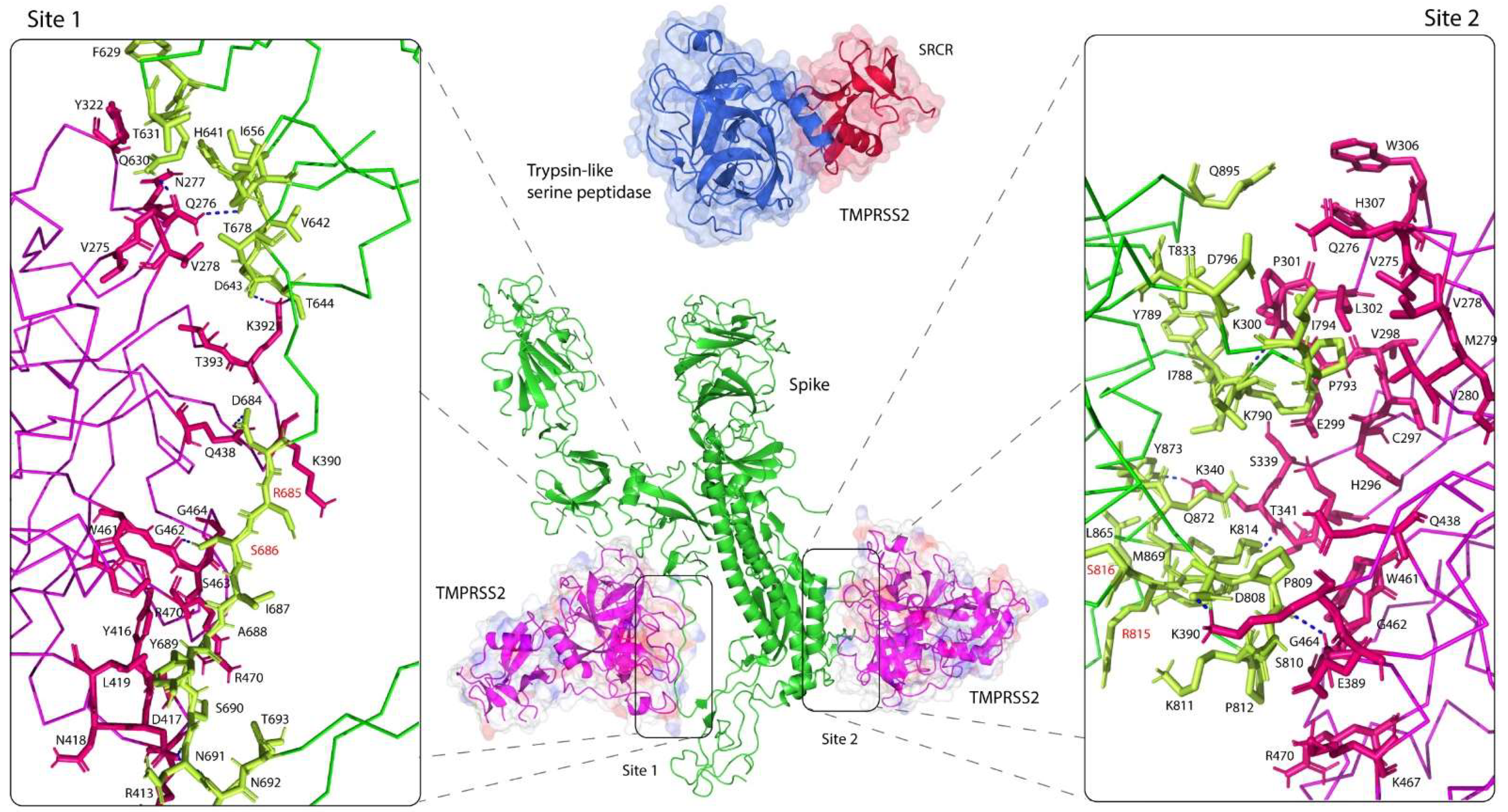
3.1. TMPRSS2 Bound in Close Proximity to Two Cleavage Sites of the S Glycoprotein
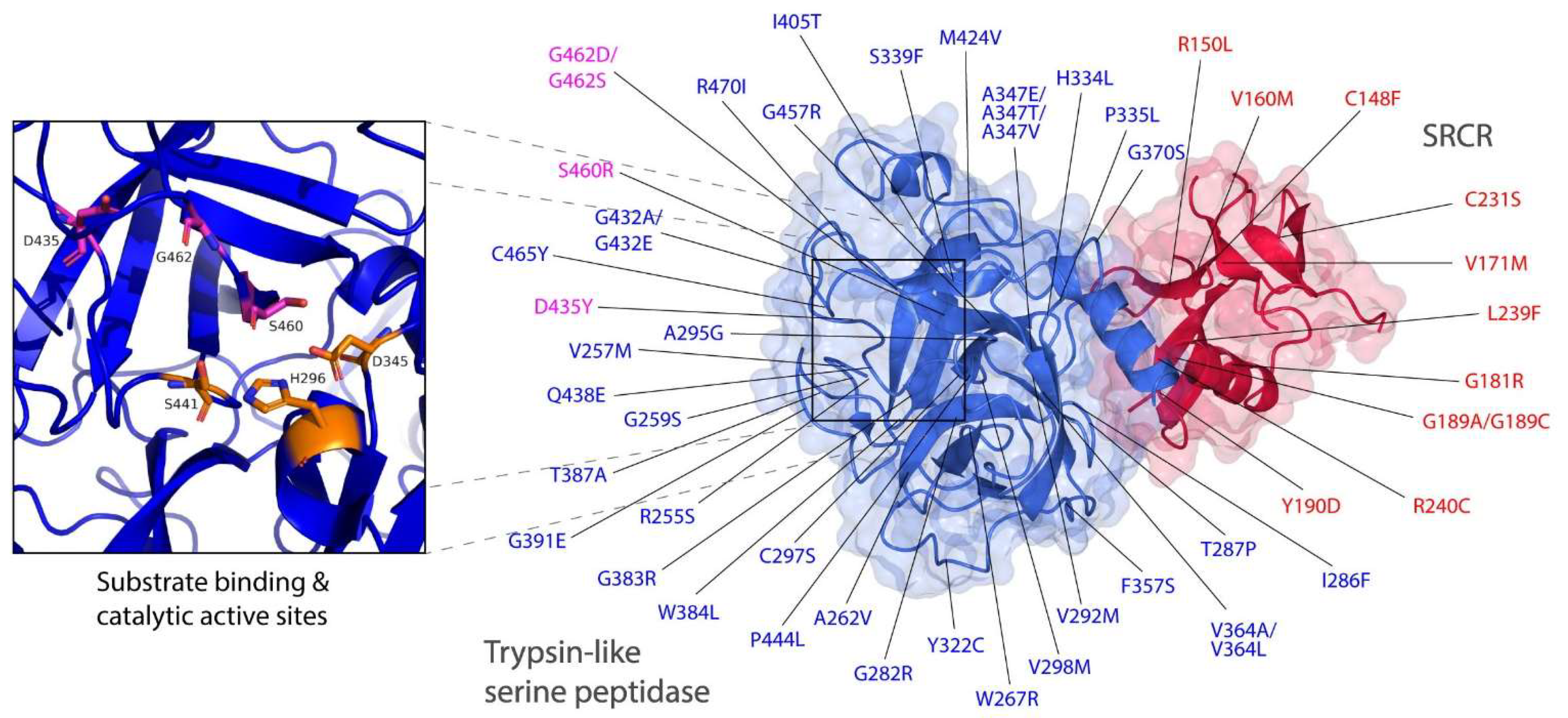
3.2. Intermolecular Interactions between TMPRSS2 Polymorphic Variants and the S Glycoprotein
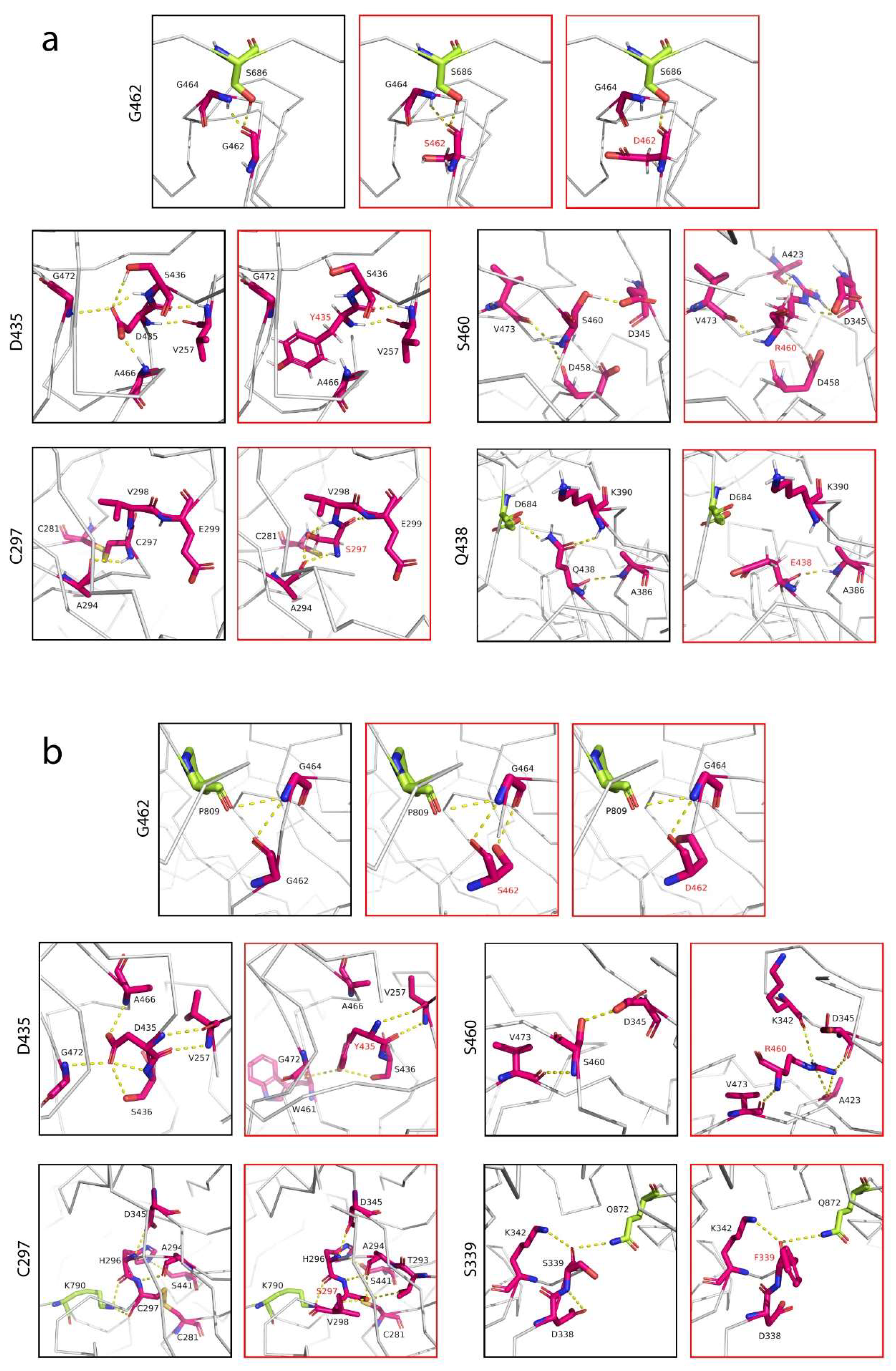
3.3. Altered Affinity of Natural TMPRSS2 Polymorphic Variants for the S Glycoprotein
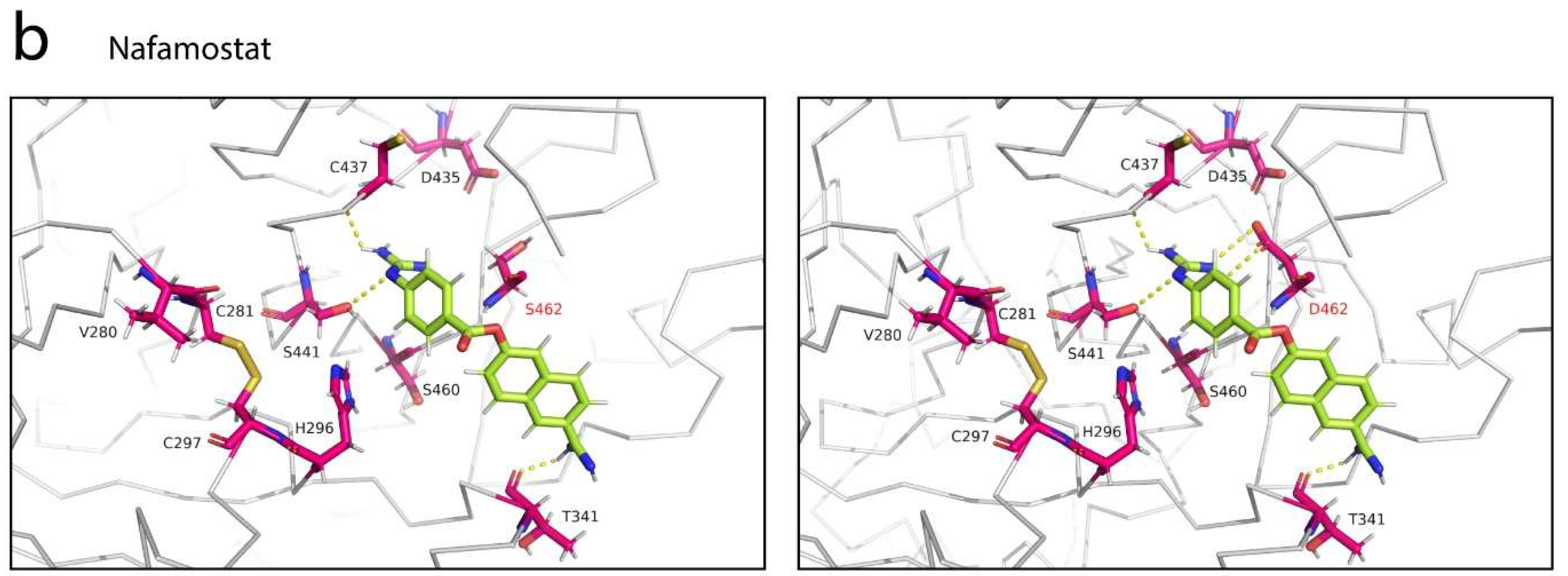
3.4. Selective Binding of Serine Protease Inhibitors to TMPRSS2
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ksiazek, T.G.; Erdman, D.; Goldsmith, C.S.; Zaki, S.R.; Peret, T.; Emery, S.; Tong, S.; Urbani, C.; Comer, J.A.; Lim, W.; et al. A Novel Coronavirus Associated with Severe Acute Respiratory Syndrome. N. Engl. J. Med. 2003, 348, 1953–1966. [Google Scholar] [CrossRef]
- Zaki, A.M.; Van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.M.E.; Fouchier, R.A.M. Isolation of a Novel Coronavirus from a Man with Pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef]
- Bar-On, Y.M.; Flamholz, A.; Phillips, R.; Milo, R. SARS-CoV-2 (COVID-19) by the numbers. eLife 2020, 9. [Google Scholar] [CrossRef]
- Wang, M.-Y.; Zhao, R.; Gao, L.-J.; Gao, X.-F.; Wang, D.-P.; Cao, J.-M. SARS-CoV-2: Structure, Biology, and Structure-Based Therapeutics Development. Front. Cell. Infect. Microbiol. 2020, 10, 587269. [Google Scholar] [CrossRef]
- Naqvi, A.A.T.; Fatima, K.; Mohammad, T.; Fatima, U.; Singh, I.K.; Singh, A.; Atif, S.M.; Hariprasad, G.; Hasan, G.M.; Hassan, I. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: Structural genomics approach. Biochim. Et Biophys. Acta (BBA) Mol. Basis Dis. 2020, 1866, 165878. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Salleh, M.; Derrick, J.; Deris, Z. Structural Evaluation of the Spike Glycoprotein Variants on SARS-CoV-2 Transmission and Immune Evasion. Int. J. Mol. Sci. 2021, 22, 7425. [Google Scholar] [CrossRef]
- Harvey, W.T.; Carabelli, A.M.; Jackson, B.; Gupta, R.K.; Thomson, E.C.; Harrison, E.M.; Ludden, C.; Reeve, R.; Rambaut, A.; COVID-19 Genomics UK (COG-UK) Consortium; et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat. Rev. Microbiol. 2021, 19, 409–424. [Google Scholar] [CrossRef]
- Dejnirattisai, W.; Zhou, D.; Supasa, P.; Liu, C.; Mentzer, A.J.; Ginn, H.M.; Zhao, Y.; Duyvesteyn, H.M.; Tuekprakhon, A.; Nutalai, R.; et al. Antibody evasion by the P.1 strain of SARS-CoV-2. Cell 2021, 184, 2939–2954. [Google Scholar] [CrossRef]
- Ramanathan, M.; Ferguson, I.D.; Miao, W.; Khavari, P.A. SARS-CoV-2 B.1.1.7 and B.1.351 spike variants bind human ACE2 with increased affinity. Lancet Infect. Dis. 2021. [Google Scholar] [CrossRef]
- Wall, E.C.; Wu, M.; Harvey, R.; Kelly, G.; Warchal, S.; Sawyer, C.; Daniels, R.; Hobson, P.; Hatipoglu, E.; Ngai, Y.; et al. Neutralising antibody activity against SARS-CoV-2 VOCs B.1.617.2 and B.1.351 by BNT162b2 vaccination. Lancet 2021, 397, 2331–2333. [Google Scholar] [CrossRef]
- Wang, P.; Nair, M.S.; Liu, L.; Iketani, S.; Luo, Y.; Guo, Y.; Wang, M.; Yu, J.; Zhang, B.; Kwong, P.D.; et al. Antibody resistance of SARS-CoV-2 variants B.1.351 and B.1.1.7. Nature 2021, 593, 130–135. [Google Scholar] [CrossRef]
- Araf, Y.; Akter, F.; Tang, Y.; Fatemi, R.; Alam Parvez, S.; Zheng, C.; Hossain, G. Omicron variant of SARS-CoV-2: Genomics, transmissibility, and responses to current COVID-19 vaccines. J. Med Virol. 2022. [Google Scholar] [CrossRef]
- Shanmugaraj, B.; Malla, A.; Khorattanakulchai, N.; Phoolcharoen, W. SARS-CoV-2 omicron variant: Could it be another threat? J. Med Virol. 2021. [Google Scholar] [CrossRef]
- Peacock, T.P.; Goldhill, D.H.; Zhou, J.; Baillon, L.; Frise, R.; Swann, O.C.; Kugathasan, R.; Penn, R.; Brown, J.C.; Sanchez-David, R.Y.; et al. The furin cleavage site in the SARS-CoV-2 spike protein is required for transmission in ferrets. Nat. Microbiol. 2021, 6, 899–909. [Google Scholar] [CrossRef]
- Anastassopoulou, C.; Gkizarioti, Z.; Patrinos, G.P.; Tsakris, A. Human genetic factors associated with susceptibility to SARS-CoV-2 infection and COVID-19 disease severity. Hum. Genom. 2020, 14, 1–8. [Google Scholar] [CrossRef]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Gao, Y.-D.; Ding, M.; Dong, X.; Zhang, J.-J.; Azkur, A.K.; Azkur, D.; Gan, H.; Sun, Y.-L.; Fu, W.; Li, W.; et al. Risk factors for severe and critically ill COVID-19 patients: A review. Allergy 2021, 76, 428–455. [Google Scholar] [CrossRef]
- Hou, Y.; Zhao, J.; Martin, W.; Kallianpur, A.; Chung, M.K.; Jehi, L.; Sharifi, N.; Erzurum, S.; Eng, C.; Cheng, F. New insights into genetic susceptibility of COVID-19: An ACE2 and TMPRSS2 polymorphism analysis. BMC Med. 2020, 18, 216. [Google Scholar] [CrossRef]
- Mauvais-Jarvis, F.; Merz, N.B.; Barnes, P.J.; Brinton, R.D.; Carrero, J.-J.; DeMeo, D.L.; De Vries, G.J.; Epperson, C.N.; Govindan, R.; Klein, S.L.; et al. Sex and gender: Modifiers of health, disease, and medicine. Lancet 2020, 396, 565–582. [Google Scholar] [CrossRef]
- Jin, J.M.; Bai, P.; He, W.; Wu, F.; Liu, X.F.; Han, D.M.; Liu, S.; Yang, J.K. Gender Differences in Patients With COVID-19: Focus on Severity and Mortality. Front. Public Health 2020, 8, 1–6. [Google Scholar] [CrossRef]
- Bienvenu, L.A.; Noonan, J.; Wang, X.; Peter, K. Higher mortality of COVID-19 in males: Sex differences in immune response and cardiovascular comorbidities. Cardiovasc. Res. 2020, 116, 2197–2206. [Google Scholar] [CrossRef]
- Cattrini, C.; Bersanelli, M.; Latocca, M.; Conte, B.; Vallome, G.; Boccardo, F. Sex Hormones and Hormone Therapy during COVID-19 Pandemic: Implications for Patients with Cancer. Cancers 2020, 12, 2325. [Google Scholar] [CrossRef]
- Yasuhara, J.; Kuno, T.; Takagi, H.; Sumitomo, N. Clinical characteristics of COVID-19 in children: A systematic review. Pediatr. Pulmonol. 2020, 55, 2565–2575. [Google Scholar] [CrossRef]
- Schuler, B.A.; Habermann, A.C.; Plosa, E.J.; Taylor, C.J.; Jetter, C.; Negretti, N.M.; Kapp, M.E.; Benjamin, J.T.; Gulleman, P.; Nichols, D.S.; et al. Age-determined expression of priming protease TMPRSS2 and localization of SARS-CoV-2 in lung epithelium. J. Clin. Investig. 2021, 131, 140766. [Google Scholar] [CrossRef]
- Li, Y.; Zhou, W.; Yang, L.; You, R. Physiological and pathological regulation of ACE2, the SARS-CoV-2 receptor. Pharmacol. Res. 2020, 157, 104833. [Google Scholar] [CrossRef]
- Felsenstein, S.; Hedrich, C.M. SARS-CoV-2 infections in children and young people. Clin. Immunol. 2020, 220, 108588. [Google Scholar] [CrossRef]
- Suryamohan, K.; Diwanji, D.; Stawiski, E.W.; Gupta, R.; Miersch, S.; Liu, J.; Chen, C.; Jiang, Y.-P.; Fellouse, F.A.; Sathirapongsasuti, J.F.; et al. Human ACE2 receptor polymorphisms and altered susceptibility to SARS-CoV-2. Commun. Biol. 2021, 4, 1–11. [Google Scholar] [CrossRef]
- Fraser, B.J.; Beldar, S.; Seitova, A.; Hutchinson, A.; Mannar, D.; Arrowsmith, C.H.; Bénard, F. Structure, activity and inhibi-tion of human TMPRSS2, a protease implicated in SARS-CoV-2 activation. bioRxiv 2021. [Google Scholar] [CrossRef]
- Hoffmann, M.; Hofmann-Winkler, H.; Smith, J.C.; Krüger, N.; Arora, P.; Sørensen, L.K.; Søgaard, O.S.; Hasselstrøm, J.B.; Winkler, M.; Hempel, T.; et al. Camostat mesylate inhibits SARS-CoV-2 activation by TMPRSS2-related proteases and its metabolite GBPA exerts antiviral activity. EBioMedicine 2021, 65, 103255. [Google Scholar] [CrossRef] [PubMed]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiser, A.; Do, R.K.; SSali, A. Modeling of loops in protein structures. Protein Sci. 2000, 9, 1753–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Honorato, R.V.; Koukos, P.I.; Jiménez-García, B.; Tsaregorodtsev, A.; Verlato, M.; Giachetti, A.; Rosato, A.; Bonvin, A.M.J.J. Structural Biology in the Clouds: The WeNMR-EOSC Ecosystem. Front. Mol. Biosci. 2021, 8, 729513. [Google Scholar] [CrossRef]
- Hussain, M.; Jabeen, N.; Amanullah, A.; Baig, A.A.; Aziz, B.; Shabbir, S.; Raza, F.; Uddin, N. Molecular docking between human TMPRSS2 and SARS-CoV-2 spike protein: Conformation and intermolecular interactions. AIMS Microbiol. 2020, 6, 350–360. [Google Scholar] [CrossRef]
- Xue, L.C.; Rodrigues, J.P.; Kastritis, P.L.; Bonvin, A.M.; Vangone, A. PRODIGY: A web server for predicting the binding affinity of protein–protein complexes. Bioinformatics 2016, 32, 3676–3678. [Google Scholar] [CrossRef]
- Li, K.; Meyerholz, D.K.; Bartlett, J.A.; McCray, P.B. The TMPRSS2 Inhibitor Nafamostat Reduces SARS-CoV-2 Pulmonary Infection in Mouse Models of COVID-19. Mbio 2021, 12, e00970-21. [Google Scholar] [CrossRef]
- Al-Horani, R.A.; Kar, S.; Aliter, K.F. Potential Anti-COVID-19 Therapeutics that Block the Early Stage of the Viral Life Cycle: Structures, Mechanisms, and Clinical Trials. Int. J. Mol. Sci. 2020, 21, 5224. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [Green Version]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daugherty, M.D.; Malik, H.S. Rules of Engagement: Molecular Insights from Host-Virus Arms Races. Annu. Rev. Genet. 2012, 46, 677–700. [Google Scholar] [CrossRef]
- Paniri, A.; Hosseini, M.M.; Akhavan-Niaki, H. First comprehensive computational analysis of functional consequences of TMPRSS2 SNPs in susceptibility to SARS-CoV-2 among different populations. J. Biomol. Struct. Dyn. 2021, 39, 3576–3593. [Google Scholar] [CrossRef] [PubMed]
- SeyedAlinaghi, S.; Mehrtak, M.; MohsseniPour, M.; Mirzapour, P.; Barzegary, A.; Habibi, P.; Moradmand-Badie, B.; Afsahi, A.M.; Karimi, A.; Heydari, M.; et al. Genetic susceptibility of COVID-19: A systematic review of current evidence. Eur. J. Med Res. 2021, 26, 46. [Google Scholar] [CrossRef] [PubMed]
- Monticelli, M.; Mele, B.H.; Benetti, E.; Fallerini, C.; Baldassarri, M.; Furini, S.; Frullanti, E.; Mari, F.; GEN-COVID Multicenter Study; Andreotti, G.; et al. Protective Role of a TMPRSS2 Variant on Severe COVID-19 Outcome in Young Males and Elderly Women. Genes 2021, 12, 596. [Google Scholar] [CrossRef]
- Andolfo, I.; Russo, R.; Lasorsa, V.A.; Cantalupo, S.; Rosato, B.E.; Bonfiglio, F.; Frisso, G.; Abete, P.; Cassese, G.M.; Servillo, G.; et al. Common variants at 21q22.3 locus influence MX1 and TMPRSS2 gene expression and susceptibility to severe COVID-19. iScience 2021, 24, 102322. [Google Scholar] [CrossRef]
- Zhou, Y.; Hou, Y.; Shen, J.; Huang, Y.; Martin, W.; Cheng, F. Network-based drug repurposing for novel coronavirus 2019-nCoV/SARS-CoV-2. Cell Discov. 2020, 6, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Beeraka, N.M.; Sadhu, S.P.; Madhunapantula, S.V.; Pragada, R.R.; Svistunov, A.A.; Nikolenko, V.N.; Mikhaleva, L.M.; Aliev, G. Strategies for Targeting SARS CoV-2: Small Molecule Inhibitors—The Current Status. Front. Immunol. 2020, 11, 552925. [Google Scholar] [CrossRef]
- Yeo, S.-J.; Liu, D.-X.; Kim, H.S.; Park, H. Anti-malarial effect of novel chloroquine derivatives as agents for the treatment of malaria. Malar. J. 2017, 16, 80. [Google Scholar] [CrossRef] [Green Version]
- White, N.J. The treatment of malaria. N. Engl. J. Med. 1996, 335, 800–806. [Google Scholar] [CrossRef]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Vincent, M.J.; Bergeron, E.; Benjannet, S.; Erickson, B.R.; Rollin, P.E.; Ksiazek, T.G.; Seidah, N.G.; Nichol, S.T. Chloroquine is a potent inhibitor of SARS coronavirus infection and spread. Virol. J. 2005, 2, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francini, E.; Miano, S.T.; Fiaschi, A.I.; Francini, G. Doxycycline or minocycline may be a viable treatment option against SARS-CoV-2. Med. Hypotheses 2020, 144, 110054. [Google Scholar] [CrossRef]
- Bharadwaj, S.; Lee, K.E.; Dwivedi, V.D.; Kang, S.G. Computational insights into tetracyclines as inhibitors against SARS-CoV-2 Mpro via combinatorial molecular simulation calculations. Life Sci. 2020, 257, 118080. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020, 10, 766–788. [Google Scholar] [CrossRef]
- Gendrot, M.; Andreani, J.; Jardot, P.; Hutter, S.; Delandre, O.; Boxberger, M.; Mosnier, J.; Le Bideau, M.; Duflot, I.; Fonta, I.; et al. In Vitro Antiviral Activity of Doxycycline against SARS-CoV-2. Molecules 2020, 25, 5064. [Google Scholar] [CrossRef] [PubMed]
- Breining, P.; Frølund, A.L.; Højen, J.F.; Gunst, J.D.; Staerke, N.B.; Saedder, E.; Cases-Thomas, M.; Little, P.; Nielsen, L.P.; Søgaard, O.S.; et al. Camostat mesylate against SARS-CoV-2 and COVID-19—Rationale, dosing and safety. Basic Clin. Pharmacol. Toxicol. 2021, 128, 204–212. [Google Scholar] [CrossRef]
- Sonawane, K.D.; Barale, S.S.; Dhanavade, M.J.; Waghmare, S.R.; Nadaf, N.H.; Kamble, S.A.; Mohammed, A.A.; Makandar, A.M.; Fandilolu, P.M.; Dound, A.S.; et al. Structural insights and inhibition mechanism of TMPRSS2 by experimentally known inhibitors Camostat mesylate, Nafamostat and Bromhexine hydrochloride to control SARS-coronavirus-2: A molecular modeling approach. Informatics Med. Unlocked 2021, 24, 100597. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Table 2 Variant | Binding Energy (∆G (kcal mol−1)) | KD (nM) at 25.0 °C | KD (nM) at 37.0 °C |
|---|---|---|---|
| WT | −8.6 | 450 | 800 |
| D435Y | −8.6 | 460 | 820 |
| S460R | −8.6 | 460 | 810 |
| G462D | −8.4 | 680 | 1200 |
| G462S | −8.5 | 620 | 1100 |
| C297S | −8.7 | 450 | 780 |
| Q438E | −8.7 | 400 | 710 |
| TMPRSS2 Variant | Binding Energy (∆G (kcal mol−1)) | KD (nM) at 25.0 °C | KD (nM) at 37.0 °C |
|---|---|---|---|
| WT | −10.3 | 27 | 53 |
| D435Y | −10.3 | 28 | 54 |
| S460R | −10.3 | 27 | 53 |
| G462D | −10.2 | 33 | 65 |
| G462S | −10.1 | 36 | 70 |
| C297S | −10.2 | 32 | 62 |
| S339F | −10.5 | 20 | 40 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salleh, M.Z.; Deris, Z.Z. In Silico Molecular Characterization of Human TMPRSS2 Protease Polymorphic Variants and Associated SARS-CoV-2 Susceptibility. Life 2022, 12, 231. https://doi.org/10.3390/life12020231
Salleh MZ, Deris ZZ. In Silico Molecular Characterization of Human TMPRSS2 Protease Polymorphic Variants and Associated SARS-CoV-2 Susceptibility. Life. 2022; 12(2):231. https://doi.org/10.3390/life12020231
Chicago/Turabian StyleSalleh, Mohd Zulkifli, and Zakuan Zainy Deris. 2022. "In Silico Molecular Characterization of Human TMPRSS2 Protease Polymorphic Variants and Associated SARS-CoV-2 Susceptibility" Life 12, no. 2: 231. https://doi.org/10.3390/life12020231
APA StyleSalleh, M. Z., & Deris, Z. Z. (2022). In Silico Molecular Characterization of Human TMPRSS2 Protease Polymorphic Variants and Associated SARS-CoV-2 Susceptibility. Life, 12(2), 231. https://doi.org/10.3390/life12020231