
The Role of Lysine Methyltransferase SET7/9 in Proliferation and Cell Stress Response

 and
and 
Abstract
:1. Introduction
2. The History of SET7/9 Discovery
3. The Substrate Specificity of SET7/9
4. Cellular Localization of SET7/9
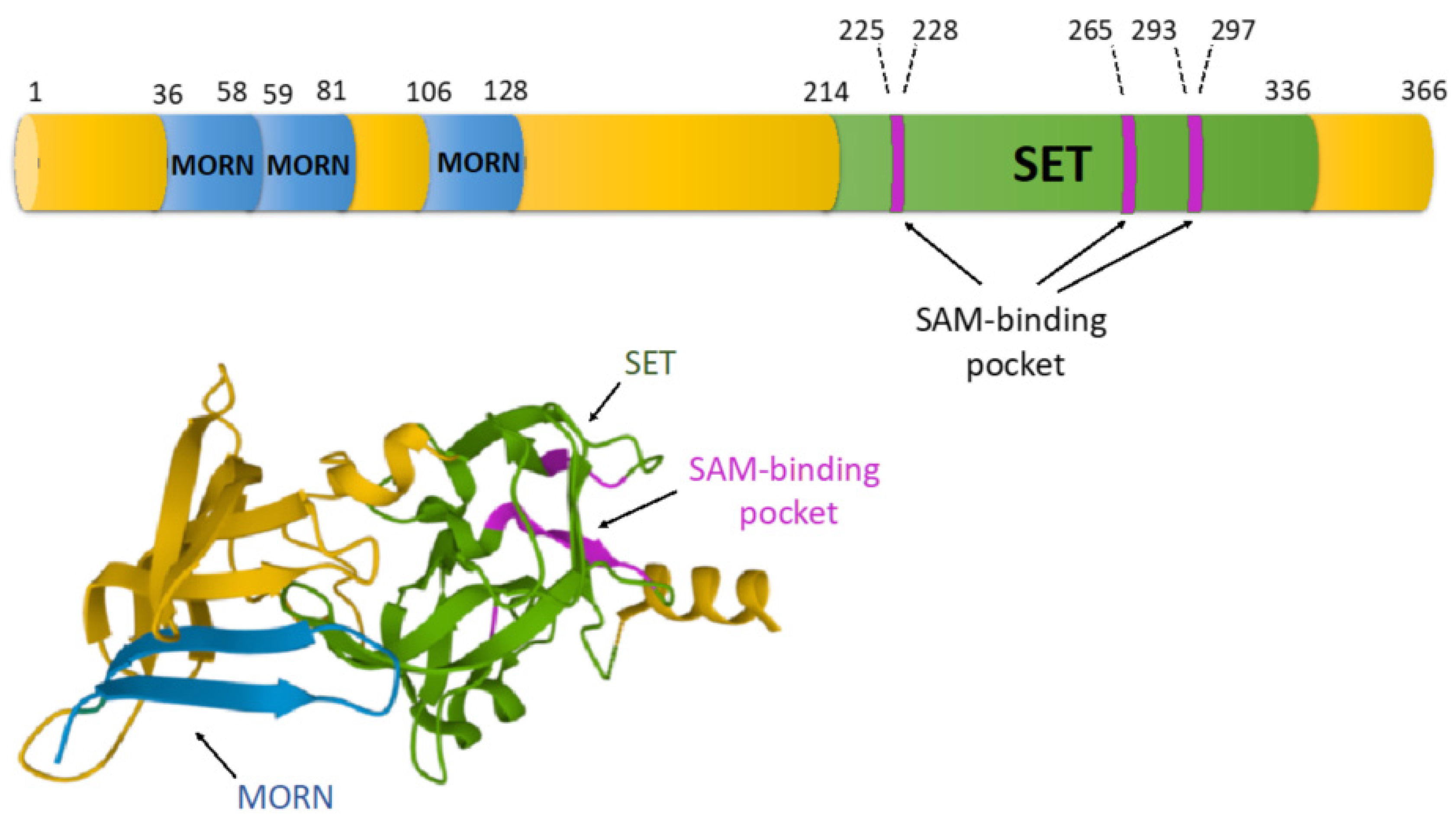
5. The Structural Organization of SET7/9 Methyltransferase
6. Histone Targets of SET7/9
7. Non-Histone Targets of SET7/9
General Effects of SET7/9 on Transcription
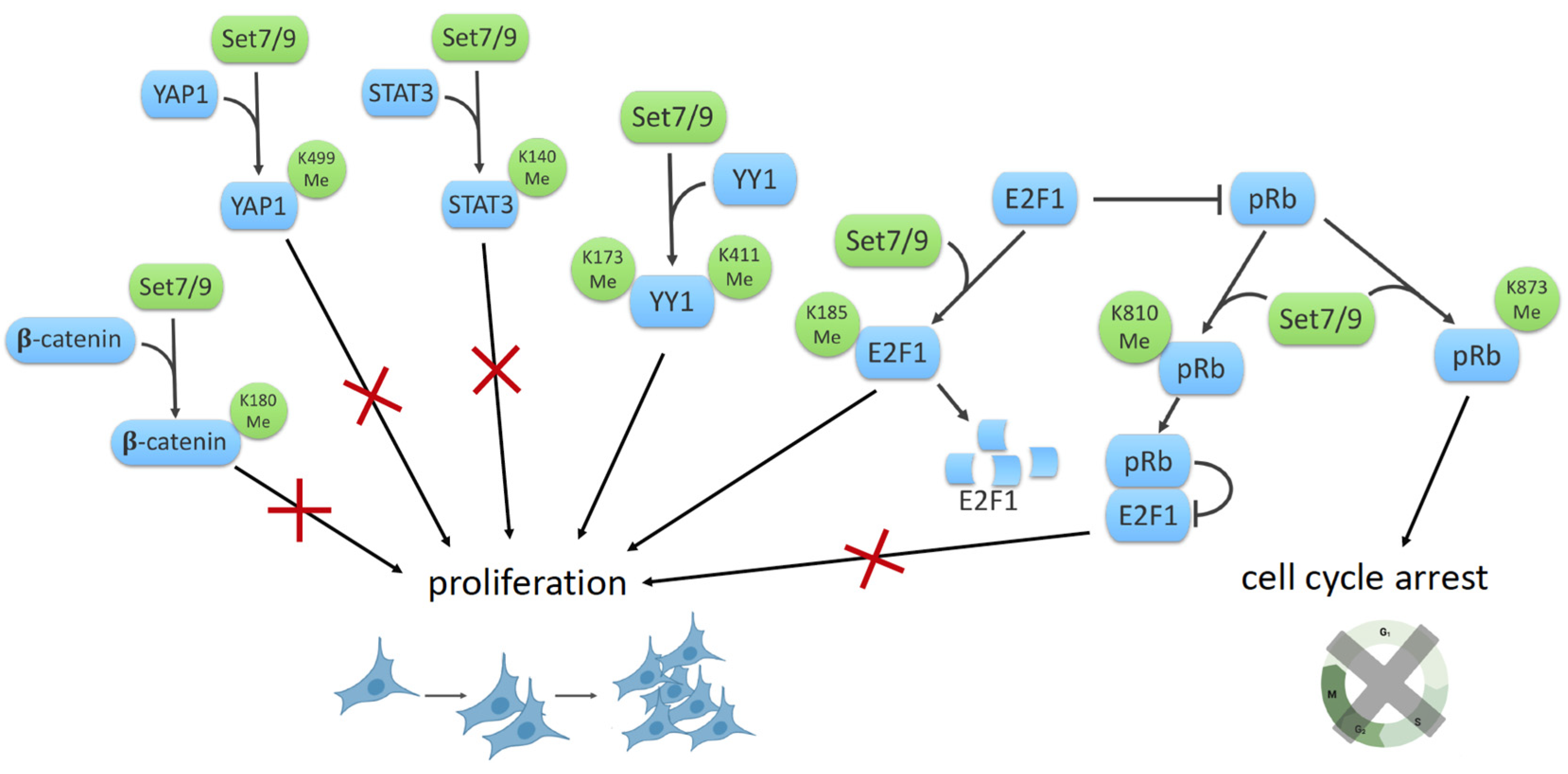
8. SET7/9 and Cell Proliferation
8.1. SET7/9, β-Catenin and YAP1
8.2. SET7/9 and STAT3
8.3. SET7/9 and YY1
8.4. SET7/9 and E2F1/Rb1
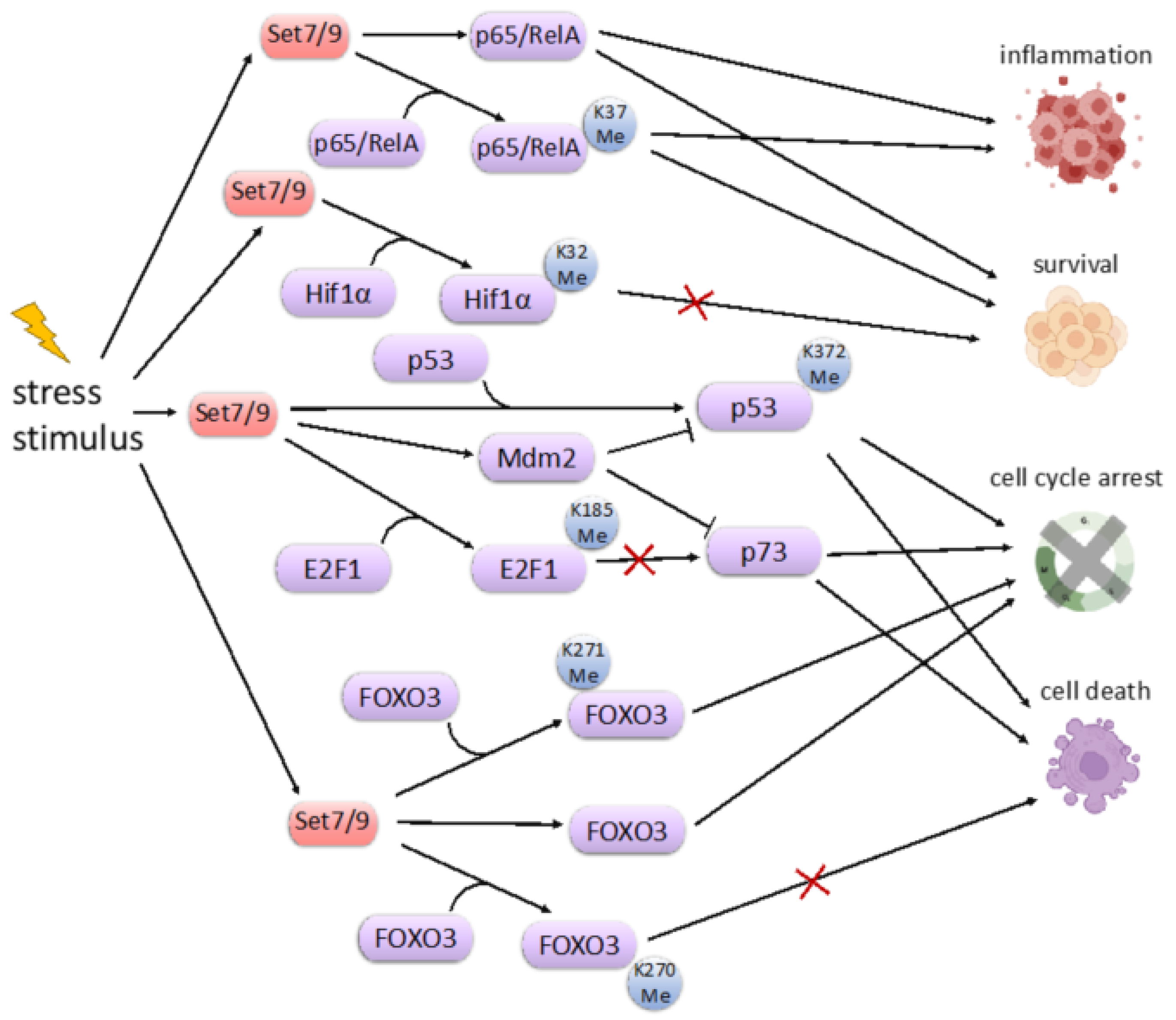
9. SET7/9 and Cell Stress Response
10. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Nishioka, K.; Chuikov, S.; Sarma, K.; Erdjument-Bromage, H.; Allis, C.D.; Tempst, P.; Reinberg, D. Set9, a Novel Histone H3 Methyltransferase that Facilitates Transcription by Precluding Histone Tail Modifications Required for Heterochromatin Formation. Genes Dev. 2002, 16, 479–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Cao, R.; Xia, L.; Erdjument-Bromage, H.; Borchers, C.; Tempst, P.; Zhang, Y. Purification and Functional Characterization of a Histone H3-Lysine 4-Specific Methyltransferase. Mol. Cell 2001, 8, 1207–1217. [Google Scholar] [CrossRef]
- Dhayalan, A.; Kudithipudi, S.; Rathert, P.; Jeltsch, A. Specificity Analysis-Based Identification of New Methylation Targets of the SET7/9 Protein Lysine Methyltransferase. Chem. Biol. 2011, 18, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Chakrabarti, S.K.; Francis, J.; Ziesmann, S.M.; Garmey, J.C.; Mirmira, R.G. Covalent Histone Modifications Underlie the Developmental Regulation of Insulin Gene Transcription in Pancreatic β Cells. J. Biol. Chem. 2003, 278, 23617–23623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans-Molina, C.; Robbins, R.D.; Kono, T.; Tersey, S.A.; Vestermark, G.L.; Nunemaker, C.S.; Garmey, J.C.; Deering, T.G.; Keller, S.R.; Maier, B.; et al. Peroxisome Proliferator-Activated Receptor γ Activation Restores Islet Function in Diabetic Mice through Reduction of Endoplasmic Reticulum Stress and Maintenance of Euchromatin Structure. Mol. Cell. Biol. 2009, 29, 2053–2067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pradhan, S.; Chin, H.G.; Estève, P.-O.; Jacobsen, S.E. SET7/9 Mediated Methylation of Non-Histone Proteins in Mammalian Cells. Epigenetics 2009, 4, 383–387. [Google Scholar] [CrossRef]
- Chuikov, S.; Kurash, J.K.; Wilson, J.; Xiao, B.; Justin, N.; Ivanov, G.S.; McKinney, K.; Tempst, P.; Prives, C.; Gamblin, S.; et al. Regulation of p53 activity through lysine methylation. Nature 2004, 432, 353–360. [Google Scholar] [CrossRef]
- Couture, J.-F.; Collazo, E.; Hauk, G.; Trievel, R.C. Structural Basis for the Methylation Site Specificity of SET7/9. Nat. Struct. Mol. Biol. 2006, 13, 140–146. [Google Scholar] [CrossRef]
- Masatsugu, T.; Yamamoto, K. Multiple Lysine Methylation of PCAF by Set9 Methyltransferase. Biochem. Biophys. Res. Commun. 2009, 381, 22–26. [Google Scholar] [CrossRef]
- Vasileva, E.; Shuvalov, O.; Petukhov, A.; Fedorova, O.; Daks, A.; Nader, R.; Barlev, N. KMT Set7/9 Is a New Regulator of Sam68 STAR-Protein. Biochem. Biophys. Res. Commun. 2020, 525, 1018–1024. [Google Scholar] [CrossRef]
- Li, Y.; Reddy, M.A.; Miao, F.; Shanmugam, N.; Yee, J.-K.; Hawkins, D.; Ren, B.; Natarajan, R. Role of the Histone H3 Lysine 4 Methyltransferase, SET7/9, in the Regulation of NF-κB-Dependent Inflammatory Genes: Relevance to Diabetes and Inflammation. J. Biol. Chem. 2008, 283, 26771–26781. [Google Scholar] [CrossRef] [Green Version]
- Del Rizzo, P.A.; Trievel, R.C. Substrate and Product Specificities of SET Domain Methyltransferases. Epigenetics 2011, 6, 1059–1067. [Google Scholar] [CrossRef]
- Okabe, J.; Orlowski, C.; Balcerczyk, A.; Tikellis, C.; Thomas, M.; Cooper, M.E.; El-Osta, A. Distinguishing Hyperglycemic Changes by Set7 in Vascular Endothelial Cells. Circ. Res. 2012, 110, 1067–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batista, I.; Helguero, L.A. Biological Processes and Signal Transduction Pathways Regulated by the Protein Methyltransferase SETD7 and Their Significance in Cancer. Signal Transduct. Target. Ther. 2018, 3, 19. [Google Scholar] [CrossRef] [PubMed]
- Keating, S.T.; El-Osta, A. Transcriptional Regulation by the Set7 Lysine Methyltransferase. Epigenetics 2013, 8, 361–372. [Google Scholar] [CrossRef] [Green Version]
- Aksenova, V.; Turoverova, L.; Khotin, M.; Magnusson, K.-E.; Tulchinsky, E.; Melino, G.; Pinaev, G.P.; Barlev, N.; Tentler, D. Actin-Binding Protein Alpha-Actinin 4 (ACTN4) Is a Transcriptional Co-Activator of RelA/p65 Sub-unit of NF-kB. Oncotarget 2013, 4, 362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oudhoff, M.J.; Braam, M.J.; Freeman, S.A.; Wong, D.; Rattray, D.G.; Wang, J.; Antignano, F.; Snyder, K.; Refaeli, I.; Hughes, M.R.; et al. SETD7 Controls Intestinal Regeneration and Tumorigenesis by Regulating Wnt/β-Catenin and Hippo/YAP Signaling. Dev. Cell 2016, 37, 47–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanov, G.S.; Ivanova, T.; Kurash, J.; Ivanov, A.; Chuikov, S.; Gizatullin, F.; Herrera-Medina, E.M.; Rauscher, F., III; Reinberg, D.; Barlev, N.A. Methylation-Acetylation Interplay Activates p53 in Response to DNA Damage. Mol. Cell. Biol. 2007, 27, 6756–6769. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Li, Z.; Yang, Q.; Liu, W.; Wan, J.; Li, J.; Zhang, M. Substrate Docking–Mediated Specific and Efficient Lysine Methylation by the SET Domain–Containing Histone Methyltransferase SETD7. J. Biol. Chem. 2019, 294, 13355–13365. [Google Scholar] [CrossRef] [Green Version]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam Protein Families Database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef]
- Mecklenburg, K.L.; Freed, S.A.; Raval, M.; Quintero, O.A.; Yengo, C.M.; O’Tousa, J.E. Invertebrate and Vertebrate Class III Myosins Interact with MORN Repeat-Containing Adaptor Proteins. PLoS ONE 2015, 10, e0122502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadano, S.; Kunita, R.; Otomo, A.; Suzuki-Utsunomiya, K.; Ikeda, J.-E. Molecular and Cellular Function of ALS2/Alsin: Implication of Membrane Dynamics in Neuronal Development and Degeneration. Neurochem. Int. 2007, 51, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Landstrom, A.; Beavers, D.L.; Wehrens, X.H. The Junctophilin Family of Proteins: From Bench to Bedside. Trends Mol. Med. 2014, 20, 353–362. [Google Scholar] [CrossRef] [Green Version]
- Guo, A.; Wang, Y.; Chen, B.; Wang, Y.; Yuan, J.; Zhang, L.; Hall, D.; Wu, J.; Shi, Y.; Zhu, Q.; et al. E-C Coupling Structural Protein Junctophilin-2 Encodes a Stress-Adaptive Transcription Regulator. Science 2018, 362, eaan3303. [Google Scholar] [CrossRef]
- Sajko, S.; Grishkovskaya, I.; Kostan, J.; Graewert, M.; Setiawan, K.; Trübestein, L.; Niedermüller, K.; Gehin, C.; Sponga, A.; Puchinger, M. Structures of Three MORN Repeat Proteins and a Re-Evaluation of the Proposed Lipid-Binding Properties of MORN Repeats. PLoS ONE 2020, 15, e0242677. [Google Scholar] [CrossRef]
- Sehnal, D.; Bittrich, S.; Deshpande, M.; Svobodová, R.; Berka, K.; Bazgier, V.; Velankar, S.; Burley, S.K.; Koča, J.; Rose, A.S. Mol* Viewer: Modern Web App for 3D Visualization and Analysis of Large Biomolecular Structures. Nucleic Acids Res. 2021, 49, W431–W437. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.R.; Jing, C.; Walker, P.A.; Martin, S.R.; Howell, S.A.; Blackburn, G.M.; Gamblin, S.J.; Xiao, B. Crystal Structure and Functional Analysis of the Histone Methyltransferase SET7/9. Cell 2002, 111, 105–115. [Google Scholar] [CrossRef]
- Vasileva, E.; Barlev, N. The World of SET-Containing Lysine Methyltransferases. eLS 2017, 1–10. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin Modifications and Their Function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Noma, K.-I.; Allis, C.D.; Grewal, S.I.S. Transitions in Distinct Histone H3 Methylation Patterns at the Heterochromatin Domain Boundaries. Science 2001, 293, 1150–1155. [Google Scholar] [CrossRef]
- Castaño, J.; Morera, C.; Sesé, B.; Boue, S.; Bonet-Costa, C.; Martí, M.; Roque, A.; Jordan, A.; Barrero, M.J. SETD7 Regulates the Differentiation of Human Embryonic Stem Cells. PLoS ONE 2016, 11, e0149502. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Zhou, J.; Liu, X.; Lu, D.; Shen, C.; Du, Y.; Wei, F.-Z.; Song, B.; Lu, X.; Yu, Y. Methylation of SUV39H1 by SET7/9 Results in Heterochromatin Relaxation and Genome Instability. Proc. Natl. Acad. Sci. USA 2013, 110, 5516–5521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estève, P.-O.; Chin, H.G.; Benner, J.; Feehery, G.R.; Samaranayake, M.; Horwitz, G.A.; Jacobsen, S.E.; Pradhan, S. Regulation of DNMT1 Stability through SET7-Mediated Lysine Methylation in Mammalian Cells. Proc. Natl. Acad. Sci. USA 2009, 106, 5076–5081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Hevi, S.; Kurash, J.K.; Lei, H.; Gay, F.; Bajko, J.; Su, H.; Sun, W.; Chang, H.; Xu, G.; et al. The Lysine Demethylase LSD1 (KDM1) Is Required for Maintenance of Global DNA Methylation. Nat. Genet. 2008, 41, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Kouskouti, A.; Scheer, E.; Staub, A.; Tora, L.; Talianidis, I. Gene-Specific Modulation of TAF10 Function by SET9-Mediated Methylation. Mol. Cell 2004, 14, 175–182. [Google Scholar] [CrossRef] [Green Version]
- Shen, C.; Wang, D.; Liu, X.; Gu, B.; Du, Y.; Wei, F.Z.; Cao, L.L.; Song, B.; Lu, X.; Yang, Q. SET7/9 Regulates Cancer Cell Proliferation by Influencing β-Catenin Stability. FASEB J. 2015, 29, 4313–4323. [Google Scholar] [CrossRef]
- Yang, J.; Huang, J.; Dasgupta, M.; Sears, N.; Miyagi, M.; Wang, B.; Chance, M.R.; Chen, X.; Du, Y.; Wang, Y.; et al. Reversible Methylation of Promoter-Bound STAT3 by Histone-Modifying Enzymes. Proc. Natl. Acad. Sci. USA 2010, 107, 21499–21504. [Google Scholar] [CrossRef] [Green Version]
- Kontaki, H.; Talianidis, I. Lysine Methylation Regulates E2F1-Induced Cell Death. Mol. Cell 2010, 39, 152–160. [Google Scholar] [CrossRef]
- Lezina, L.; Aksenova, V.; Ivanova, T.; Purmessur, N.; Antonov, A.; Tentler, D.; Fedorova, O.; Garabadgiu, A.; Talianidis, I.; Melino, G. KMTase Set7/9 Is a Critical Regulator of E2F1 Activity upon Genotoxic Stress. Cell Death Differ. 2014, 21, 1889–1899. [Google Scholar] [CrossRef] [PubMed]
- Munro, S.; Khaire, N.; Inche, A.; Carr, S.; La Thangue, N.B. Lysine Methylation Regulates the pRb Tumour Suppressor Protein. Oncogene 2010, 29, 2357–2367. [Google Scholar] [CrossRef] [Green Version]
- Carr, S.M.; Munro, S.; Kessler, B.; Oppermann, U.; La Thangue, N.B. Interplay between Lysine Methylation and Cdk Phosphorylation in Growth Control by the Retinoblastoma Protein. EMBO J. 2010, 30, 317–327. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.-J.; Wu, X.-N.; Shi, T.-T.; Xu, H.-T.; Yi, J.; Shen, H.-F.; Huang, M.-F.; Shu, X.-Y.; Wang, F.-F.; Peng, B.-L. Regulation of Transcription Factor Yin Yang 1 by SET7/9-Mediated Lysine Methylation. Sci. Rep. 2016, 6, 21718. [Google Scholar] [CrossRef] [Green Version]
- Ea, C.-K.; Baltimore, D. Regulation of NF-κB Activity through Lysine Monomethylation of p65. Proc. Natl. Acad. Sci. USA 2009, 106, 18972–18977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.-D.; Huang, B.; Li, M.; Lamb, A.; Kelleher, N.L.; Chen, L.-F. Negative Regulation of NF-κB Action by Set9-Mediated Lysine Methylation of the RelA Subunit. EMBO J. 2009, 28, 1055–1066. [Google Scholar] [CrossRef] [Green Version]
- Xie, Q.; Hao, Y.; Tao, L.; Peng, S.; Rao, C.; Chen, H.; You, H.; Dong, M.; Yuan, Z. Lysine Methylation of FOXO3 Regulates Oxidative Stress-Induced Neuronal Cell Death. EMBO Rep. 2012, 13, 371–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calnan, D.R.; Webb, A.E.; White, J.L.; Stowe, T.R.; Goswami, T.; Shi, X.; Espejo, A.; Bedford, M.T.; Gozani, O.; Gygi, S.P.; et al. Methylation by Set9 Modulates FoxO3 Stability and Transcriptional Activity. Aging 2012, 4, 462–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Nam, H.J.; Lee, J.; Park, D.Y.; Kim, C.; Yu, Y.S.; Kim, D.; Park, S.W.; Bhin, J.; Hwang, D.; et al. Methylation-Dependent Regulation of HIF-1α Stability Restricts Retinal and Tumour Angiogenesis. Nat. Commun. 2016, 7, 10347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Chenxi, X.; Xu, C.; Leng, X.; Cao, H.; Ouyang, G.; Xiaoqian, L. Repression of Hypoxia-Inducible Factor α Signaling by Set7-Mediated Methylation. Nucleic Acids Res. 2015, 43, 5081–5098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Wang, D.; Zhao, Y.; Tu, B.; Zheng, Z.; Wang, L.; Wang, H.; Gu, W.; Roeder, R.G.; Zhu, W.-G. Methyltransferase Set7/9 Regulates p53 Activity by Interacting with Sirtuin 1 (SIRT1). Proc. Natl. Acad. Sci. USA 2011, 108, 1925–1930. [Google Scholar] [CrossRef] [Green Version]
- Kassner, I.; Barandun, M.; Fey, M.; Rosenthal, F.; Hottiger, M.O. Crosstalk between SET7/9-Dependent Methylation and ARTD1-Mediated ADP-Ribosylation of Histone H1. 4. Epigenet. Chromatin 2013, 6, 1. [Google Scholar] [CrossRef] [Green Version]
- Gaughan, L.; Stockley, J.; Wang, N.; McCracken, S.R.; Treumann, A.; Armstrong, K.; Shaheen, F.; Watt, K.; McEwan, I.J.; Wang, C. Regulation of the Androgen Receptor by SET9-Mediated Methylation. Nucleic Acids Res. 2011, 39, 1266–1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, K.; Jia, D.; Kapoor-Vazirani, P.; Powell, D.R.; Collins, R.; Sharma, D.; Peng, J.; Cheng, X.; Vertino, P.M. Regulation of Estrogen Receptor α by the SET7 Lysine Methyltransferase. Mol. Cell 2008, 30, 336–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heintzman, N.D.; Stuart, R.K.; Hon, G.; Fu, Y.; Ching, C.W.; Hawkins, R.D.; Barrera, L.O.; Van Calcar, S.; Qu, C.; Ching, K.A.; et al. Distinct and Predictive Chromatin Signatures of Transcriptional Promoters and Enhancers in the Human Genome. Nat. Genet. 2007, 39, 311–318. [Google Scholar] [CrossRef]
- Lehnertz, B.; Rogalski, J.C.; Schulze, F.M.; Yi, L.; Lin, S.; Kast, J.; Rossi, F.M. P53-Dependent Transcription and Tumor Suppression Are Not Affected in Set7/9-Deficient Mice. Mol. Cell 2011, 43, 673–680. [Google Scholar] [CrossRef] [Green Version]
- Sun, G.; Reddy, M.A.; Yuan, H.; Lanting, L.; Kato, M.; Natarajan, R. Epigenetic Histone Methylation Modulates Fibrotic Gene Expression. J. Am. Soc. Nephrol. 2010, 21, 2069–2080. [Google Scholar] [CrossRef]
- Nagashimada, M.; Ueda, T.; Ishita, Y.; Sakurai, H. TAF7 Is a Heat-Inducible Unstable Protein and Is Required for Sustained Expression of Heat Shock Protein Genes. FEBS J. 2018, 285, 3215–3224. [Google Scholar] [CrossRef] [PubMed]
- Estève, P.-O.; Chang, Y.; Samaranayake, M.; Upadhyay, A.K.; Horton, J.R.; Feehery, G.R.; Cheng, X.; Pradhan, S. A Methylation and Phosphorylation Switch between an Adjacent Lysine and Serine Determines Human DNMT1 Stability. Nat. Struct. Mol. Biol. 2010, 18, 42–48. [Google Scholar] [CrossRef] [Green Version]
- Konsavage, W.M.; Kyler, S.L.; Rennoll, S.A.; Jin, G.; Yochum, G.S. Wnt/β-Catenin Signaling Regulates Yes-associated Protein (YAP) Gene Expression in Colorectal Carcinoma Cells. J. Biol. Chem. 2012, 287, 11730–11739. [Google Scholar] [CrossRef] [Green Version]
- Azzolin, L.; Panciera, T.; Soligo, S.; Enzo, E.; Bicciato, S.; Dupont, S.; Bresolin, S.; Frasson, C.; Basso, G.; Guzzardo, V.; et al. YAP/TAZ Incorporation in the β-Catenin Destruction Complex Orchestrates the Wnt Response. Cell 2014, 158, 157–170. [Google Scholar] [CrossRef] [Green Version]
- Weinberg, R.A. The Retinoblastoma Protein and Cell Cycle Control. Cell 1995, 81, 323–330. [Google Scholar] [CrossRef] [Green Version]
- Stevens, C.; La Thangue, N.B. E2F and Cell Cycle Control: A Double-Edged Sword. Arch. Biochem. Biophys. 2003, 412, 157–169. [Google Scholar] [CrossRef]
- Classon, M.; Harlow, E. The Retinoblastoma Tumour Suppressor in Development and Cancer. Nat. Cancer 2002, 2, 910–917. [Google Scholar] [CrossRef]
- Giacinti, C.; Giordano, A. RB and Cell Cycle Progression. Oncogene 2006, 25, 5220–5227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kastan, M.B.; Onyekwere, O.; Sidransky, D.; Vogelstein, B.; Craig, R.W. Participation of p53 Protein in the Cellular Response to DNA Damage. Cancer Res. 1991, 51, 6304–6311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daks, A.; Melino, D.; Barlev, N.A. The Role of Different E3 Ubiquitin Ligases in Regulation of the P53 Tumor Suppressor Protein. Tsitologiia 2013, 55, 673–687. [Google Scholar]
- Fedorova, O.; Daks, A.; Petrova, V.; Petukhov, A.; Lezina, L.; Shuvalov, O.; Davidovich, P.; Kriger, D.; Lomert, E.; Tentler, D.; et al. Novel Isatin-Derived Molecules Activate p53 via Interference with Mdm2 to Promote Apoptosis. Cell Cycle 2018, 17, 1917–1930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vassilev, L.T.; Vu, B.T.; Graves, B.; Carvajal, D.; Podlaski, F.; Filipovic, Z.; Kong, N.; Kammlott, U.; Lukacs, C.; Klein, C.; et al. In Vivo Activation of the p53 Pathway by Small-Molecule Antagonists of MDM2. Science 2004, 303, 844–848. [Google Scholar] [CrossRef] [Green Version]
- Davidovich, P.B.; Aksenova, V.; Petrova, V.; Tentler, D.; Orlova, D.; Smirnov, S.V.; Gurzhiy, V.; Okorokov, A.; Garabadzhiu, A.V.; Melino, G.; et al. Discovery of Novel Isatin-Based p53 Inducers. ACS Med. Chem. Lett. 2015, 6, 856–860. [Google Scholar] [CrossRef] [Green Version]
- Lezina, L.; Aksenova, V.; Fedorova, O.; Malikova, D.; Shuvalov, O.; Antonov, A.V.; Tentler, D.; Garabadgiu, A.V.; Melino, G.; Barlev, N.A. KMT Set7/9 Affects Genotoxic Stress Response via the Mdm2 Axis. Oncotarget 2015, 6, 25843. [Google Scholar] [CrossRef] [Green Version]
- Greer, E.; Brunet, A. FOXO Transcription Factors at the Interface between Longevity and Tumor Suppression. Oncogene 2005, 24, 7410–7425. [Google Scholar] [CrossRef] [Green Version]
- Hwangbo, D.S.; Gersham, B.; Tu, M.-P.; Palmer, M.; Tatar, M. Drosophila dFOXO Controls Lifespan and Regulates Insulin Signalling in Brain and Fat Body. Nature 2004, 429, 562–566. [Google Scholar] [CrossRef] [PubMed]
- Castrillon, D.H.; Miao, L.; Kollipara, R.; Horner, J.W.; DePinho, R.A. Suppression of Ovarian Follicle Activation in Mice by the Transcription Factor Foxo3a. Science 2003, 301, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.; Tsai, W.-B.; Cheng, C.-J.; Hsu, C.; Chung, Y.M.; Li, P.-C.; Lin, S.-H.; Hu, M.C.-T. Forkhead Box Transcription Factor FOXO3a Suppresses Estrogen-Dependent Breast Cancer Cell Proliferation and Tumorigenesis. Breast Cancer Res. 2008, 10, R21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bullock, M.D.; Bruce, A.; Sreekumar, R.; Curtis, N.; Cheung, T.; Reading, I.; Primrose, J.N.; Ottensmeier, C.; Packham, G.K.; Thomas, G.; et al. FOXO3 Expression during Colorectal Cancer Progression: Biomarker Potential Reflects a Tumour Suppressor Role. Br. J. Cancer 2013, 109, 387–394. [Google Scholar] [CrossRef]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.-W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The Tumour Suppressor Protein VHL Targets Hypoxia-Inducible Factors for Oxygen-Dependent Proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef]
- Barsyte-Lovejoy, D.; Li, F.; Oudhoff, M.; Tatlock, J.H.; Dong, A.; Zeng, H.; Wu, H.; Freeman, S.A.; Schapira, M.; Senisterra, G.A.; et al. (R)-PFI-2 Is a Potent and Selective Inhibitor of SETD7 Methyltransferase Activity in Cells. Proc. Natl. Acad. Sci. USA 2014, 111, 12853–12858. [Google Scholar] [CrossRef] [Green Version]
- Daks, A.; Mamontova, V.; Fedorova, O.; Petukhov, A.; Shuvalov, O.; Parfenyev, S.; Netsvetay, S.; Venina, A.; Kizenko, A.; Imyanitov, E. Set7/9 Controls Proliferation and Genotoxic Drug Resistance of NSCLC Cells. Biochem. Biophys. Res. Commun. 2021, 572, 41–48. [Google Scholar] [CrossRef]
- Mori, S.; Iwase, K.; Iwanami, N.; Tanaka, Y.; Kagechika, H.; Hirano, T. Development of Novel Bisubstrate-Type Inhibitors of Histone Methyltransferase SET7/9. Bioorg. Med. Chem. 2010, 18, 8158–8166. [Google Scholar] [CrossRef]
- Hu, H.-Y.; Li, K.-P.; Wang, X.-J.; Liu, Y.; Lu, Z.-G.; Dong, R.-H.; Guo, H.-B.; Zhang, M.-X. Set9, NF-κB, and MicroRNA-21 Mediate Berberine-Induced Apoptosis of Human Multiple Myeloma Cells. Acta Pharmacol. Sin. 2012, 34, 157–166. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| SET7/9 Target Protein | Methylation Sites | Effect of the Modification | Reference |
|---|---|---|---|
| Histone H1 | K12, K14, K17, K20, K21, K27, K111 | Modulation of the affinity of histone H1 to chromatin during human pluripotent cells differentiation | [31] |
| Histone H1.4 | K34, K127, K129, K130 | Prevention of acetylation at the same sites, heterochromatin formation | [31] |
| Histone H2.A | K5, K13, K15 | Unknown | [3] |
| Histone H2.B | K15 | Unknown | [3] |
| Histone H3 | K4 | Activation of transcription | [1] |
| Suv39H1 | K25, K123 | Heterohromatin relaxation, genome instability | [32] |
| DNMT1 | K142 | Promotion of DNMT1 ubiquitination and proteasomal degradation | [33] |
| K1094 | Decrease of the DNMT1 level | [34] | |
| TAF7 | K5 | Enhancement of TAF7 activity as co-factor of RNA polymerase II | [8] |
| TAF10 | K189 | Enhancement of TAF10 activity as co-factor of RNA polymerase II, activation of transcription of TAF10 target genes | [35] |
| YAP1 | K494 | Retention of YAP1 in the cytoplasm | [17] |
| β-catenin | K180 | Promotion of β-catenin ubiquitination by (GSK)-3b and its subsequent proteasomal degradation | [36] |
| STAT3 | K140 | Dissociation of STAT3 from promoter elements, downregulation of STAT3-dependent genes expression | [37] |
| E2F1 | K185 | Promotion of E2F1 ubiquitination and subsequent proteasomal degradation | [38] |
| Unknown | Enhancement of E2F transactivation of its target genes | [39] | |
| pRb | K873 | Enhancement of pRB-dependent repression of transcription | [40] |
| K810 | Promotion of p65/RelA ubiquitination and its subsequent proteasomal degradation | [41] | |
| YY1 | K173, K411 | Retention of YY1 in the cytoplasm | [42] |
| p65/RelA | K37 | Translocation to the nucleus and transactivation of target genes | [43] |
| K314, K315 | Promotion of p65/RelA ubiquitination and subsequent proteasomal degradation | [44] | |
| FOXO3 | K270 | Downregulation of FOXO3-dependent transactivation of BIM | [45] |
| K271 | Increase of the FOXO3 transactivation potential | [46] | |
| Hif1α | K32 | Suppression of Hif1α transactivation of its target genes | [47,48] |
| p53 | K372 | Stabilization, translocation to the nucleus and transactivation of target genes | [7] |
| SIRT1 | K233, K235, K236, K238 | Enhancement of SIRT1-dependent p53 acetylation and activation | [49] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daks, A.; Vasileva, E.; Fedorova, O.; Shuvalov, O.; Barlev, N.A. The Role of Lysine Methyltransferase SET7/9 in Proliferation and Cell Stress Response. Life 2022, 12, 362. https://doi.org/10.3390/life12030362
Daks A, Vasileva E, Fedorova O, Shuvalov O, Barlev NA. The Role of Lysine Methyltransferase SET7/9 in Proliferation and Cell Stress Response. Life. 2022; 12(3):362. https://doi.org/10.3390/life12030362
Chicago/Turabian StyleDaks, Alexandra, Elena Vasileva, Olga Fedorova, Oleg Shuvalov, and Nickolai A. Barlev. 2022. "The Role of Lysine Methyltransferase SET7/9 in Proliferation and Cell Stress Response" Life 12, no. 3: 362. https://doi.org/10.3390/life12030362
APA StyleDaks, A., Vasileva, E., Fedorova, O., Shuvalov, O., & Barlev, N. A. (2022). The Role of Lysine Methyltransferase SET7/9 in Proliferation and Cell Stress Response. Life, 12(3), 362. https://doi.org/10.3390/life12030362