
Targeting Specific Checkpoints in the Management of SARS-CoV-2 Induced Cytokine Storm

 , , , ,
, , , ,  , , , , ,
, , , , ,  , ,
, ,  , , ,
, , ,  , ,
, ,  and
and
Abstract
:1. Introduction
2. Objectives of the Study
3. Materials and Methods
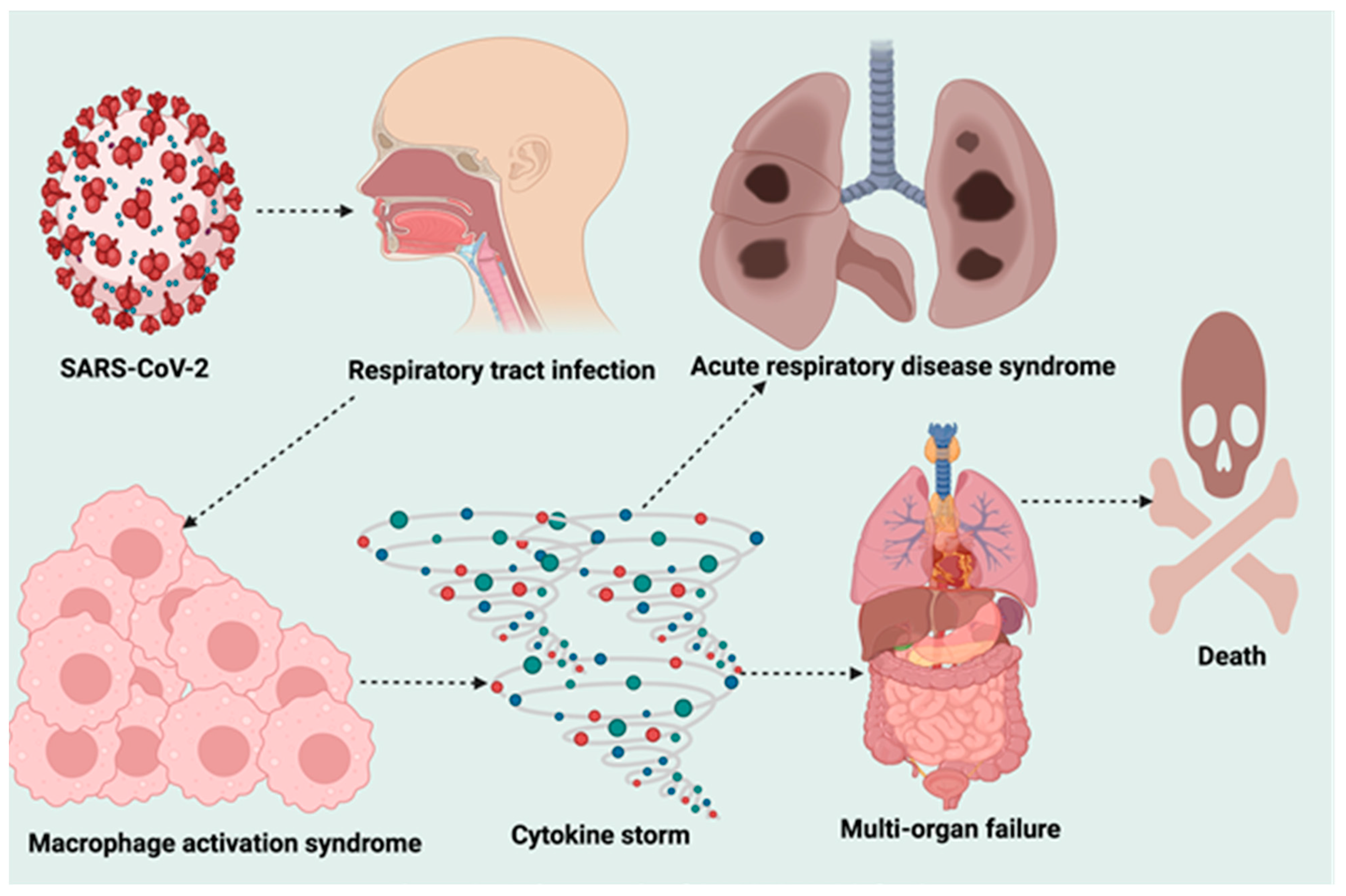
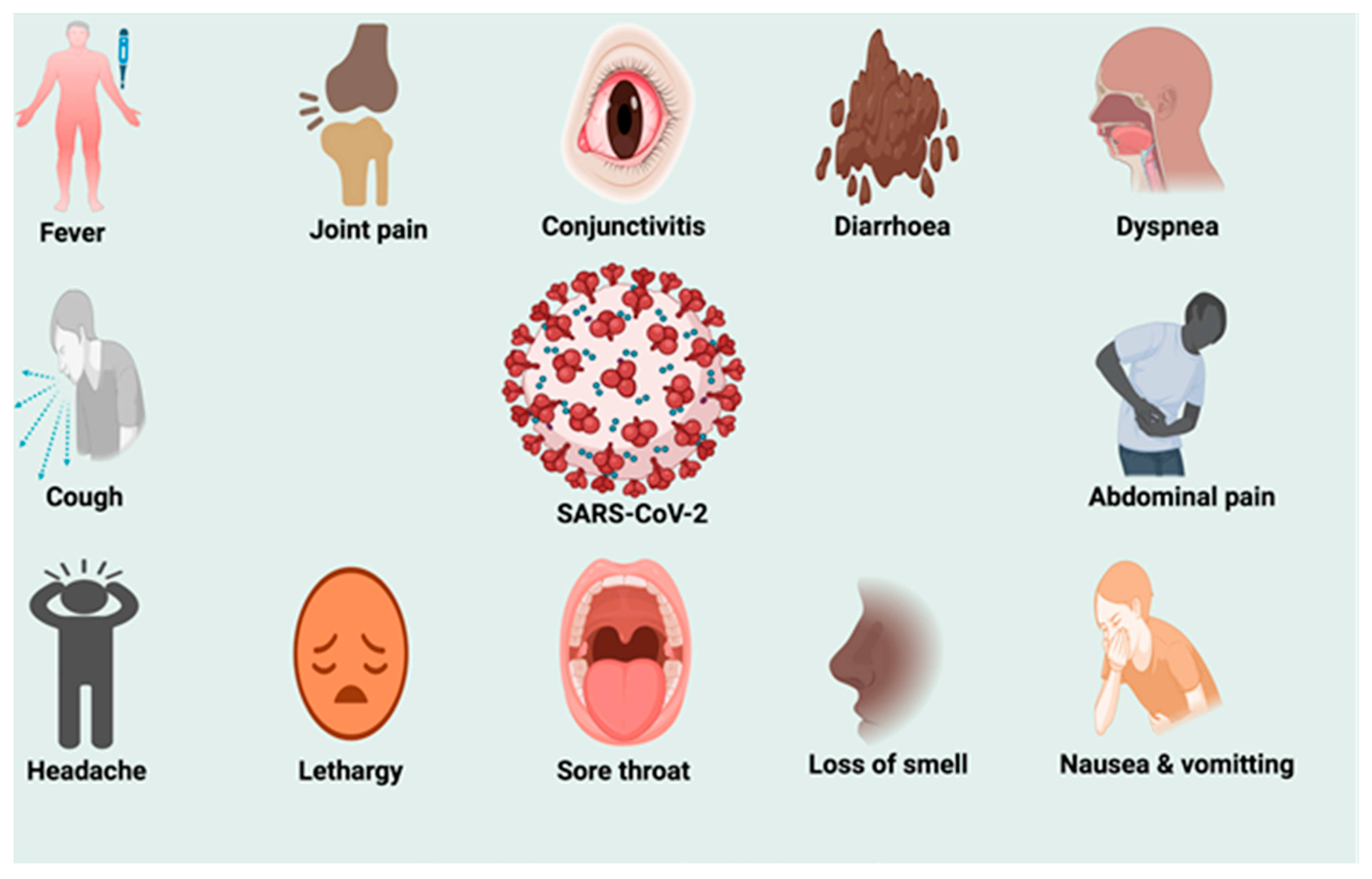
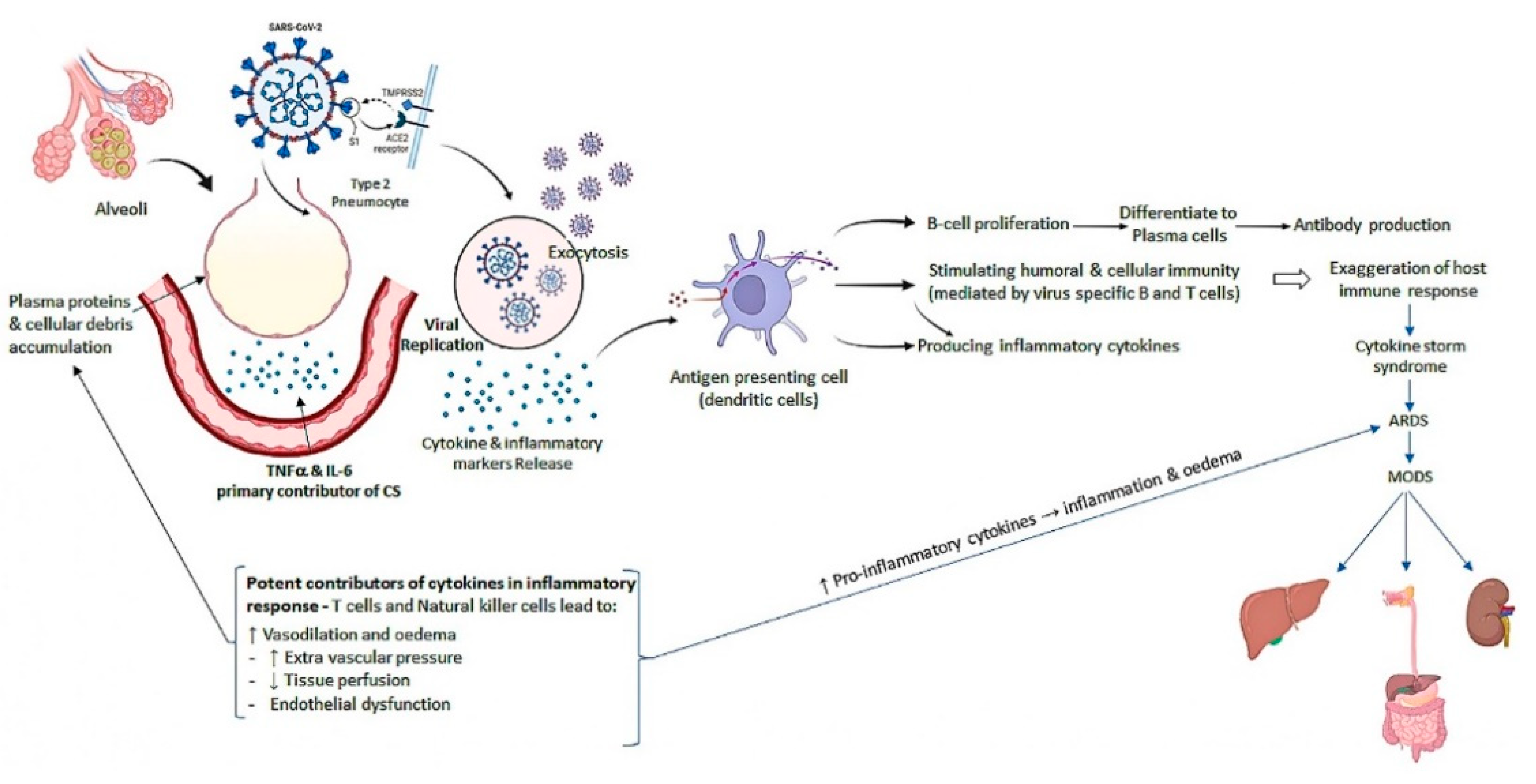
4. Pathogenesis of SARS-CoV-2 Infection
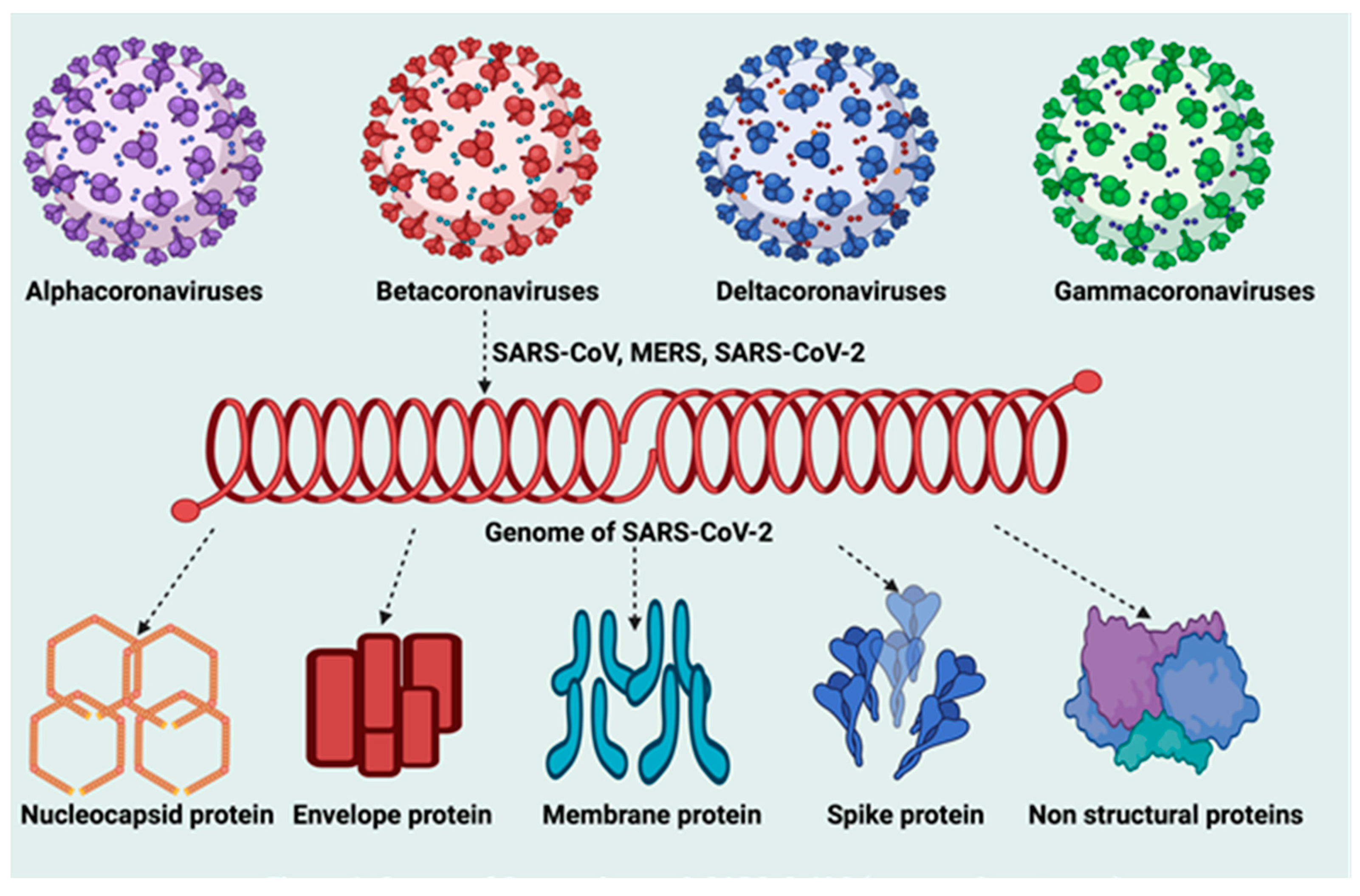
5. Molecular Anonymity of SARS-CoV-2 Infection
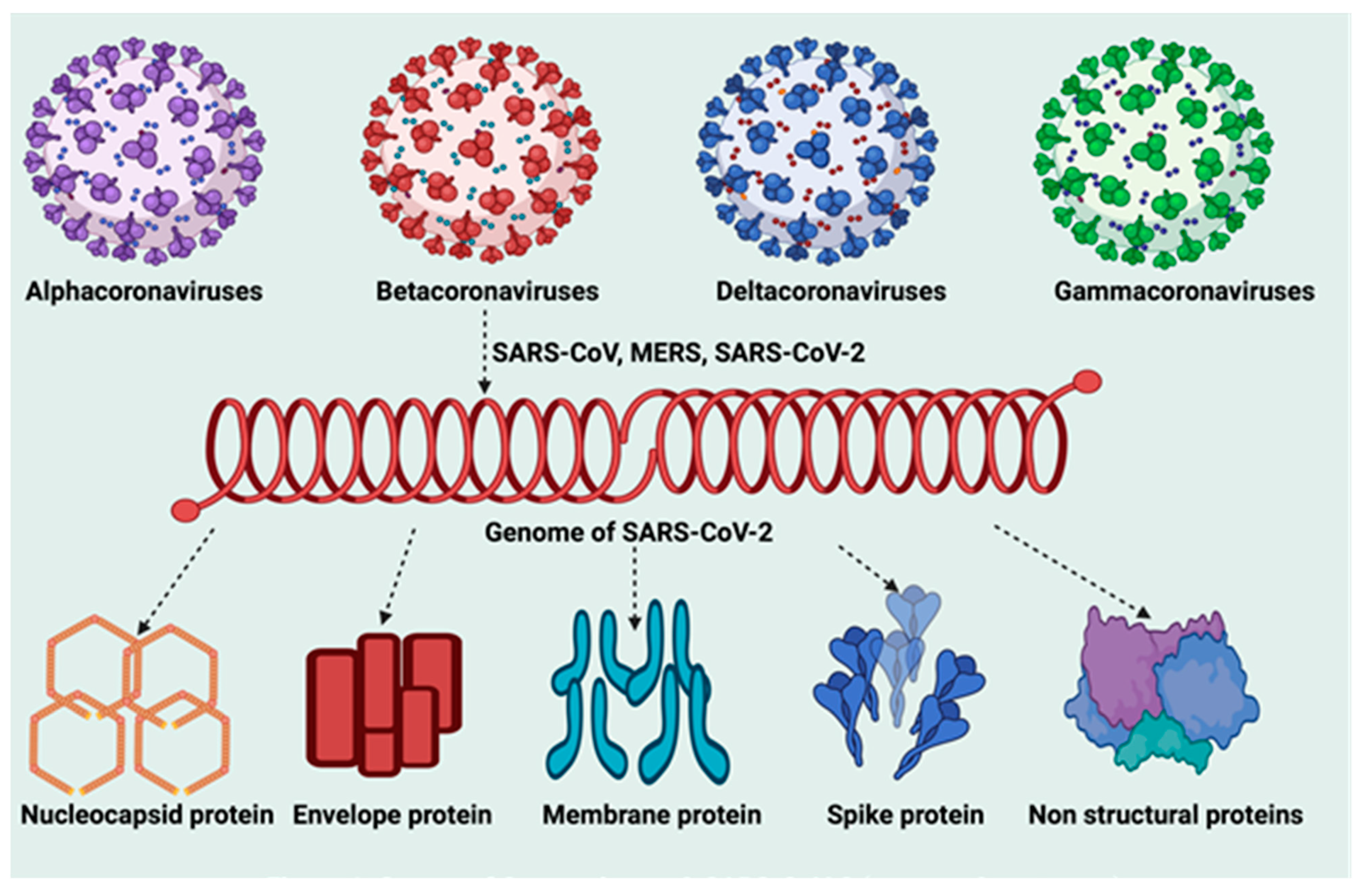
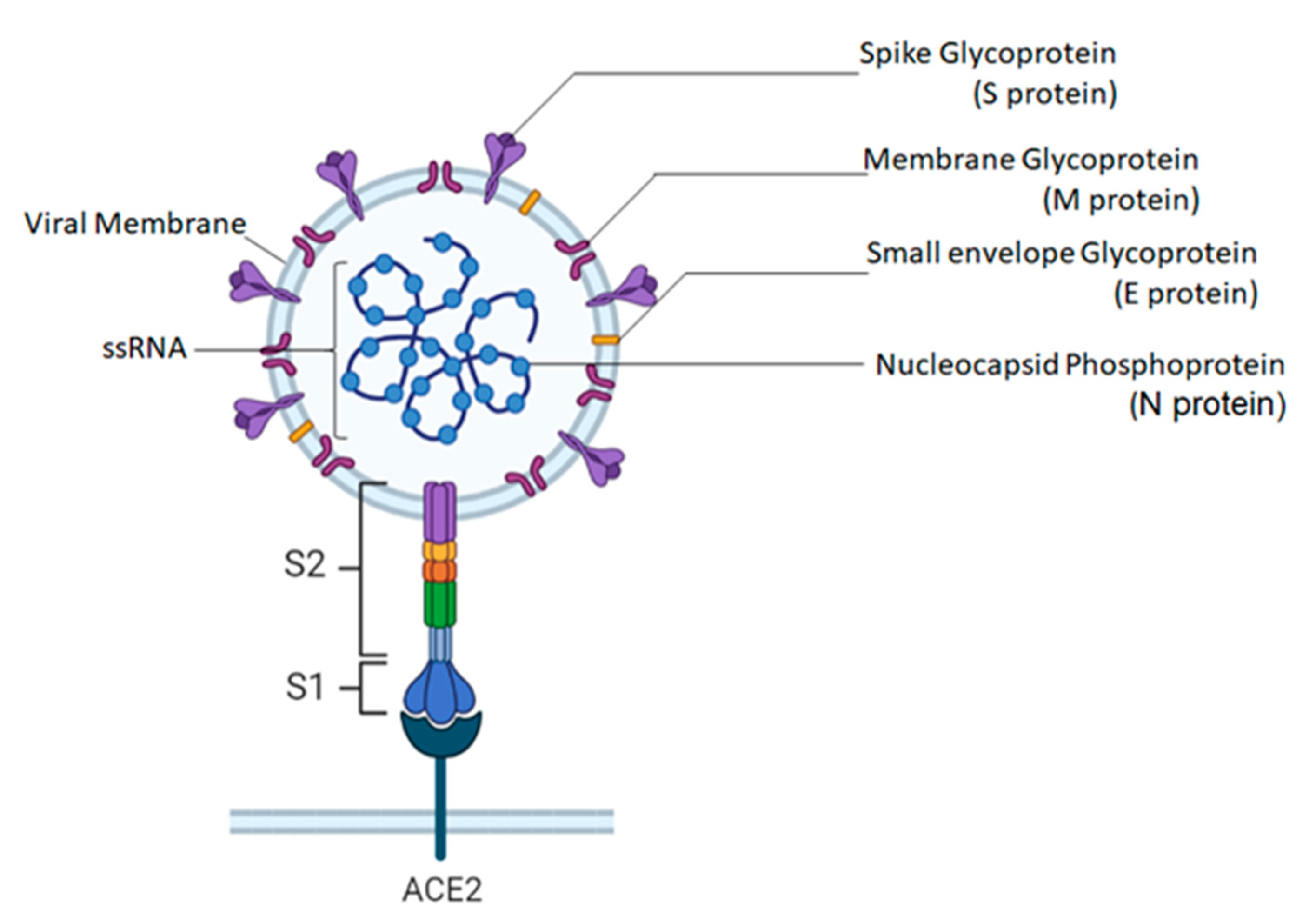
5.1. Molecular Structure of SARS-CoV-2
5.2. Viral Replication of SARS-CoV-2
5.3. Molecular Components of Cytokine Storm (CS)
5.4. Cytokine Storm Molecular Mechanism in SARS-CoV-2
6. SARS-CoV-2 Viral Load and Cytokine Storm
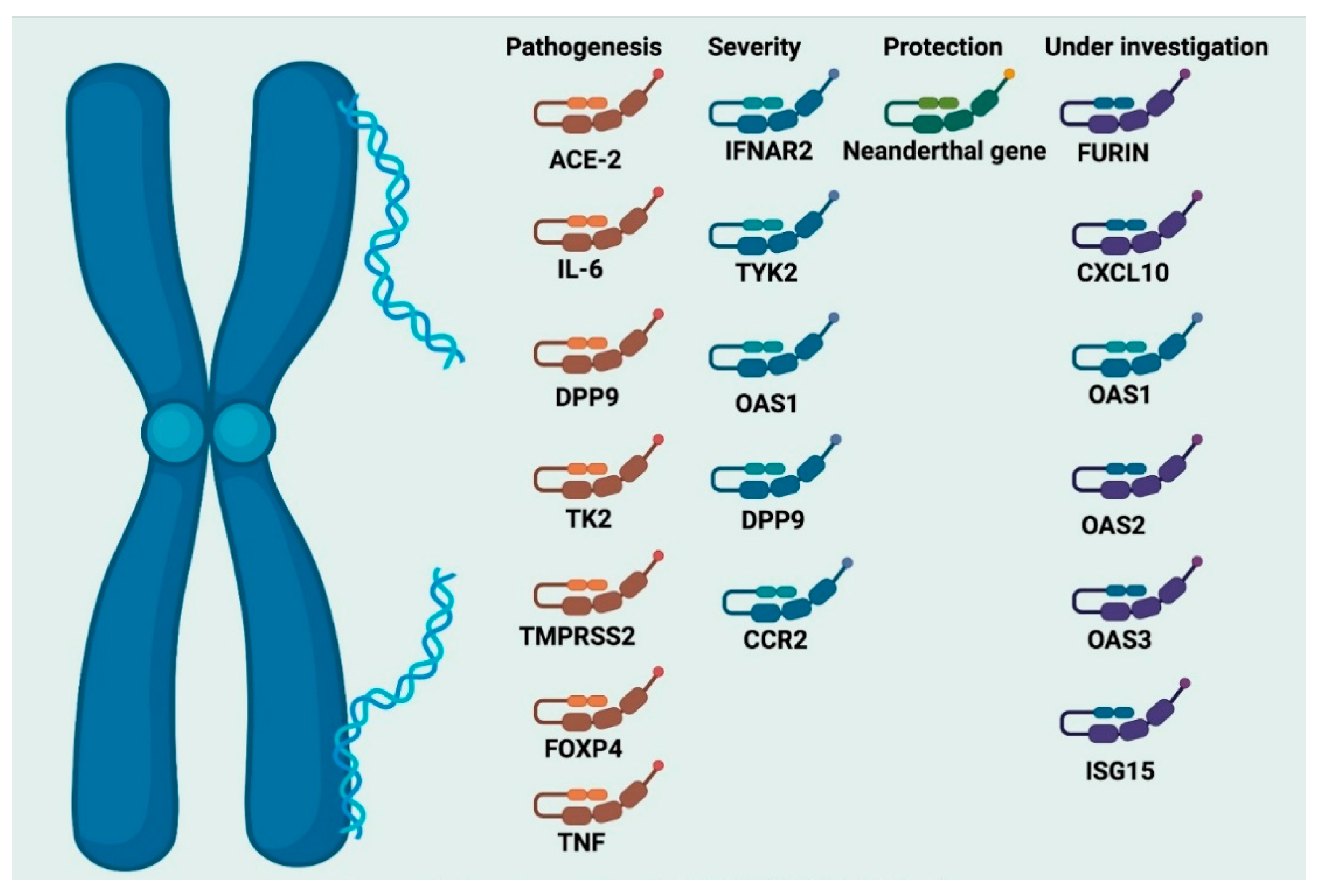
7. Genetic and Molecular Susceptibility to SARS-CoV-2 Infection
7.1. The Major Genetic Risk Factors for SARS-CoV-2 Infection
7.2. Genetic Fingerprints for Critical Illness in COVID-19

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GENES | Full Name | Polymorphism | Specific Role | Reference |
|---|---|---|---|---|
| IFNAR2 | Interferon alpha and beta receptor 2 | NM_000874:exon9:c.C966A:p.Y322X | Severe COVID-19-risk | Schmiedel et al., 2021 [156] |
| IFITM3 | Interferon-induced transmembrane protein 3 | rs12252-C/C | It is a risk factor for developing severe influenza | Kaidashev et al., 2021. [157] |
| OAS1 | Oligoadenylate synthase 1 | rs2057778 | Increased risk of infection | Schmiedel et al., 2021 [156] |
| CCR2 | Chemokine receptor | rs11385942-GA | Respiratory failure | Anastassopoulou et al., 2020 [158] |
| ACE2 | Angiotensin Converting Enzyme 2 | p.Arg514-Gly | Increase in pulmonary and cardiovascular complications in the African American population | Anastassopoulou et al., 2020 [158] |
| IL6 | Interleukin-6 | rs180079 | Associated with the increase in susceptibility and severity | Kaidashev et al., 2021. [157] |
| TMPRSS2 | Transmembrane serine protease 2 | rs12329760 | Increased susceptibility to disease | Anastassopoulou et al., 2020 [158] |
| HLA | Human leukocyte antigen (HLA) system | HLA-B*46:01 | Exhibit the lowest binding cap | Pollitt et al., 2020. [159] |
| TNF | Tumor necrosis factor | rs1800629 | Increase in pulmonary complications | Fishchuk et al., 2021. [160] |
| FURIN | Furin | rs16944971 | Promotes entry of the virus into the cell | Kucher et al., 2021 [161] |
| CXCL10 | Chemokine ligand 10 | rs11385942-GA | Respiratory failure | Anastassopoulou et al., 2020 [158] |
7.3. The Neanderthal Gene Variant and Coronavirus Disease-19
7.4. Resistance to Coronavirus Disease-19
8. SARS-COV-2 Induced Thromboinflammation
9. SARS-CoV-2 Oxidative Stress
10. SARS-CoV-2 and Defective Immune Response
10.1. Innate Immune Response
10.2. Adaptive Immune Response
10.3. Antibody Response
11. SARS-CoV-2 Hyperinflammation
11.1. SARS-CoV-2-Induced Hyperinflammation in Children
11.2. Diagnosis of SARS-CoV-2-Induced Hyperinflammation
12. Therapeutic Options for SARS-CoV-2-Induced Hyperinflammation
12.1. Corticosteroids
12.2. Interleukin-6 (IL-6) Antagonists
12.3. Interleuckin-1 (IL-1) Inhibitors
12.3.1. Canakinumab
12.3.2. Anakinra
12.4. Janus Kinase (JAK) Inhibitors
12.5. Quercetin
13. Conclusions
14. Recommendation
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DC | dendritic cells |
| ICU | Intensive care units |
| MPS | mononuclear phagocyte system |
| mtROS | increasing mitochondrial ROS |
| NETs | neutrophil extracellular traps |
| NOXs | NADPH oxidases |
| RONS | reactive oxygen/nitrogen species |
| OS | oxidative stress |
| RNS | reactive nitrogen species |
| ROS | oxygen-reactive species |
| SARS-CoV | Severe Acute Respiratory Syndrome Coronavirus |
References
- WHO Timeline—COVID-19. Cited on 8 November 2021. Available online: https://www.who.int/news/item/27-04-2020-who-timeline---covid-19 (accessed on 30 December 2021).
- WHO Coronavirus (COVID-19) Dashboard. Cited on 11 February 2022. Available online: https://covid19.who.int/ (accessed on 30 December 2021).
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Nile, S.H.; Nile, A.; Qiu, J.; Li, L.; Jia, X.; Kai, G. COVID-19: Pathogenesis, cytokine storm and therapeutic potential of interferons. Cytokine Growth Factor Rev. 2020, 53, 66–70. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Wang, B.; Yuan, T.; Chen, X.; Ao, Y.; Fitzpatrick, T.; Li, P.; Zhou, Y.; Lin, Y.-F.; Duan, Q.; et al. Clinical characteristics of coronavirus disease 2019 (COVID-19) in China: A systematic review and meta-analysis. J. Infect. 2020, 80, 656–665. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus—Infected Pneumonia in Wuhan, China. JAMA 2020, 323, 1061–1069. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Zaim, S.; Chong, J.H.; Sankaranarayanan, V.; Harky, A. COVID-19 and Multiorgan Response. Curr. Probl. Cardiol. 2020, 45, 100618. [Google Scholar] [CrossRef]
- Synowiec, A.; Szczepański, A.; Barreto-Duran, E.; Lie, L.K.; Pyrc, K. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): A Systemic Infection. Clin. Microbiol. Rev. 2021, 34, e00133-20. [Google Scholar] [CrossRef]
- Mehta, O.P.; Bhandari, P.; Raut, A.; Kacimi, S.E.O.; Huy, N.T. Coronavirus Disease (COVID-19): Comprehensive Review of Clinical Presentation. Front. Public Health 2021, 8, 582932. [Google Scholar] [CrossRef]
- Hamming, I.; Timens, W.; Bulthuis, M.L.C.; Lely, A.T.; Navis, G.J.; van Goor, H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J. Pathol. 2004, 203, 631–637. [Google Scholar] [CrossRef]
- Yang, L.; Xie, X.; Tu, Z.; Fu, J.; Xu, D.; Zhou, Y. The signal pathways and treatment of cytokine storm in COVID-19. Signal Transduct. Target. Ther. 2021, 6, 255. [Google Scholar] [CrossRef]
- Charan, J.; Dutta, S.; Kaur, R.; Bhardwaj, P.; Sharma, P.; Ambwani, S.; Jahan, I.; Abubakar, A.R.; Islam, S.; Hardcastle, T.C.; et al. Tocilizumab in COVID-19: A study of adverse drug events reported in the WHO database. Expert Opin. Drug Saf. 2021, 20, 1125–1136. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Wu, D.; Guo, W.; Cao, Y.; Huang, D.; Wang, H.; Wang, T.; Zhang, X.; Chen, H.; Yu, H.; et al. Clinical and immunological features of severe and moderate coronavirus disease 2019. J. Clin. Investig. 2020, 130, 2620–2629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ragab, D.; Eldin, H.S.; Taeimah, M.; Khattab, R.; Salem, R. The COVID-19 Cytokine Storm; What We Know So Far. Front. Immunol. 2020, 11, 1446. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M. Clinical Features of Cytokine Storm Syndrome. In Cytokine Storm Syndrome; Cron, R., Behrens, E., Eds.; Springer: Cham, Switzerland, 2019. [Google Scholar] [CrossRef]
- Tufan, A.; GÜLER, A.A.; Matucci-Cerinic, M. COVID-19, immune system response, hyperinflammation and repurposing antirheumatic drugs. Turk. J. Med. Sci. 2020, 50, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Mangalmurti, N.; Hunter, C.A. Cytokine Storms: Understanding COVID-19. Immunity 2020, 53, 19–25. [Google Scholar] [CrossRef]
- Dutta, S.; Kaur, R.J.; Bhardwaj, P.; Charan, J.; Bist, S.K.S.; Detha, M.D.; Kanchan, T.; Sharma, P.; Misra, S. Household Transmission of COVID-19: A Cross-Sectional Study. Infect. Drug Resist. 2020, 13, 4637–4642. [Google Scholar] [CrossRef]
- Dutta, S.; Ambwani, S.; Lal, H.; Ram, K.; Mishra, G.; Kumar, T.; Varthya, S.B. The Satisfaction Level of Undergraduate Medical and Nursing Students Regarding Distant Preclinical and Clinical Teaching amidst COVID-19 across India. Adv. Med. Educ. Pract. 2021, 12, 113–122. [Google Scholar] [CrossRef]
- Kaur, R.J.; Charan, J.; Dutta, S.; Sharma, P.; Bhardwaj, P.; Sharma, P.; Lugova, H.; Krishnapillai, A.; Islam, S.; Haque, M.; et al. Favipiravir Use in COVID-19: Analysis of Suspected Adverse Drug Events Reported in the WHO Database. Infect. Drug Resist. 2020, 13, 4427–4438. [Google Scholar] [CrossRef]
- Dutta, S.; Kaur, R.; Bhardwaj, P.; Deora, S.; Singh, K.; Ambwani, S.; Charan, J.; Abubakar, A.R.; Jahan, I.; Lugova, H.; et al. Hydroxychloroquine as Therapeutic Option in COVID-19: Analysis of Suspected Cardiovascular Adverse Drug Events Reported in the VigiBase. Bangladesh J. Med Sci. 2021, 20, 897–910. [Google Scholar] [CrossRef]
- Charan, J.; Bhardwaj, P.; Dutta, S.; Kaur, R.; Bist, S.K.; Detha, M.D.; Kanchan, T.; Yadav, D.; Mitra, P.; Sharma, P. Use of Complementary and Alternative Medicine (CAM) and Home Remedies by COVID-19 Patients: A Telephonic Survey. Indian J. Clin. Biochem. 2020, 36, 108–111. [Google Scholar] [CrossRef]
- Alam, S.; Kamal, T.B.; Sarker, M.R.; Zhou, J.-R.; Rahman, S.M.A.; Mohamed, I.N. Therapeutic Effectiveness and Safety of Repurposing Drugs for the Treatment of COVID-19: Position Standing in 2021. Front. Pharmacol. 2021, 12, 659577. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.A. Lack of Effectiveness of Repurposed Drugs for COVID-19 Treatment. Front. Immunol. 2021, 12, 635371. [Google Scholar] [CrossRef] [PubMed]
- Abubakar, A.R.; Sani, I.H.; Godman, B.; Kumar, S.; Islam, S.; Jahan, I.; Haque, M. Systematic Review on the Therapeutic Options for COVID-19: Clinical Evidence of Drug Efficacy and Implications. Infect. Drug Resist. 2020, 13, 4673–4695. [Google Scholar] [CrossRef] [PubMed]
- Kaur, R.J.; Dutta, S.; Bhardwaj, P.; Charan, J.; Dhingra, S.; Mitra, P.; Singh, K.; Yadav, D.; Sharma, P.; Misra, S. Adverse Events Reported From COVID-19 Vaccine Trials: A Systematic Review. Indian J. Clin. Biochem. 2021, 36, 427–439. [Google Scholar] [CrossRef]
- Dutta, S.; Kaur, R.J.; Bhardwaj, P.; Sharma, P.; Ambwani, S.; Islam, S.; Tandon, A.; Abhayanand, J.P.; Sukhija, S.; Venkatesh, S.S.; et al. Adverse events reported from the COVID-19 vaccines: A descriptive study based on the WHO database (VigiBase®). J. Appl. Pharm. Sci. 2021, 11, 001–009. [Google Scholar] [CrossRef]
- Jeet Kaur, R.; Dutta, S.; Charan, J.; Bhardwaj, P.; Tandon, A.; Yadav, D.; Islam, S.; Haque, M. Cardiovascular Adverse Events Reported from COVID-19 Vaccines: A Study Based on WHO Database. Int. J. Gen. Med. 2021, 14, 3909–3927. [Google Scholar] [CrossRef]
- World Health Organization. Weekly Epidemiological Update on COVID-19—9 November 2021 (Edition 65). Cited on 10 November 2021. Available online: https://www.who.int/publications/m/item/weekly-epidemiological-update-on-covid-19---9-november-2021 (accessed on 30 December 2021).
- Zalzala, H. Diagnosis of COVID-19: Facts and challenges. New Microbes New Infect. 2020, 38, 100761. [Google Scholar] [CrossRef]
- Pal, M.; Berhanu, G.; Desalegn, C.; Kandi, V. Severe Acute Respiratory Syndrome Coronavirus-2 (SARS-CoV-2): An Update. Cureus 2020, 12, e7423. [Google Scholar] [CrossRef] [Green Version]
- Contini, C.; Di Nuzzo, M.; Barp, N.; Bonazza, A.; de Giorgio, R.; Tognon, M.; Rubino, S. The novel zoonotic COVID-19 pandemic: An expected global health concern. J. Infect. Dev. Ctries. 2020, 14, 254–264. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Shin, W.I.; Pang, Y.X.; Meng, Y.; Lai, J.; You, C.; Zhao, H.; Lester, E.; Wu, T.; Pang, C.H. The First 75 Days of Novel Coronavirus (SARS-CoV-2) Outbreak: Recent Advances, Prevention, and Treatment. Int. J. Environ. Res. Public Health 2020, 17, 2323. [Google Scholar] [CrossRef] [Green Version]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Perlman, S. Another Decade, Another Coronavirus. N. Engl. J. Med. 2020, 382, 760–762. [Google Scholar] [CrossRef] [PubMed]
- Stadler, K.; Masignani, V.; Eickmann, M.; Becker, S.; Abrignani, S.; Klenk, H.-D.; Rappuoli, R. SARS—Beginning to understand a new virus. Nat. Rev. Genet. 2003, 1, 209–218. [Google Scholar] [CrossRef] [PubMed]
- McBride, R.; Van Zyl, M.; Fielding, B.C. The Coronavirus Nucleocapsid Is a Multifunctional Protein. Viruses 2014, 6, 2991–3018. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.; Khan, H.; Khan, S.; Nawaz, M. Epidemiological and clinical characteristics of coronavirus disease (COVID-19) cases at a screening clinic during the early outbreak period: A single-centre study. J. Med. Microbiol. 2020, 69, 1114–1123. [Google Scholar] [CrossRef]
- Rowaiye, A.B.; Okpalefe, O.A.; Adejoke, O.O.; Ogidigo, J.O.; Oladipo, O.H.; Ogu, A.C.; Oli, A.N.; Olofinsae, S.; Onyekwere, O.; Abubakar, A.R.; et al. Attenuating the Effects of Novel COVID-19 (SARS-CoV-2) Infection-Induced Cytokine Storm and the Implications. J. Inflamm. Res. 2021, 14, 1487–1510. [Google Scholar] [CrossRef]
- Yang, P.; Wang, X. COVID-19: A new challenge for human beings. Cell. Mol. Immunol. 2020, 17, 555–557. [Google Scholar] [CrossRef] [Green Version]
- Pascarella, G.; Strumia, A.; Piliego, C.; Bruno, F.; Del Buono, R.; Costa, F.; Scarlata, S.; Agrò, F.E. COVID-19 diagnosis and management: A comprehensive review. J. Intern. Med. 2020, 288, 192–206. [Google Scholar] [CrossRef]
- Rowaiye, A.B.; Onuh, O.A.; Asala, T.M.; Ogu, A.C.; Bur, D.; Nwankwo, E.J.; Orji, U.M.; Ibrahim, Z.R.; Hamza, J.; Ugorji, A.L. In Silico Identification of Potential Allosteric Inhibitors of the SARS-CoV-2 Helicase. Trop. J. Nat. Prod. Res. 2021, 5, 165–177. [Google Scholar]
- Rahman, S.; Montero, M.T.V.; Rowe, K.; Kirton, R.; Kunik, F. Epidemiology, pathogenesis, clinical presentations, diagnosis and treatment of COVID-19: A review of current evidence. Expert Rev. Clin. Pharmacol. 2021, 14, 601–621. [Google Scholar] [CrossRef]
- Millet, J.K.; Whittaker, G.R. Host cell proteases: Critical determinants of coronavirus tropism and pathogenesis. Virus Res. 2015, 202, 120–134. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.-L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Lee, J.-Y.; Yang, J.-S.; Kim, J.W.; Kim, V.N.; Chang, H. The Architecture of SARS-CoV-2 Transcriptome. Cell 2020, 181, 914–921.e10. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.X.; Fung, T.S.; Chong, K.K.-L.; Shukla, A.; Hilgenfeld, R. Accessory proteins of SARS-CoV and other coronaviruses. Antivir. Res. 2014, 109, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.; Chaudhary, J.; Jain, N.; Chaudhary, P.; Khanra, S.; Dhamija, P.; Sharma, A.; Kumar, A.; Handu, S. Role of Structural and Non-Structural Proteins and Therapeutic Targets of SARS-CoV-2 for COVID-19. Cells 2021, 10, 821. [Google Scholar] [CrossRef]
- Thiel, V.; Ivanov, K.A.; Putics, A.; Hertzig, T.; Schelle, B.; Bayer, S.; Weißbrich, B.; Snijder, E.J.; Rabenau, H.; Doerr, H.W.; et al. Mechanisms and enzymes involved in SARS coronavirus genome expression. J. Gen. Virol. 2003, 84, 2305–2315. [Google Scholar] [CrossRef]
- Marra, M.A.; Jones, S.J.M.; Astell, C.R.; Holt, R.A.; Brooks-Wilson, A.; Butterfield, Y.S.N.; Khattra, J.; Asano, J.K.; Barber, S.A.; Chan, S.Y.; et al. The Genome Sequence of the SARS-Associated Coronavirus. Science 2003, 300, 1399–1404. [Google Scholar] [CrossRef] [Green Version]
- Letko, M.; Marzi, A.; Munster, V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Microbiol. 2020, 5, 562–569. [Google Scholar] [CrossRef] [Green Version]
- Pang, X.C.; Zhang, H.X.; Zhang, Z.; Rinkiko, S.; Cui, Y.M.; Zhu, Y.Z. The Two-Way Switch Role of ACE2 in the Treatment of Novel Coronavirus Pneumonia and Underlying Comorbidities. Molecules 2020, 26, 142. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94, e00127-20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Y.-R.; Cao, Q.-D.; Hong, Z.-S.; Tan, Y.-Y.; Chen, S.-D.; Jin, H.-J.; Tan, K.-S.; Wang, D.-Y.; Yan, Y. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak—An update on the status. Mil. Med. Res. 2020, 7, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Bosch, B.J.; van der Zee, R.; de Haan, C.A.; Rottier, P.J.M. The Coronavirus Spike Protein Is a Class I Virus Fusion Protein: Structural and Functional Characterization of the Fusion Core Complex. J. Virol. 2003, 77, 8801–8811. [Google Scholar] [CrossRef] [Green Version]
- Astuti, I.; Ysrafil. Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2): An overview of viral structure and host response. Diabetes Metab. Syndr. 2020, 14, 407–412. [Google Scholar] [CrossRef] [PubMed]
- Naqvi, A.A.T.; Fatima, K.; Mohammad, T.; Fatima, U.; Singh, I.K.; Singh, A.; Atif, S.M.; Hariprasad, G.; Hasan, G.M.; Hassan, I. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: Structural genomics approach. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2020, 1866, 165878. [Google Scholar] [CrossRef] [PubMed]
- Sadarangani, M.; Marchant, A.; Kollmann, T.R. Immunological mechanisms of vaccine-induced protection against COVID-19 in humans. Nat. Rev. Immunol. 2021, 21, 475–484. [Google Scholar] [CrossRef]
- Bakhshandeh, B.; Jahanafrooz, Z.; Abbasi, A.; Goli, M.B.; Sadeghi, M.; Mottaqi, M.S.; Zamani, M. Mutations in SARS-CoV-2; Consequences in structure, function, and pathogenicity of the virus. Microb. Pathog. 2021, 154, 104831. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Hilton, S.K.; Ellis, D.; Crawford, K.H.; Dingens, A.S.; Navarro, M.J.; Bowen, J.E.; Tortorici, M.A.; Walls, A.C.; et al. Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding. Cell 2020, 182, 1295–1310.e20. [Google Scholar] [CrossRef]
- Schoeman, D.; Fielding, B.C. Coronavirus envelope protein: Current knowledge. Virol. J. 2019, 16, 69. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, S.; Bhattacharyya, D.; Bhunia, A. Host-membrane interacting interface of the SARS coronavirus envelope protein: Immense functional potential of C-terminal domain. Biophys. Chem. 2020, 266, 106452. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-K.; Hou, M.-H.; Chang, C.-F.; Hsiao, C.-D.; Huang, T.-H. The SARS coronavirus nucleocapsid protein—Forms and functions. Antivir. Res. 2014, 103, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Alharbi, S.N.; Alrefaei, A.F. Comparison of the SARS-CoV-2 (2019-nCoV) M protein with its counterparts of SARS-CoV and MERS-CoV species. J. King Saud Univ. Sci. 2021, 33, 101335. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, A.; Naujokat, C. Structural features of coronavirus SARS-CoV-2 spike protein: Targets for vaccination. Life Sci. 2020, 257, 118056. [Google Scholar] [CrossRef]
- Ke, Z.; Oton, J.; Qu, K.; Cortese, M.; Zila, V.; McKeane, L.; Nakane, T.; Zivanov, J.; Neufeldt, C.J.; Cerikan, B.; et al. Structures and distributions of SARS-CoV-2 spike proteins on intact virions. Nature 2020, 588, 498–502. [Google Scholar] [CrossRef]
- Verdecchia, P.; Cavallini, C.; Spanevello, A.; Angeli, F. The pivotal link between ACE2 deficiency and SARS-CoV-2 infection. Eur. J. Intern. Med. 2020, 76, 14–20. [Google Scholar] [CrossRef]
- Rabi, F.A.; Al Zoubi, M.S.; Kasasbeh, G.A.; Salameh, D.M.; Al-Nasser, A.D. SARS-CoV-2 and Coronavirus Disease 2019: What We Know So Far. Pathogens 2020, 9, 231. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, Y.; Wang, M.; Cheng, A.; Yang, Q.; Wu, Y.; Jia, R.; Liu, M.; Zhu, D.; Chen, S.; et al. Structures and Functions of the 3′ Untranslated Regions of Positive-Sense Single-Stranded RNA Viruses Infecting Humans and Animals. Front. Cell. Infect. Microbiol. 2020, 10, 453. [Google Scholar] [CrossRef]
- Neuman, B.W.; Kiss, G.; Kunding, A.H.; Bhella, D.; Baksh, M.F.; Connelly, S.; Droese, B.; Klaus, J.P.; Makino, S.; Sawicki, S.G.; et al. A structural analysis of M protein in coronavirus assembly and morphology. J. Struct. Biol. 2011, 174, 11–22. [Google Scholar] [CrossRef]
- Boson, B.; Legros, V.; Zhou, B.; Siret, E.; Mathieu, C.; Cosset, F.L.; Lavillette, D.; Denolly, S. The SARS-CoV-2 envelope and membrane proteins modulate maturation and retention of the spike protein, allowing assembly of virus-like particles. J. Biol. Chem. 2021, 296, 100111. [Google Scholar] [CrossRef]
- Lu, S.; Ye, Q.; Singh, D.; Cao, Y.; Diedrich, J.K.; Yates JR 3rd Villa, E.; Cleveland, D.W.; Corbett, K.D. The SARS-CoV-2 nucleocapsid phosphoprotein forms mutually exclusive condensates with RNA and the membrane-associated M protein. Nat. Commun. 2021, 12, 502. [Google Scholar] [CrossRef] [PubMed]
- de Breyne, S.; Vindry, C.; Guillin, O.; Condé, L.; Mure, F.; Gruffat, H.; Chavatte, L.; Ohlmann, T. Translational control of coronaviruses. Nucleic Acids Res. 2020, 48, 12502–12522. [Google Scholar] [CrossRef] [PubMed]
- V’Kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Malone, B.; Urakova, N.; Snijder, E.J.; Campbell, E.A. Structures and functions of coronavirus replication–transcription complexes and their relevance for SARS-CoV-2 drug design. Nat. Rev. Mol. Cell Biol. 2022, 23, 21–39. [Google Scholar] [CrossRef] [PubMed]
- Florindo, H.F.; Kleiner, R.; Vaskovich-Koubi, D.; Acúrcio, R.C.; Carreira, B.; Yeini, E.; Tiram, G.; Liubomirski, Y.; Satchi-Fainaro, R. Immune-mediated approaches against COVID-19. Nat. Nanotechnol. 2020, 15, 630–645. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.M.; An, J. Cytokines, inflammation, and pain. Int. Anesthesiol. Clin. 2007, 45, 27–37. [Google Scholar] [CrossRef] [Green Version]
- Zdravkovic, N.; Rosic, M.; Lutovac, M.; Zdravkovic, V. Physiology and Pathology of Cytokine: Commercial Production and Medical Use, Physiology and Pathology of Immunology. Nima Rezaei. IntechOpen. Available online: https://www.intechopen.com/chapters/57932 (accessed on 25 December 2021).
- Institute of Medicine (US) Committee on Military Nutrition Research. Military Strategies for Sustainment of Nutrition and Immune Function in the Field; National Academies Press: Washington, DC, USA, 1999. Available online: https://www.ncbi.nlm.nih.gov/books/NBK230989/ (accessed on 25 December 2021).
- Caldwell, A.B.; Cheng, Z.; Vargas, J.D.; Birnbaum, H.A.; Hoffmann, A. Network dynamics determine the autocrine and paracrine signaling functions of TNF. Genes Dev. 2014, 28, 2120–2133. [Google Scholar] [CrossRef] [Green Version]
- Jaffer, U.; Wade, R.G.; Gourlay, T. Cytokines in the systemic inflammatory response syndrome: A review. HSR Proc. Intensive Care Cardiovasc. Anesth. 2010, 2, 161–175. [Google Scholar]
- Palomino, D.C.T.; Marti, L.C. CChemokines and immunity. Einstein 2015, 13, 469–473. [Google Scholar] [CrossRef] [Green Version]
- Zav'ialov, V.P. Strukturno-funktsional’naia klassifikatsiia i évoliutsiia tsitokinov [The structural and functional classification and evolution of cytokines]. Vestn. Ross Akad. Med. Nauk. 1993, 2, 8–10. (In Russian) [Google Scholar]
- Fara, A.; Mitrev, Z.; Rosalia, R.A.; Assas, B.M. Cytokine storm and COVID-19: A chronicle of pro-inflammatory cytokines. Open Biol. 2020, 10, 200160. [Google Scholar] [CrossRef] [PubMed]
- Anoop, U.R.; Kavita, V. Cytokine Storm in COVID19: A Neural Hypothesis. ACS Chem. Neurosci. 2020, 11, 1868–1870. [Google Scholar] [CrossRef]
- Kosyreva, A.; Dzhalilova, D.; Lokhonina, A.; Vishnyakova, P.; Fatkhudinov, T. The Role of Macrophages in the Pathogenesis of SARS-CoV-2-Associated Acute Respiratory Distress Syndrome. Front. Immunol. 2021, 12, 682871. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Wu, L.; Yan, G.; Chen, Y.; Zhou, M.; Wu, Y.; Li, Y. Inflammation and tumor progression: Signaling pathways and targeted intervention. Signal Transduct. Target. Ther. 2021, 6, 263. [Google Scholar] [CrossRef]
- Devasthanam, A.S. Mechanisms underlying the inhibition of interferon signaling by viruses. Virulence 2014, 5, 270–277. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.J.; Ashkar, A.A. The Dual Nature of Type I and Type II Interferons. Front. Immunol. 2018, 9, 2061. [Google Scholar] [CrossRef] [Green Version]
- Samuel, C.E. Antiviral actions of interferons. Clin. Microbiol. Rev. 2001, 14, 778–809. [Google Scholar] [CrossRef] [Green Version]
- Rong, L.; Perelson, A.S. Treatment of Hepatitis C Virus Infection with Interferon and Small Molecule Direct Antivirals: Viral Kinetics and Modeling. Crit. Rev. Immunol. 2010, 30, 131–148. [Google Scholar] [CrossRef] [Green Version]
- Mu, X.; Liu, K.; Li, H.; Wang, F.-S.; Xu, R. Granulocyte-macrophage colony-stimulating factor: An immunotarget for sepsis and COVID-19. Cell. Mol. Immunol. 2021, 18, 2057–2058. [Google Scholar] [CrossRef]
- Takatsu, K. Cytokines involved in B-cell differentiation and their sites of action. Proc. Soc. Exp. Biol. Med. 1997, 215, 121–133. [Google Scholar] [CrossRef]
- Robb, L. Cytokine receptors and hematopoietic differentiation. Oncogene 2007, 26, 6715–6723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fajgenbaum, D.C.; June, C.H. Cytokine Storm. N. Engl. J. Med. 2020, 383, 2255–2273. [Google Scholar] [CrossRef] [PubMed]
- Rabaan, A.; Al-Ahmed, S.; Muhammad, J.; Khan, A.; Sule, A.; Tirupathi, R.; Mutair, A.; Alhumaid, S.; Al-Omari, A.; Dhawan, M.; et al. Role of Inflammatory Cytokines in COVID-19 Patients: A Review on Molecular Mechanisms, Immune Functions, Immunopathology and Immunomodulatory Drugs to Counter Cytokine Storm. Vaccines 2021, 9, 436. [Google Scholar] [CrossRef] [PubMed]
- Soto, J.A.; Gálvez, N.M.S.; Andrade, C.A.; Pacheco, G.A.; Bohmwald, K.; Berrios, R.V.; Bueno, S.M.; Kalergis, A.M. The Role of Dendritic Cells during Infections Caused by Highly Prevalent Viruses. Front. Immunol. 2020, 11, 1513. [Google Scholar] [CrossRef]
- López, C.B.; Moran, T.M.; Schulman, J.L.; Fernandez-Sesma, A. Antiviral immunity and the role of dendritic cells. Int. Rev. Immunol. 2002, 21, 339–353. [Google Scholar] [CrossRef]
- Cron, R.Q.; Chatham, W.W. The Rheumatologist’s Role in COVID-19. J. Rheumatol. 2020, 47, 639–642. [Google Scholar] [CrossRef]
- Björkström, N.K.; Ponzetta, A. Natural killer cells and unconventional T cells in COVID-19. Curr. Opin. Virol. 2021, 49, 176–182. [Google Scholar] [CrossRef]
- Copaescu, A.; Smibert, O.; Gibson, A.; Phillips, E.J.; Trubiano, J.A. The role of IL-6 and other mediators in the cytokine storm associated with SARS-CoV-2 infection. J. Allergy Clin. Immunol. 2020, 146, 518–534.e1. [Google Scholar] [CrossRef]
- Catanzaro, M.; Fagiani, F.; Racchi, M.; Corsini, E.; Govoni, S.; Lanni, C. Immune response in COVID-19: Addressing a pharmacological challenge by targeting pathways triggered by SARS-CoV-2. Signal Transduct. Target. Ther. 2020, 5, 84. [Google Scholar] [CrossRef]
- Caricchio, R.; Abbate, A.; Gordeev, I.; Meng, J.; Hsue, P.Y.; Neogi, T.; Arduino, R.; Fomina, D.; Bogdanov, R.; Stepanenko, T.; et al. Effect of Canakinumab vs Placebo on Survival Without Invasive Mechanical Ventilation in Patients Hospitalized With Severe COVID-19. JAMA 2021, 326, 230–239. [Google Scholar] [CrossRef]
- Gheblawi, M.; Wang, K.; Viveiros, A.; Nguyen, Q.; Zhong, J.-C.; Turner, A.J.; Raizada, M.K.; Grant, M.B.; Oudit, G.Y. Angiotensin-Converting Enzyme 2: SARS-CoV-2 Receptor and Regulator of the Renin-Angiotensin System: Celebrating the 20th Anniversary of the Discovery of ACE2. Circ. Res. 2020, 126, 1456–1474. [Google Scholar] [CrossRef] [PubMed]
- Tay, M.Z.; Poh, C.M.; Rénia, L.; Macary, P.A.; Ng, L.F.P. The trinity of COVID-19: Immunity, inflammation and intervention. Nat. Rev. Immunol. 2020, 20, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Ko, J.H.; Kim, Y.; Kim, Y.J.; Kim, J.M.; Chung, Y.S.; Kim, H.M.; Han, M.G.; Kim, S.Y.; Chin, B.S. Viral Load Kinetics of SARS-CoV-2 Infection in First Two Patients in Korea. J. Korean Med. Sci. 2020, 35, e86. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Zhang, D.; Yang, P.; Poon, L.L.M.; Wang, Q. Viral load of SARS-CoV-2 in clinical samples. Lancet Infect. Dis. 2020, 20, 411–412. [Google Scholar] [CrossRef]
- Lauer, S.A.; Grantz, K.H.; Bi, Q.; Jones, F.K.; Zheng, Q.; Meredith, H.R.; Azman, A.S.; Reich, N.G.; Lessler, J. The Incubation Period of Coronavirus Disease 2019 (COVID-19) From Publicly Reported Confirmed Cases: Estimation and Application. Ann. Intern. Med. 2020, 172, 577–582. [Google Scholar] [CrossRef] [Green Version]
- Peiris, J.S.M.; Chu, C.M.; Cheng, V.; Chan, K.; Hung, I.F.N.; Poon, L.; Law, K.; Tang, B.; Hon, T.; Chan, C.; et al. Clinical progression and viral load in a community outbreak of coronavirus-associated SARS pneumonia: A prospective study. Lancet 2003, 361, 1767–1772. [Google Scholar] [CrossRef] [Green Version]
- Zou, L.; Ruan, F.; Huang, M.; Liang, L.; Huang, H.; Hong, Z.; Yu, J.; Kang, M.; Song, Y.; Xia, J.; et al. SARS-CoV-2 Viral Load in Upper Respiratory Specimens of Infected Patients. N. Engl. J. Med. 2020, 382, 1177–1179. [Google Scholar] [CrossRef]
- Chu, H.; Chan, J.F.-W.; Wang, Y.; Yuen, T.T.-T.; Chai, Y.; Hou, Y.; Shuai, H.; Yang, D.; Hu, B.; Huang, X.; et al. Comparative Replication and Immune Activation Profiles of SARS-CoV-2 and SARS-CoV in Human Lungs: An Ex Vivo Study With Implications for the Pathogenesis of COVID-19. Clin. Infect. Dis. 2020, 71, 1400–1409. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Shi, L.; Wang, Y.; Zhang, J.; Huang, L.; Zhang, C.; Liu, S.; Zhao, P.; Liu, H.; Zhu, L.; et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir. Med. 2020, 8, 420–422. [Google Scholar] [CrossRef]
- Hui, K.P.Y.; Cheung, M.C.; Perera, R.A.P.M.; Ng, K.-C.; Bui, C.H.T.; Ho, J.C.W.; Ng, M.M.T.; Kuok, D.I.T.; Shih, K.C.; Tsao, S.-W.; et al. Tropism, replication competence, and innate immune responses of the coronavirus SARS-CoV-2 in human respiratory tract and conjunctiva: An analysis in ex-vivo and in-vitro cultures. Lancet Respir. Med. 2020, 8, 687–695. [Google Scholar] [CrossRef]
- Fajnzylber, J.; The Massachusetts Consortium for Pathogen Readiness; Regan, J.; Coxen, K.; Corry, H.; Wong, C.; Rosenthal, A.; Worrall, D.; Giguel, F.; Piechocka-Trocha, A.; et al. SARS-CoV-2 viral load is associated with increased disease severity and mortality. Nat. Commun. 2020, 11, 5493. [Google Scholar] [CrossRef] [PubMed]
- Castelli, V.; Cimini, A.; Ferri, C. Cytokine Storm in COVID-19: “When You Come Out of the Storm, You Won’t Be the Same Person Who Walked in”. Front. Immunol. 2020, 11, 2132. [Google Scholar] [CrossRef] [PubMed]
- Chousterman, B.G.; Swirski, F.; Weber, G.F. Cytokine storm and sepsis disease pathogenesis. Semin. Immunopathol. 2017, 39, 517–528. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Ye, F.; Wu, A.; Yang, R.; Pan, M.; Sheng, J.; Zhu, W.; Mao, L.; Wang, M.; Xia, Z.; et al. Comparative Transcriptome Analysis Reveals the Intensive Early-Stage Responses of Host Cells to SARS-CoV-2 Infection. Front. Microbiol. 2020, 11, 593857. [Google Scholar] [CrossRef] [PubMed]
- Olbei, M.; Hautefort, I.; Modos, D.; Treveil, A.; Poletti, M.; Gul, L.; Shannon-Lowe, C.D.; Korcsmaros, T. SARS-CoV-2 Causes a Different Cytokine Response Compared to Other Cytokine Storm-Causing Respiratory Viruses in Severely Ill Patients. Front. Immunol. 2021, 12, 629193. [Google Scholar] [CrossRef] [PubMed]
- Hojyo, S.; Uchida, M.; Tanaka, K.; Hasebe, R.; Tanaka, Y.; Murakami, M.; Hirano, T. How COVID-19 induces cytokine storm with high mortality. Inflamm. Regen. 2020, 40, 37. [Google Scholar] [CrossRef]
- Hirano, T.; Murakami, M. COVID-19: A New Virus, but a Familiar Receptor and Cytokine Release Syndrome. Immunity 2020, 52, 731–733. [Google Scholar] [CrossRef]
- Mahmudpour, M.; Roozbeh, J.; Keshavarz, M.; Farrokhi, S.; Nabipour, I. COVID-19 cytokine storm: The anger of inflammation. Cytokine 2020, 133, 155151. [Google Scholar] [CrossRef]
- McGonagle, D.; Sharif, K.; O’Regan, A.; Bridgewood, C. The Role of Cytokines including Interleukin-6 in COVID-19 induced Pneumonia and Macrophage Activation Syndrome-Like Disease. Autoimmun. Rev. 2020, 19, 102537. [Google Scholar] [CrossRef]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Liu, B.; Li, M.; Zhou, Z.; Guan, X.; Xiang, Y. Can we use interleukin-6 (IL-6) blockade for coronavirus disease 2019 (COVID-19)-induced cytokine release syndrome (CRS)? J. Autoimmun. 2020, 111, 102452. [Google Scholar] [CrossRef] [PubMed]
- Merad, M.; Martin, J.C. Pathological inflammation in patients with COVID-19: A key role for monocytes and macrophages. Nat. Rev. Immunol. 2020, 20, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Van Der Poll, T.; Van De Veerdonk, F.L.; Scicluna, B.; Netea, M.G. The immunopathology of sepsis and potential therapeutic targets. Nat. Rev. Immunol. 2017, 17, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Li, X.; Chen, M.; Feng, Y.; Xiong, C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc. Res. 2020, 116, 1097–1100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sedding, D.G.; Boyle, E.C.; Demandt, J.A.F.; Sluimer, J.C.; Dutzmann, J.; Haverich, A.; Bauersachs, J. Vasa Vasorum Angiogenesis: Key Player in the Initiation and Progression of Atherosclerosis and Potential Target for the Treatment of Cardiovascular Disease. Front. Immunol. 2018, 9, 706. [Google Scholar] [CrossRef] [Green Version]
- Rodrigue-Gervais, I.G.; Labbé, K.; Dagenais, M.; Dupaul-Chicoine, J.; Champagne, C.; Morizot, A.; Skeldon, A.; Brincks, E.L.; Vidal, S.M.; Griffith, T.S.; et al. Cellular Inhibitor of Apoptosis Protein cIAP2 Protects against Pulmonary Tissue Necrosis during Influenza Virus Infection to Promote Host Survival. Cell Host Microbe 2014, 15, 23–35. [Google Scholar] [CrossRef] [Green Version]
- Burgner, D.; Jamieson, S.E.; Blackwell, J.M. Genetic susceptibility to infectious diseases: Big is beautiful, but will bigger be even better? Lancet Infect. Dis. 2006, 6, 653–663. [Google Scholar] [CrossRef] [Green Version]
- Institute of Medicine (US) Committee on Assessing Interactions among Social, Behavioral, and Genetic Factors in Health. Genes, Behavior, and the Social Environment: Moving Beyond the Nature/Nurture Debate; Hernandez, L.M., Blazer, D.G., Eds.; National Academies Press: Washington, DC, USA, 2006. Available online: https://www.ncbi.nlm.nih.gov/books/NBK19932/ (accessed on 6 November 2021).
- Casanova, J.-L. Human genetic basis of interindividual variability in the course of infection. Proc. Natl. Acad. Sci. USA 2015, 112, E7118–E7127. [Google Scholar] [CrossRef] [Green Version]
- Casanova, J.-L.; Abel, L. Human genetics of infectious diseases: A unified theory. EMBO J. 2007, 26, 915–922. [Google Scholar] [CrossRef] [Green Version]
- Wijsman, E.M. Family-based approaches: Design, imputation, analysis, and beyond. BMC Genet. 2016, 17, 9. [Google Scholar] [CrossRef] [Green Version]
- Kazma, R.; Bailey, J.N. Population-based and family-based designs to analyze rare variants in complex diseases. Genet. Epidemiology 2011, 35, S41–S47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, K.M.; Machalaba, C.C.; Seifman, R.; Feferholtz, Y.; Karesh, W.B. Infectious disease and economics: The case for considering multi-sectoral impacts. One Health 2019, 7, 10008. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Vega, A.M.; Bueno, M.M.; Oparina, N.Y.; Herráez, D.L.; Kristjansdottir, H.; Steinsson, K.; Kozyrev, S.V.; Alarcón-Riquelme, M.E. Whole Exome Sequencing of Patients from Multicase Families with Systemic Lupus Erythematosus Identifies Multiple Rare Variants. Sci. Rep. 2018, 8, 8775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frodsham, A.J. Genetics of infectious diseases. Hum. Mol. Genet. 2004, 13, R187–R194. [Google Scholar] [CrossRef] [Green Version]
- MacArthur, J.; Bowler-Barnett, E.; Cerezo, M.; Gil, L.; Hall, P.; Hastings, E.; Junkins, H.; McMahon, A.; Milano, A.; Morales, J.; et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 2017, 45, D896–D901. [Google Scholar] [CrossRef]
- Hill, A.V.S. Aspects of Genetic Susceptibility to Human Infectious Diseases. Annu. Rev. Genet. 2006, 40, 469–486. [Google Scholar] [CrossRef]
- Mboowa, G.; Sserwadda, I.; Amujal, M.; Namatovu, N. Human Genomic Loci Important in Common Infectious Diseases: Role of High-Throughput Sequencing and Genome-Wide Association Studies. Can. J. Infect. Dis. Med Microbiol. 2018, 2018, 1875217. [Google Scholar] [CrossRef]
- Kwok, A.J.; Mentzer, A.; Knight, J.C. Host genetics and infectious disease: New tools, insights and translational opportunities. Nat. Rev. Genet. 2021, 22, 137–153. [Google Scholar] [CrossRef]
- Deng, H.; Yan, X.; Yuan, L. Human genetic basis of coronavirus disease 2019. Signal Transduct. Target. Ther. 2021, 6, 344. [Google Scholar] [CrossRef]
- Pairo-Castineira, E.; Clohisey, S.; Klaric, L.; Bretherick, A.D.; Rawlik, K.; Pasko, D.; Walker, S.; Parkinson, N.; Fourman, M.H.; Russell, C.D.; et al. Genetic mechanisms of critical illness in COVID-19. Nature 2021, 591, 92–98. [Google Scholar] [CrossRef]
- Novelli, A.; Andreani, M.; Biancolella, M.; Liberatoscioli, L.; Passarelli, C.; Colona, V.L.; Rogliani, P.; Leonardis, F.; Campana, A.; Carsetti, R.; et al. HLA allele frequencies and susceptibility to COVID -19 in a group of 99 Italian patients. HLA 2020, 96, 610–614. [Google Scholar] [CrossRef] [PubMed]
- COVID-19 Host Genetics Initiative. Mapping the human genetic architecture of COVID-19. Nature 2021, 600, 472–477. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.-C.; Chen, C.-H.; Wang, J.-H.; Liao, H.-C.; Yang, C.-T.; Chen, C.-W.; Lin, Y.-C.; Kao, C.-H.; Lu, M.-Y.J.; Liao, J.C. Analysis of genomic distributions of SARS-CoV-2 reveals a dominant strain type with strong allelic associations. Proc. Natl. Acad. Sci. USA 2020, 117, 30679–30686. [Google Scholar] [CrossRef] [PubMed]
- Shkurnikov, M.; Nersisyan, S.; Jankevic, T.; Galatenko, A.; Gordeev, I.; Vechorko, V.; Tonevitsky, A. Association of HLA Class I Genotypes with Severity of Coronavirus Disease-19. Front. Immunol. 2021, 12, 641900. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Zhang, Y.; Grosser, M.; Tipper, S.; Venter, D.; Lin, H.; Lu, J. Profiling COVID-19 Genetic Research: A Data-Driven Study Utilizing Intelligent Bibliometrics. Front. Res. Metrics Anal. 2021, 6, 683212. [Google Scholar] [CrossRef] [PubMed]
- Mahamat-Saleh, Y.; Fiolet, T.; Rebeaud, M.E.; Mulot, M.; Guihur, A.; El Fatouhi, D.; Laouali, N.; Peiffer-Smadja, N.; Aune, D.; Severi, G. Diabetes, hypertension, body mass index, smoking and COVID-19-related mortality: A systematic review and meta-analysis of observational studies. BMJ Open 2021, 11, e052777. [Google Scholar] [CrossRef]
- Leite, M.D.M.; Gonzalez-Galarza, F.F.; da Silva, B.C.C.; Middleton, D.; dos Santos, E.J.M. Predictive immunogenetic markers in COVID-19. Hum. Immunol. 2021, 82, 247–254. [Google Scholar] [CrossRef]
- Sakuraba, A.; Haider, H.; Sato, T. Population Difference in Allele Frequency of HLA-C*05 and Its Correlation with COVID-19 Mortality. Viruses 2020, 12, 1333. [Google Scholar] [CrossRef]
- Schmiedel, B.J.; Rocha, J.; Gonzalez-Colin, C.; Bhattacharyya, S.; Madrigal, A.; Ottensmeier, C.H.; Ay, F.; Chandra, V.; Vijayanand, P. COVID-19 genetic risk variants are associated with expression of multiple genes in diverse immune cell types. Nat. Commun. 2021, 12, 1–12. [Google Scholar] [CrossRef]
- Kaidashev, I.; Shlykova, O.; Izmailova, O.; Torubara, O.; Yushchenko, Y.; Tyshkovska, T.; Kyslyi, V.; Belyaeva, A.; Maryniak, D. Host gene variability and SARS-CoV-2 infection: A review article. Heliyon 2021, 7, e07863. [Google Scholar] [CrossRef]
- Anastassopoulou, C.; Gkizarioti, Z.; Patrinos, G.P.; Tsakris, A. Human genetic factors associated with susceptibility to SARS-CoV-2 infection and COVID-19 disease severity. Hum. Genom. 2020, 14, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Pollitt, K.J.G.; Peccia, J.; Ko, A.; Kaminski, N.; Cruz, C.S.D.; Nebert, D.W.; Reichardt, J.; Thompson, D.C.; Vasiliou, V. COVID-19 vulnerability: The potential impact of genetic susceptibility and airborne transmission. Hum. Genom. 2020, 14, 1–7. [Google Scholar]
- Fishchuk, L.; Rossokha, Z.; Pokhylko, V.; Cherniavska, Y.; Tsvirenko, S.; Kovtun, S.; Medvedieva, N.; Vershyhora, V.; Gorovenko, N. Modifying effects of TNF-α, IL-6 and VDR genes on the development risk and the course of COVID-19. Pilot study. Drug Metab. Pers. Ther. 2021. [Google Scholar] [CrossRef] [PubMed]
- Kucher, A.N.; Babushkina, N.P.; Sleptcov, A.A.; Nazarenko, M.S. Genetic control of human infection with SARS-CoV-2. Russ. J. Genet. 2021, 57, 627–641. [Google Scholar] [CrossRef]
- Zeberg, H.; Pääbo, S. A genomic region associated with protection against severe COVID-19 is inherited from Neandertals. Proc. Natl. Acad. Sci. USA 2021, 118, e2026309118. [Google Scholar] [CrossRef]
- Zeberg, H.; Pääbo, S. The major genetic risk factor for severe COVID-19 is inherited from Neanderthals. Nature 2020, 587, 610–612. [Google Scholar] [CrossRef]
- Karolinska Institutet. Neandertal Gene Variants both Increase and Decrease the Risk for Severe COVID-19. ScienceDaily. 16 February 2021. Available online: www.sciencedaily.com/releases/2021/02/210216144328.htm (accessed on 28 November 2021).
- Barmania, F.; Mellet, J.; Ryder, M.A.; Ford, G.; Herd, C.L.; Tamuhla, T.; Hendricks, C.; Giles, R.; Kalua, T.; Joubert, F.; et al. Coronavirus Host Genetics South Africa (COHG-SA) database-a variant database for gene regions associated with SARS-CoV-2 outcomes. Eur. J. Hum. Genet. 2022, 29, 1–9. [Google Scholar] [CrossRef]
- Andreakos, E.; Abel, L.; Vinh, D.C.; Kaja, E.; Drolet, B.A.; Zhang, Q.; O’Farrelly, C.; Novelli, G.; Rodríguez-Gallego, C.; Haerynck, F.; et al. A global effort to dissect the human genetic basis of resistance to SARS-CoV-2 infection. Nat. Immunol. 2021, 23, 159–164. [Google Scholar] [CrossRef]
- Shelton, J.F.; Shastri, A.J.; Ye, C.; Weldon, C.H.; Filshtein-Sonmez, T.; Coker, D.; Symons, A.; Esparza-Gordillo, J.; Aslibekyan, S.; Auton, A.; et al. Trans-ancestry analysis reveals genetic and nongenetic associations with COVID-19 susceptibility and severity. Nat. Genet. 2021, 53, 801–808. [Google Scholar] [CrossRef]
- Severe Covid-19 GWAS Group; Ellinghaus, D.; Degenhardt, F.; Bujanda, L.; Buti, M.; Albillos, A.; Invernizzi, P.; Fernández, J.; Prati, D. Genomewide Association Study of Severe Covid-19 with Respiratory Failure. N. Engl. J. Med. 2020, 383, 1522–1534. [Google Scholar] [CrossRef]
- Liu, J.; Li, S.; Liu, J.; Liang, B.; Wang, X.; Wang, H.; Li, W.; Tong, Q.; Yi, J.; Zhao, L.; et al. Longitudinal characteristics of lymphocyte responses and cytokine profiles in the peripheral blood of SARS-CoV-2 infected patients. EBioMedicine 2020, 55, 102763. [Google Scholar] [CrossRef]
- Lin, G.-L.; McGinley, J.P.; Drysdale, S.B.; Pollard, A.J. Epidemiology and Immune Pathogenesis of Viral Sepsis. Front. Immunol. 2018, 9, 2147. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; McGoogan, J.M. Characteristics of and Important Lessons from the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72 314 Cases from the Chinese Center for Disease Control and Prevention. JAMA 2020, 323, 1239–1242. [Google Scholar] [CrossRef] [PubMed]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.-D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Maiese, A.; Manetti, A.C.; La Russa, R.; Di Paolo, M.; Turillazzi, E.; Frati, P.; Fineschi, V. Autopsy findings in COVID-19-related deaths: A literature review. Forensic Sci. Med. Pathol. 2021, 17, 279–296. [Google Scholar] [CrossRef]
- Gu, S.X.; Tyagi, T.; Jain, K.; Gu, V.W.; Lee, S.H.; Hwa, J.M.; Kwan, J.M.; Krause, D.S.; Lee, A.I.; Halene, S.; et al. Thrombocytopathy and endotheliopathy: Crucial contributors to COVID-19 thromboinflammation. Nat. Rev. Cardiol. 2021, 18, 194–209. [Google Scholar] [CrossRef]
- Maiese, A.; Passaro, G.; De Matteis, A.; Fazio, V.; Raffaele, L.R.; Di Paolo, M. Thromboinflammatory response in SARS-CoV-2 sepsis. Medico-Legal J. 2020, 88, 78–80. [Google Scholar] [CrossRef]
- Rahal, A.; Kumar, A.; Singh, V.; Yadav, B.; Tiwari, R.; Chakraborty, S.; Dhama, K. Oxidative Stress, Prooxidants, and Antioxidants: The Interplay. BioMed Res. Int. 2014, 2014, 761264. [Google Scholar] [CrossRef] [Green Version]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell. Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Snezhkina, A.V.; Kudryavtseva, A.V.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS Generation and Antioxidant Defense Systems in Normal and Malignant Cells. Oxidative Med. Cell. Longev. 2019, 2019, 6175804. [Google Scholar] [CrossRef]
- Li, R.; Jia, Z.; Trush, M.A. Defining ROS in biology and medicine. React. Oxyg. Species 2016, 1, 9–21. [Google Scholar] [CrossRef] [Green Version]
- Suhail, S.; Zajac, J.; Fossum, C.; Lowater, H.; McCracken, C.; Severson, N.; Laatsch, B.; Narkiewicz-Jodko, A.; Johnson, B.; Liebau, J.; et al. Role of Oxidative Stress on SARS-CoV (SARS) and SARS-CoV-2 (COVID-19) Infection: A Review. J. Protein Chem. 2020, 39, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Bazhin, A.V.; Werner, J.; Karakhanova, S. Reactive Oxygen Species in the Immune System. Int. Rev. Immunol. 2013, 32, 249–270. [Google Scholar] [CrossRef] [PubMed]
- Chernyak, B.V.; Popova, E.N.; Prikhodko, A.S.; Grebenchikov, O.A.; Zinovkina, L.A.; Zinovkin, R.A. COVID-19 and Oxidative Stress. Biochemistry 2020, 85, 1543–1553. [Google Scholar] [CrossRef] [PubMed]
- Jafarzadeh, A.; Chauhan, P.; Saha, B.; Jafarzadeh, S.; Nemati, M. Contribution of monocytes and macrophages to the local tissue inflammation and cytokine storm in COVID-19: Lessons from SARS and MERS, and potential therapeutic interventions. Life Sci. 2020, 257, 118102. [Google Scholar] [CrossRef] [PubMed]
- Hanidziar, D.; Robson, S.C. Hyperoxia and modulation of pulmonary vascular and immune responses in COVID-19. Am. J. Physiol. Cell. Mol. Physiol. 2021, 320, L12–L16. [Google Scholar] [CrossRef]
- Fernandes, I.G.; De Brito, C.A.; Dos Reis, V.M.S.; Sato, M.N.; Pereira, N.Z. SARS-CoV-2 and Other Respiratory Viruses: What Does Oxidative Stress Have to Do with It? Oxidative Med. Cell. Longev. 2020, 2020, 8844280. [Google Scholar] [CrossRef]
- Liu, K.; Chen, Y.; Lin, R.; Han, K. Clinical features of COVID-19 in elderly patients: A comparison with young and middle-aged patients. J. Infect. 2020, 80, e14–e18. [Google Scholar] [CrossRef] [Green Version]
- Williamson, E.J.; Walker, A.J.; Bhaskaran, K.; Bacon, S.; Bates, C.; Morton, C.E.; Curtis, H.J.; Mehrkar, A.; Evans, D.; Inglesby, P.; et al. Factors associated with COVID-19-related death using OpenSAFELY. Nature 2020, 584, 430–436. [Google Scholar] [CrossRef]
- Diamond, M.S.; Kanneganti, T.D. Innate immunity: The first line of defense against SARS-CoV-2. Nat. Immunol. 2022, 23, 165–176. [Google Scholar] [CrossRef]
- Kanneganti, T.D. Intracellular innate immune receptors: Life inside the cell. Immunol Rev. 2020, 297, 5–12. [Google Scholar] [CrossRef]
- Jang, J.H.; Shin, H.W.; Lee, J.M.; Lee, H.W.; Kim, E.C.; Park, S.H. An Overview of Pathogen Recognition Receptors for Innate Immunity in Dental Pulp. Mediat. Inflamm. 2015, 2015, 794143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suresh, R.; Mosser, D.M. Pattern recognition receptors in innate immunity, host defense, and immunopathology. Adv. Physiol. Educ. 2013, 37, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labarrere, C.A.; Kassab, G.S. Pattern Recognition Proteins: First Line of Defense against Coronaviruses. Front. Immunol. 2021, 12, 652252. [Google Scholar] [CrossRef]
- Kim, W.Y.; Kweon, O.J.; Cha, M.J.; Baek, M.S.; Choi, S.H. Dexamethasone may improve severe COVID-19 via ameliorating endothelial injury and inflammation: A preliminary pilot study. PLoS ONE. 2021, 16, e0254167. [Google Scholar] [CrossRef]
- Saeedi-Boroujeni, A.; Mahmoudian-Sani, M.-R. Anti-inflammatory potential of Quercetin in COVID-19 treatment. J. Inflamm. 2021, 18, 3. [Google Scholar] [CrossRef]
- Pelaia, C.; Calabrese, C.; Garofalo, E.; Bruni, A.; Vatrella, A.; Pelaia, G. Therapeutic Role of Tocilizumab in SARS-CoV-2-Induced Cytokine Storm: Rationale and Current Evidence. Int. J. Mol. Sci. 2021, 22, 3059. [Google Scholar] [CrossRef]
- Heimfarth, L.; Serafini, M.R.; Martins-Filho, P.R.; Quintans, L.; Quintans-Júnior, L.J. Drug repurposing and cytokine management in response to COVID-19: A review. Int. Immunopharmacol. 2020, 88, 106947. [Google Scholar] [CrossRef]
- Ali, L. Cytokines mediated hyperinflammation in SARS-CoV2: An Overview. Life Sci. 2020, 1, 57–63. [Google Scholar] [CrossRef]
- Mishra, K.P.; Singh, A.K.; Singh, S.B. Hyperinflammation and Immune Response Generation in COVID-19. Neuroimmunomodulation 2020, 27, 80–86. [Google Scholar] [CrossRef]
- Cardone, M.; Yano, M.; Rosenberg, A.S.; Puig, M. Lessons Learned to Date on COVID-19 Hyperinflammatory Syndrome: Considerations for Interventions to Mitigate SARS-CoV-2 Viral Infection and Detrimental Hyperinflammation. Front. Immunol. 2020, 11, 1131. [Google Scholar] [CrossRef] [PubMed]
- Long, Q.-X.; Liu, B.-Z.; Deng, H.-J.; Wu, G.-C.; Deng, K.; Chen, Y.-K.; Liao, P.; Qiu, J.-F.; Lin, Y.; Cai, X.-F.; et al. Antibody responses to SARS-CoV-2 in patients with COVID-19. Nat. Med. 2020, 26, 845–848. [Google Scholar] [CrossRef] [PubMed]
- Iyer, A.S.; Jones, F.K.; Nodoushani, A.; Kelly, M.; Becker, M.; Slater, D.; Mills, R.; Teng, E.; Kamruzzaman, M.; Garcia-Beltran, W.F.; et al. Persistence and decay of human antibody responses to the receptor binding domain of SARS-CoV-2 spike protein in COVID-19 patients. Sci. Immunol. 2020, 5, 367. [Google Scholar] [CrossRef] [PubMed]
- Marchi, S.; Viviani, S.; Remarque, E.J.; Ruello, A.; Bombardieri, E.; Bollati, V.; Milani, G.P.; Manenti, A.; Lapini, G.; Rebuffat, A.; et al. Characterization of antibody response in asymptomatic and symptomatic SARS-CoV-2 infection. PLoS ONE. 2021, 16, e0253977. [Google Scholar] [CrossRef]
- Bao, C.; Tao, X.; Cui, W.; Hao, Y.; Zheng, S.; Yi, B.; Pan, T.; Young, K.H.; Qian, W. Natural killer cells associated with SARS-CoV-2 viral RNA shedding, antibody response and mortality in COVID-19 patients. Exp. Hematol. Oncol. 2021, 10, 5. [Google Scholar] [CrossRef]
- Kim, J.Y.; Kwon, J.-S.; Bae, S.; Cha, H.H.; Lim, J.S.; Kim, M.-C.; Chung, J.-W.; Park, S.Y.; Lee, M.J.; Kim, B.-N.; et al. SARS-CoV-2-Specific Antibody and T Cell Response Kinetics According to Symptom Severity. Am. J. Trop. Med. Hygiene 2021, 105, 395–400. [Google Scholar] [CrossRef]
- Boonyaratanakornkit, J.; Morishima, C.; Selke, S.; Zamora, D.; McGuffin, S.A.; Shapiro, A.E.; Campbell, V.L.; McClurkan, C.L.; Jing, L.; Gross, R.; et al. Clinical, laboratory, and temporal predictors of neutralizing antibodies against SARS-CoV-2 among COVID-19 convalescent plasma donor candidates. J. Clin. Investig. 2021, 131, 144930. [Google Scholar] [CrossRef]
- Shirin, T.; Bhuiyan, T.R.; Charles, R.C.; Amin, S.; Bhuiyan, I.; Kawser, Z.; Rahat, A.; Alam, A.N.; Sultana, S.; Aleem, M.A.; et al. Antibody responses after COVID-19 infection in patients who are mildly symptomatic or asymptomatic in Bangladesh. Int. J. Infect Dis. 2020, 101, 220–225. [Google Scholar] [CrossRef]
- Wan, Y.; Shang, J.; Sun, S.; Tai, W.; Chen, J.; Geng, Q.; He, L.; Chen, Y.; Wu, J.; Shi, Z.; et al. Molecular Mechanism for Antibody-Dependent Enhancement of Coronavirus Entry. J Virol. 2020, 94, e02015-19. [Google Scholar] [CrossRef] [Green Version]
- Chow, E.J. The Multisystem Inflammatory Syndrome in Adults with SARS-CoV-2 Infection-Another Piece of an Expanding Puzzle. JAMA Netw. Open. 2021, 4, e2110344. [Google Scholar] [CrossRef]
- Sui, J.; Noubouossie, D.F.; Gandotra, S.; Cao, L. Elevated Plasma Fibrinogen Is Associated With Excessive Inflammation and Disease Severity in COVID-19 Patients. Front. Cell Infect Microbiol. 2021, 11, 734005. [Google Scholar] [CrossRef] [PubMed]
- Nugroho, J.; Wardhana, A.; Mulia, E.P.; Maghfirah, I.; Rachmi, D.A.; A’yun, M.Q.; Septianda, I. Elevated fibrinogen and fibrin degradation product are associated with poor outcome in COVID-19 patients: A meta-analysis. Clin. Hemorheol. Microcirc. 2021, 77, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Landewé, R.B.M.; Ramiro, S.; Mostard, R.L.M. COVID-19-induced hyper inflammation, immunosuppression, recovery, and survival: How causal inference may help draw robust conclusions. RMD Open 2021, 7, e001638. [Google Scholar] [CrossRef] [PubMed]
- Puzyrenko, A.; Felix, J.C.; Sun, Y.; Rui, H.; Sheinin, Y. Acute SARS-CoV-2 pneumonitis with cytotoxic CD8 positive T-lymphocytes: Case report and review of the literature. Pathol. Res. Pract. 2021, 220, 153380. [Google Scholar] [CrossRef]
- Kaneko, N.; Boucau, J.; Kuo, H.H.; Perugino, C.; Mahajan, V.S.; Farmer, J.R.; Liu, H.; Diefenbach, T.J.; Piechocka-Trocha, A.; Lefteri, K.; et al. Expansion of Cytotoxic CD4+ T cells in the lungs in severe COVID-19. medRxiv 2021. [Google Scholar] [CrossRef]
- Westmeier, J.; Paniskaki, K.; Karaköse, Z.; Werner, T.; Sutter, K.; Dolff, S.; Overbeck, M.; Limmer, A.; Liu, J.; Zheng, X.; et al. Impaired Cytotoxic CD8+ T Cell Response in Elderly COVID-19 Patients. mBio 2020, 11, e02243-20. [Google Scholar] [CrossRef]
- Nienhold, R.; Ciani, Y.; Koelzer, V.H.; Tzankov, A.; Haslbauer, J.D.; Menter, T.; Schwab, N.; Henkel, M.; Frank, A.; Zsikla, V.; et al. Two distinct immunopathological profiles in autopsy lungs of COVID-19. Nat. Commun. 2020, 11, 5086. [Google Scholar] [CrossRef]
- Wang, C.; Xie, J.; Zhao, L.; Fei, X.; Zhang, H.; Tan, Y.; Nie, X.; Zhou, L.; Liu, Z.; Ren, Y.; et al. Alveolar macrophage dysfunction and cytokine storm in the pathogenesis of two severe COVID-19 patients. EBioMedicine 2020, 57, 102833. [Google Scholar] [CrossRef]
- Janssen, M.T.H.F.; Ramiro, S.; Landewé, R.B.M.; Magro-Checa, C.; Mostard, R.L.M. Antibody response to SARS-CoV-2 in patients receiving glucocorticoids with or without tocilizumab for COVID-19-associated hyperinflammation. Ann. Rheum. Dis. 2021, 80, 1362–1363. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rabiu Abubakar, A.; Ahmad, R.; Rowaiye, A.B.; Rahman, S.; Iskandar, K.; Dutta, S.; Oli, A.N.; Dhingra, S.; Tor, M.A.; Etando, A.; et al. Targeting Specific Checkpoints in the Management of SARS-CoV-2 Induced Cytokine Storm. Life 2022, 12, 478. https://doi.org/10.3390/life12040478
Rabiu Abubakar A, Ahmad R, Rowaiye AB, Rahman S, Iskandar K, Dutta S, Oli AN, Dhingra S, Tor MA, Etando A, et al. Targeting Specific Checkpoints in the Management of SARS-CoV-2 Induced Cytokine Storm. Life. 2022; 12(4):478. https://doi.org/10.3390/life12040478
Chicago/Turabian StyleRabiu Abubakar, Abdullahi, Rahnuma Ahmad, Adekunle Babajide Rowaiye, Sayeeda Rahman, Katia Iskandar, Siddhartha Dutta, Angus Nnamdi Oli, Sameer Dhingra, Maryam Abba Tor, Ayukafangha Etando, and et al. 2022. "Targeting Specific Checkpoints in the Management of SARS-CoV-2 Induced Cytokine Storm" Life 12, no. 4: 478. https://doi.org/10.3390/life12040478
APA StyleRabiu Abubakar, A., Ahmad, R., Rowaiye, A. B., Rahman, S., Iskandar, K., Dutta, S., Oli, A. N., Dhingra, S., Tor, M. A., Etando, A., Kumar, S., Irfan, M., Gowere, M., Chowdhury, K., Akter, F., Jahan, D., Schellack, N., & Haque, M. (2022). Targeting Specific Checkpoints in the Management of SARS-CoV-2 Induced Cytokine Storm. Life, 12(4), 478. https://doi.org/10.3390/life12040478