
The Platelet-Derived Growth Factor Pathway in Pulmonary Arterial Hypertension: Still an Interesting Target?


 , and
, and 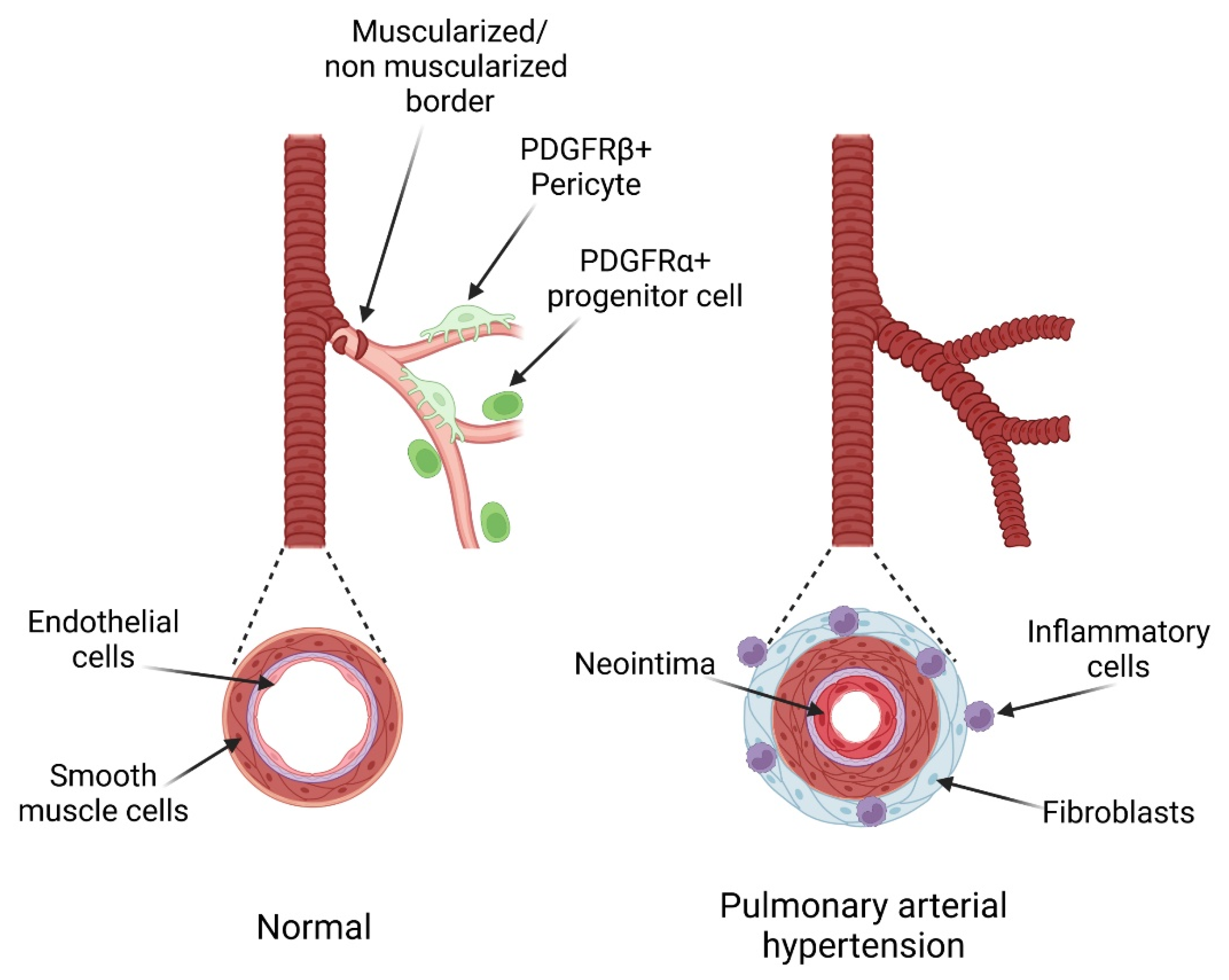{kind=link}
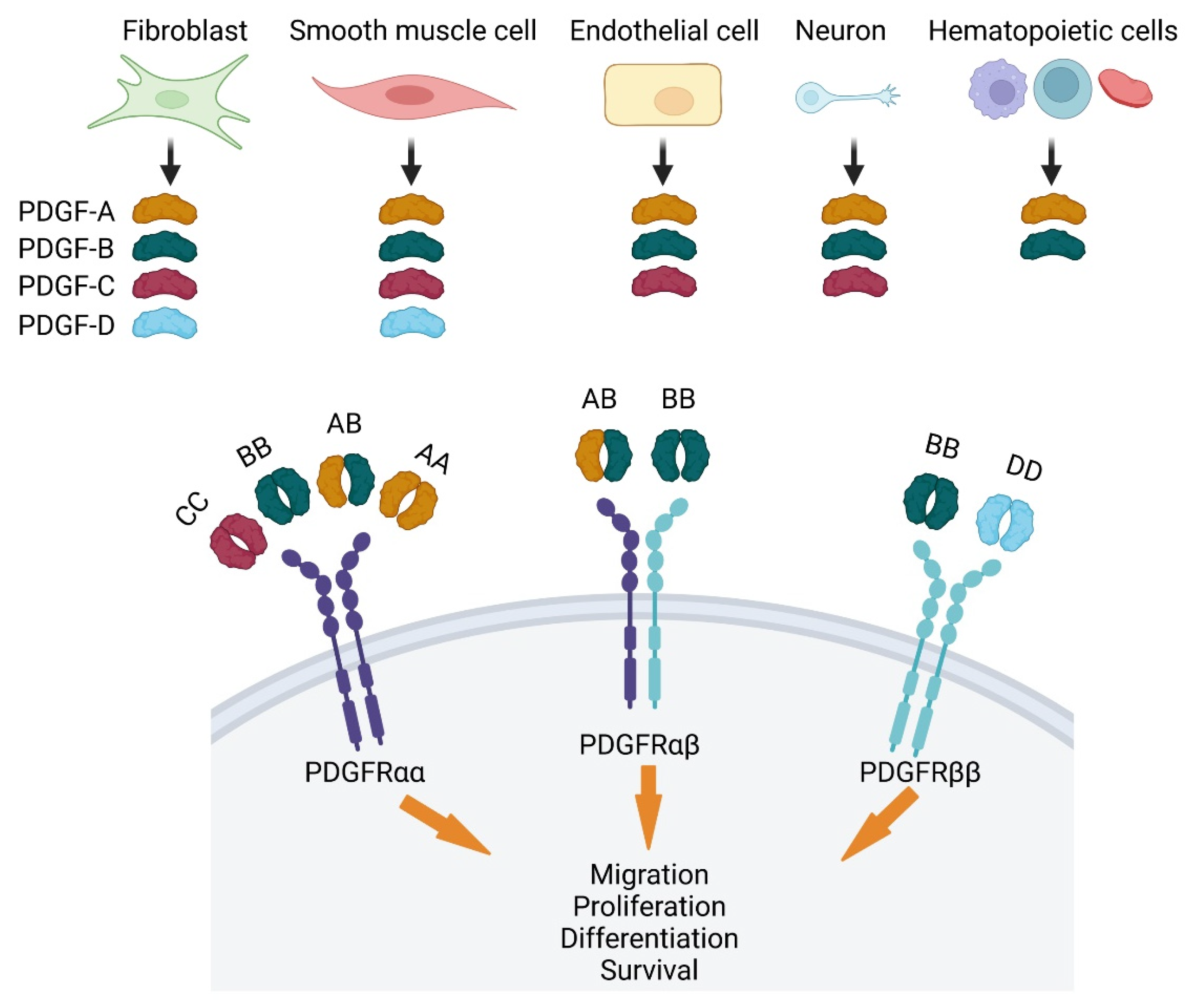{kind=link}
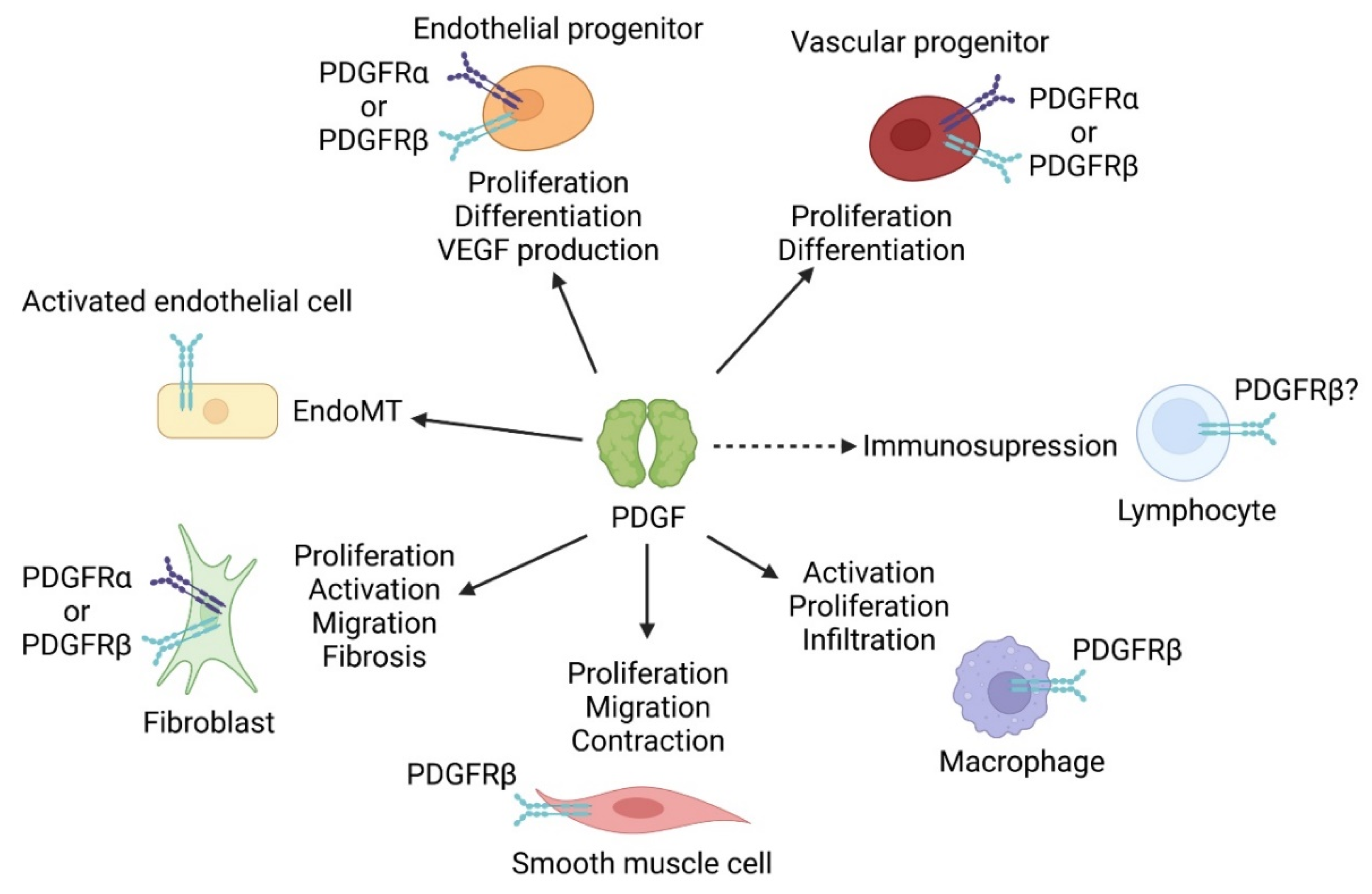{kind=link}
{kind=link}
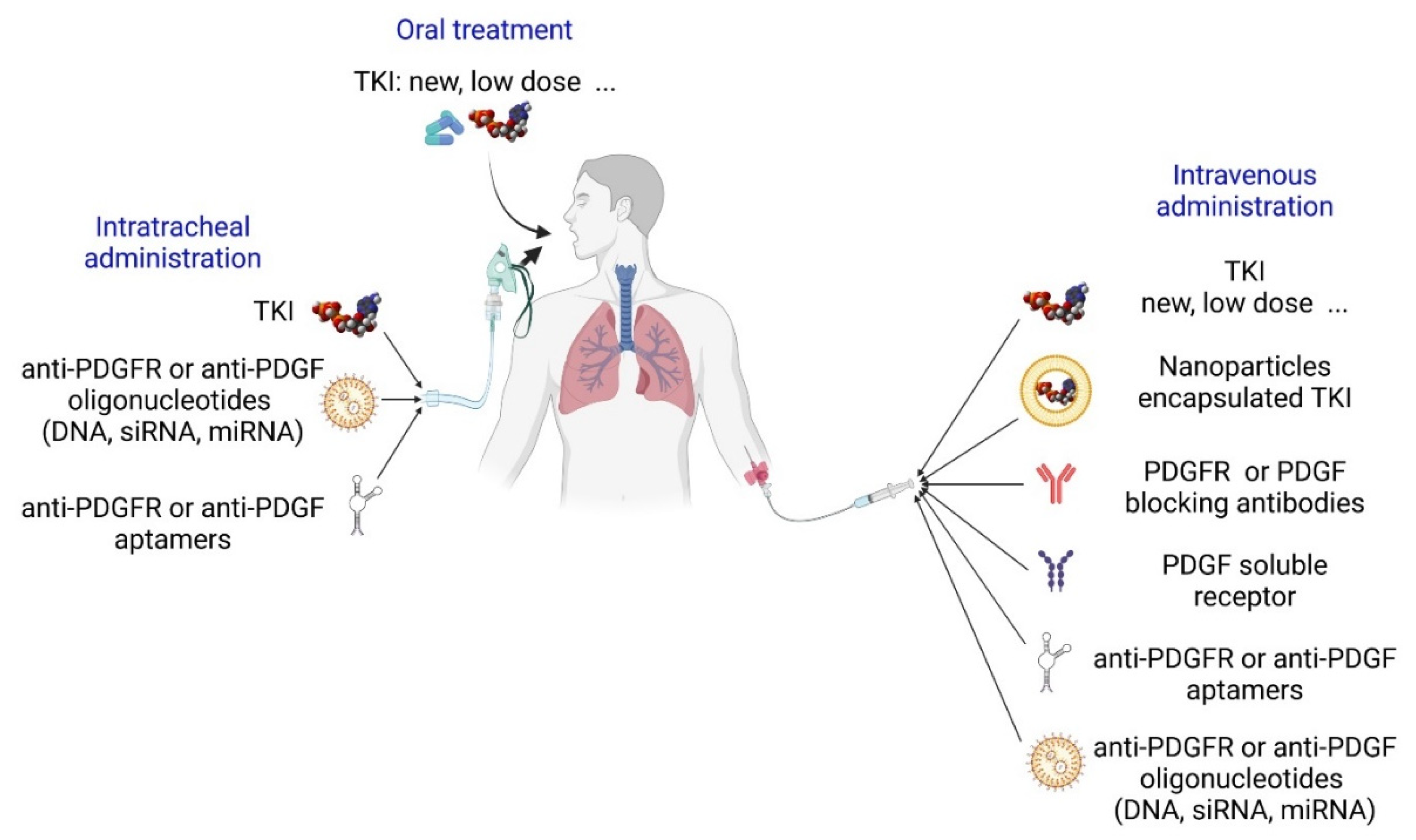{kind=link}
Abstract
:1. Introduction
2. PDGF Ligands and Their Receptors PDGFR
3. PDGF Effects on Pulmonary Cells
3.1. PDGF and Pulmonary Endothelial Cells
3.2. PDGF and Pulmonary Arterial Smooth Muscle Cells
3.3. PDGF and Pulmonary Fibroblasts
3.4. PDGF and Pulmonary Vascular Smooth Muscle Progenitor Cells
3.5. PDGF and Inflammatory Cells
4. Role and Regulation of the PDGF Pathway in PH
5. Clinical Assessment of Therapies Targeting the PDGF Pathway in PAH Patients
6. Potential Future Therapies Targeting the PDGF Pathway in PAH Patients
6.1. Receptor Tyrosine Kinase Inhibitors
6.2. Specific PDGF/PDGFR Inhibitors
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ma, L.; Chung, W.K. The Genetic Basis of Pulmonary Arterial Hypertension. Hum. Genet. 2014, 133, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Humbert, M.; Guignabert, C.; Bonnet, S.; Dorfmüller, P.; Klinger, J.R.; Nicolls, M.R.; Olschewski, A.J.; Pullamsetti, S.S.; Schermuly, R.T.; Stenmark, K.R.; et al. Pathology and Pathobiology of Pulmonary Hypertension: State of the Art and Research Perspectives. Eur. Respir. J. 2019, 53, 1801887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schermuly, R.T. Reversal of Experimental Pulmonary Hypertension by PDGF Inhibition. J. Clin. Investig. 2005, 115, 2811–2821. [Google Scholar] [CrossRef] [Green Version]
- Perros, F.; Montani, D.; Dorfmüller, P.; Durand-Gasselin, I.; Tcherakian, C.; Le Pavec, J.; Mazmanian, M.; Fadel, E.; Mussot, S.; Mercier, O.; et al. Platelet-Derived Growth Factor Expression and Function in Idiopathic Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2008, 178, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Frost, A.E.; Barst, R.J.; Hoeper, M.M.; Chang, H.-J.; Frantz, R.P.; Fukumoto, Y.; Galié, N.; Hassoun, P.M.; Klose, H.; Matsubara, H.; et al. Long-Term Safety and Efficacy of Imatinib in Pulmonary Arterial Hypertension. J. Heart Lung Transplant. 2015, 34, 1366–1375. [Google Scholar] [CrossRef] [Green Version]
- Zhu, N.; Swietlik, E.M.; Welch, C.L.; Pauciulo, M.W.; Hagen, J.J.; Zhou, X.; Guo, Y.; Karten, J.; Pandya, D.; Tilly, T.; et al. Rare Variant Analysis of 4241 Pulmonary Arterial Hypertension Cases from an International Consortium Implicates FBLN2, PDGFD, and Rare de Novo Variants in PAH. Genome Med. 2021, 13, 80. [Google Scholar] [CrossRef]
- Donovan, J.; Abraham, D.; Norman, J. Platelet-Derived Growth Factor Signaling in Mesenchymal Cells. Front. Biosci. 2013, 18, 106–119. [Google Scholar] [CrossRef] [Green Version]
- Andrae, J.; Gallini, R.; Betsholtz, C. Role of Platelet-Derived Growth Factors in Physiology and Medicine. Genes Dev. 2008, 22, 1276–1312. [Google Scholar] [CrossRef] [Green Version]
- Kaminski, W.E.; Lindahl, P.; Lin, N.L.; Broudy, V.C.; Crosby, J.R.; Hellström, M.; Swolin, B.; Bowen-Pope, D.F.; Martin, P.J.; Ross, R.; et al. Basis of Hematopoietic Defects in Platelet-Derived Growth Factor (PDGF)-B and PDGF Beta-Receptor Null Mice. Blood 2001, 97, 1990–1998. [Google Scholar] [CrossRef] [Green Version]
- Noskovičová, N.; Petřek, M.; Eickelberg, O.; Heinzelmann, K. Platelet-Derived Growth Factor Signaling in the Lung. From Lung Development and Disease to Clinical Studies. Am. J. Respir. Cell Mol. Biol. 2015, 52, 263–284. [Google Scholar] [CrossRef]
- Fredriksson, L.; Li, H.; Eriksson, U. The PDGF Family: Four Gene Products Form Five Dimeric Isoforms. Cytokine Growth Factor Rev. 2004, 15, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Li, X. Platelet-Derived Growth Factor-C and -D in the Cardiovascular System and Diseases. Mol. Asp. Med. 2018, 62, 12–21. [Google Scholar] [CrossRef]
- Bergsten, E.; Uutela, M.; Li, X.; Pietras, K.; Ostman, A.; Heldin, C.H.; Alitalo, K.; Eriksson, U. PDGF-D Is a Specific, Protease-Activated Ligand for the PDGF Beta-Receptor. Nat. Cell Biol. 2001, 3, 512–516. [Google Scholar] [CrossRef] [PubMed]
- Ustach, C.V.; Kim, H.-R.C. Platelet-Derived Growth Factor D Is Activated by Urokinase Plasminogen Activator in Prostate Carcinoma Cells. Mol. Cell. Biol. 2005, 25, 6279–6288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazlauskas, A. PDGFs and Their Receptors. Gene 2017, 614, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.-H. Targeting the PDGF Signaling Pathway in Tumor Treatment. Cell Commun. Signal. CCS 2013, 11, 97. [Google Scholar] [CrossRef] [Green Version]
- Heldin, C.H.; Westermark, B. Mechanism of Action and in Vivo Role of Platelet-Derived Growth Factor. Physiol. Rev. 1999, 79, 1283–1316. [Google Scholar] [CrossRef]
- Montero, P.; Milara, J.; Roger, I.; Cortijo, J. Role of JAK/STAT in Interstitial Lung Diseases; Molecular and Cellular Mechanisms. Int. J. Mol. Sci. 2021, 22, 6211. [Google Scholar] [CrossRef]
- Rogers, M.A.; Fantauzzo, K.A. The Emerging Complexity of PDGFRs: Activation, Internalization and Signal Attenuation. Biochem. Soc. Trans. 2020, 48, 1167–1176. [Google Scholar] [CrossRef]
- Pennock, S.; Haddock, L.J.; Eliott, D.; Mukai, S.; Kazlauskas, A. Is Neutralizing Vitreal Growth Factors a Viable Strategy to Prevent Proliferative Vitreoretinopathy? Prog. Retin. Eye Res. 2014, 40, 16–34. [Google Scholar] [CrossRef]
- Lei, H.; Kazlauskas, A. Growth Factors Outside of the Platelet-Derived Growth Factor (PDGF) Family Employ Reactive Oxygen Species/Src Family Kinases to Activate PDGF Receptor Alpha and Thereby Promote Proliferation and Survival of Cells. J. Biol. Chem. 2009, 284, 6329–6336. [Google Scholar] [CrossRef] [Green Version]
- Pennock, S.; Kazlauskas, A. Vascular Endothelial Growth Factor A Competitively Inhibits Platelet-Derived Growth Factor (PDGF)-Dependent Activation of PDGF Receptor and Subsequent Signaling Events and Cellular Responses. Mol. Cell. Biol. 2012, 32, 1955–1966. [Google Scholar] [CrossRef] [Green Version]
- Rolny, C.; Nilsson, I.; Magnusson, P.; Armulik, A.; Jakobsson, L.; Wentzel, P.; Lindblom, P.; Norlin, J.; Betsholtz, C.; Heuchel, R.; et al. Platelet-Derived Growth Factor Receptor-Beta Promotes Early Endothelial Cell Differentiation. Blood 2006, 108, 1877–1886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, S.; Yu, H.; Chen, X.; Shen, T.; Cui, Z.; Si, G.; Zhang, J.; Cheng, Y.; Jia, S.; Song, S.; et al. PDGF-BB/KLF4/VEGF Signaling Axis in Pulmonary Artery Endothelial Cell Angiogenesis. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2017, 41, 2333–2349. [Google Scholar] [CrossRef] [PubMed]
- Kemp, S.S.; Aguera, K.N.; Cha, B.; Davis, G.E. Defining Endothelial Cell-Derived Factors That Promote Pericyte Recruitment and Capillary Network Assembly. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2632–2648. [Google Scholar] [CrossRef]
- Gerhardt, H.; Golding, M.; Fruttiger, M.; Ruhrberg, C.; Lundkvist, A.; Abramsson, A.; Jeltsch, M.; Mitchell, C.; Alitalo, K.; Shima, D.; et al. VEGF Guides Angiogenic Sprouting Utilizing Endothelial Tip Cell Filopodia. J. Cell Biol. 2003, 161, 1163–1177. [Google Scholar] [CrossRef]
- Bjarnegård, M.; Enge, M.; Norlin, J.; Gustafsdottir, S.; Fredriksson, S.; Abramsson, A.; Takemoto, M.; Gustafsson, E.; Fässler, R.; Betsholtz, C. Endothelium-Specific Ablation of PDGFB Leads to Pericyte Loss and Glomerular, Cardiac and Placental Abnormalities. Development 2004, 131, 1847–1857. [Google Scholar] [CrossRef] [Green Version]
- Gianni-Barrera, R.; Butschkau, A.; Uccelli, A.; Certelli, A.; Valente, P.; Bartolomeo, M.; Groppa, E.; Burger, M.G.; Hlushchuk, R.; Heberer, M.; et al. PDGF-BB Regulates Splitting Angiogenesis in Skeletal Muscle by Limiting VEGF-Induced Endothelial Proliferation. Angiogenesis 2018, 21, 883–900. [Google Scholar] [CrossRef] [Green Version]
- Song, S.; Zhang, M.; Yi, Z.; Zhang, H.; Shen, T.; Yu, X.; Zhang, C.; Zheng, X.; Yu, L.; Ma, C.; et al. The Role of PDGF-B/TGF-Β1/Neprilysin Network in Regulating Endothelial-to-Mesenchymal Transition in Pulmonary Artery Remodeling. Cell. Signal. 2016, 28, 1489–1501. [Google Scholar] [CrossRef]
- Paulin, R.; Meloche, J.; Jacob, M.H.; Bisserier, M.; Courboulin, A.; Bonnet, S. Dehydroepiandrosterone Inhibits the Src/STAT3 Constitutive Activation in Pulmonary Arterial Hypertension. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1798–H1809. [Google Scholar] [CrossRef]
- Solinc, J.; Raimbault-Machado, J.; Dierick, F.; El Bernoussi, L.; Tu, L.; Thuillet, R.; Mougenot, N.; Hoareau-Coudert, B.; Monceau, V.; Pavoine, C.; et al. Platelet-Derived Growth Factor Receptor Type α Activation Drives Pulmonary Vascular Remodeling Via Progenitor Cell Proliferation and Induces Pulmonary Hypertension. J. Am. Heart Assoc. 2022, 11, e023021. [Google Scholar] [CrossRef] [PubMed]
- Ren, W.; Watts, S.W.; Fanburg, B.L. Serotonin Transporter Interacts with the PDGFβ Receptor in PDGF-BB-Induced Signaling and Mitogenesis in Pulmonary Artery Smooth Muscle Cells. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2011, 300, L486–L497. [Google Scholar] [CrossRef] [Green Version]
- Yamamura, A.; Nayeem, M.J.; Mamun, A.A.; Takahashi, R.; Hayashi, H.; Sato, M. Platelet-Derived Growth Factor up-Regulates Ca2+-Sensing Receptors in Idiopathic Pulmonary Arterial Hypertension. FASEB J. 2019, 33, 7363–7374. [Google Scholar] [CrossRef]
- Zhao, F.-Y.; Xu, S.-L.; Zhang, C.-F.; Liu, J.; Zhang, Y.; Yang, J.; Xing, X.-Q. PDGF Mediates Pulmonary Arterial Smooth Muscle Cell Proliferation and Migration by Regulating NFATc2. Mol. Med. Rep. 2021, 23, 39. [Google Scholar] [CrossRef]
- Bonnet, S.; Rochefort, G.; Sutendra, G.; Archer, S.L.; Haromy, A.; Webster, L.; Hashimoto, K.; Bonnet, S.N.; Michelakis, E.D. The Nuclear Factor of Activated T Cells in Pulmonary Arterial Hypertension Can Be Therapeutically Targeted. Proc. Natl. Acad. Sci. USA 2007, 104, 11418–11423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Zhang, Q.; Yan, X.; Liu, L.; Zhai, C.; Wang, Q.; Chai, L.; Li, M. Ubiquitin-Specific Protease 7 Mediates Platelet-Derived Growth Factor-Induced Pulmonary Arterial Smooth Muscle Cells Proliferation. Pulm. Circ. 2021, 11, 20458940211046132. [Google Scholar] [CrossRef]
- Rieg, A.D.; Suleiman, S.; Anker, C.; Verjans, E.; Rossaint, R.; Uhlig, S.; Martin, C. PDGF-BB Regulates the Pulmonary Vascular Tone: Impact of Prostaglandins, Calcium, MAPK- and PI3K/AKT/MTOR Signalling and Actin Polymerisation in Pulmonary Veins of Guinea Pigs. Respir. Res. 2018, 19, 120. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, A.; Firth, A.L.; Smith, K.A.; Maliakal, M.V.; Yuan, J.X.-J. PDGF Enhances Store-Operated Ca2+ Entry by Upregulating STIM1/Orai1 via Activation of Akt/MTOR in Human Pulmonary Arterial Smooth Muscle Cells. Am. J. Physiol. Cell Physiol. 2012, 302, C405–C411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, H.; Yamamura, A.; Yamamura, H.; Song, S.; Fraidenburg, D.R.; Chen, J.; Gu, Y.; Pohl, N.M.; Zhou, T.; Jiménez-Pérez, L.; et al. Pathogenic Role of Calcium-Sensing Receptors in the Development and Progression of Pulmonary Hypertension. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2016, 310, L846–L859. [Google Scholar] [CrossRef] [Green Version]
- Plikus, M.V.; Wang, X.; Sinha, S.; Forte, E.; Thompson, S.M.; Herzog, E.L.; Driskell, R.R.; Rosenthal, N.; Biernaskie, J.; Horsley, V. Fibroblasts: Origins, Definitions, and Functions in Health and Disease. Cell 2021, 184, 3852–3872. [Google Scholar] [CrossRef]
- Aono, Y.; Nishioka, Y.; Inayama, M.; Ugai, M.; Kishi, J.; Uehara, H.; Izumi, K.; Sone, S. Imatinib as a Novel Antifibrotic Agent in Bleomycin-Induced Pulmonary Fibrosis in Mice. Am. J. Respir. Crit. Care Med. 2005, 171, 1279–1285. [Google Scholar] [CrossRef]
- Bonner, J.C.; Lindroos, P.M.; Rice, A.B.; Moomaw, C.R.; Morgan, D.L. Induction of PDGF Receptor-Alpha in Rat Myofibroblasts during Pulmonary Fibrogenesis in Vivo. Am. J. Physiol. 1998, 274, L72–L80. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhai, C.; Pan, Y.; Zhu, Y.; Shi, W.; Wang, J.; Yan, X.; Su, X.; Song, Y.; Gao, L.; et al. Sphingosine-1-Phosphate Induces Airway Smooth Muscle Cell Proliferation, Migration, and Contraction by Modulating Hippo Signaling Effector YAP. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2018, 315, L609–L621. [Google Scholar] [CrossRef]
- Rock, J.R.; Barkauskas, C.E.; Cronce, M.J.; Xue, Y.; Harris, J.R.; Liang, J.; Noble, P.W.; Hogan, B.L.M. Multiple Stromal Populations Contribute to Pulmonary Fibrosis without Evidence for Epithelial to Mesenchymal Transition. Proc. Natl. Acad. Sci. USA 2011, 108, E1475–E1483. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Urabe, G.; Huang, Y.; Zhang, M.; Wang, B.; Marcho, L.; Shen, H.; Kent, K.C.; Guo, L.-W. A Role for Polo-Like Kinase 4 in Vascular Fibroblast Cell-Type Transition. JACC Basic Transl. Sci. 2021, 6, 257–283. [Google Scholar] [CrossRef]
- Olson, L.E.; Soriano, P. Increased PDGFRalpha Activation Disrupts Connective Tissue Development and Drives Systemic Fibrosis. Dev. Cell 2009, 16, 303–313. [Google Scholar] [CrossRef] [Green Version]
- Yi, E.S.; Lee, H.; Yin, S.; Piguet, P.; Sarosi, I.; Kaufmann, S.; Tarpley, J.; Wang, N.S.; Ulich, T.R. Platelet-Derived Growth Factor Causes Pulmonary Cell Proliferation and Collagen Deposition In Vivo. Am. J. Pathol. 1996, 149, 539–548. [Google Scholar] [PubMed]
- Zhuo, Y.; Hoyle, G.W.; Shan, B.; Levy, D.R.; Lasky, J.A. Over-Expression of PDGF-C Using a Lung Specific Promoter Results in Abnormal Lung Development. Transgenic Res. 2006, 15, 543–555. [Google Scholar] [CrossRef] [PubMed]
- Green, J.; Endale, M.; Auer, H.; Perl, A.-K.T. Diversity of Interstitial Lung Fibroblasts Is Regulated by Platelet-Derived Growth Factor Receptor α Kinase Activity. Am. J. Respir. Cell Mol. Biol. 2016, 54, 532–545. [Google Scholar] [CrossRef] [Green Version]
- Chandran, R.R.; Xie, Y.; Gallardo-Vara, E.; Adams, T.; Garcia-Milian, R.; Kabir, I.; Sheikh, A.Q.; Kaminski, N.; Martin, K.A.; Herzog, E.L.; et al. Distinct Roles of KLF4 in Mesenchymal Cell Subtypes during Lung Fibrogenesis. Nat. Commun. 2021, 12, 7179. [Google Scholar] [CrossRef]
- Dierick, F.; Solinc, J.; Bignard, J.; Soubrier, F.; Nadaud, S. Progenitor/Stem Cells in Vascular Remodeling during Pulmonary Arterial Hypertension. Cells 2021, 10, 1338. [Google Scholar] [CrossRef]
- Sheikh, A.Q.; Misra, A.; Rosas, I.O.; Adams, R.H.; Greif, D.M. Smooth Muscle Cell Progenitors Are Primed to Muscularize in Pulmonary Hypertension. Sci. Transl. Med. 2015, 7, 308ra159. [Google Scholar] [CrossRef] [Green Version]
- Dierick, F.; Héry, T.; Hoareau-Coudert, B.; Mougenot, N.; Monceau, V.; Claude, C.; Crisan, M.; Besson, V.; Dorfmüller, P.; Marodon, G.; et al. Resident PW1+ Progenitor Cells Participate in Vascular Remodeling During Pulmonary Arterial Hypertension. Circ. Res. 2016, 118, 822–833. [Google Scholar] [CrossRef] [Green Version]
- Betsholtz, C. Insight into the Physiological Functions of PDGF through Genetic Studies in Mice. Cytokine Growth Factor Rev. 2004, 15, 215–228. [Google Scholar] [CrossRef]
- Lindahl, P.; Johansson, B.R.; Levéen, P.; Betsholtz, C. Pericyte Loss and Microaneurysm Formation in PDGF-B-Deficient Mice. Science 1997, 277, 242–245. [Google Scholar] [CrossRef]
- Ando, K.; Wang, W.; Peng, D.; Chiba, A.; Lagendijk, A.K.; Barske, L.; Crump, J.G.; Stainier, D.Y.R.; Lendahl, U.; Koltowska, K.; et al. Peri-Arterial Specification of Vascular Mural Cells from Naïve Mesenchyme Requires Notch Signaling. Dev. Camb. Engl. 2019, 146, dev165589. [Google Scholar] [CrossRef] [Green Version]
- Chow, K.; Fessel, J.P.; Stansbury, K.I.; Schmidt, E.P.; Gaskill, C.; Alvarez, D.; Graham, B.; Harrison, D.G.; Wagner, D.H.; Nozik-Grayck, E.; et al. Dysfunctional Resident Lung Mesenchymal Stem Cells Contribute to Pulmonary Microvascular Remodeling. Pulm. Circ. 2013, 3, 31–49. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Bernau, K.; Sandbo, N.; Gu, J.; Preissl, S.; Sun, X. Pdgfra Marks a Cellular Lineage with Distinct Contributions to Myofibroblasts in Lung Maturation and Injury Response. eLife 2018, 7, e36865. [Google Scholar] [CrossRef]
- Boström, H.; Gritli-Linde, A.; Betsholtz, C. PDGF-A/PDGF Alpha-Receptor Signaling Is Required for Lung Growth and the Formation of Alveoli but Not for Early Lung Branching Morphogenesis. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2002, 223, 155–162. [Google Scholar] [CrossRef]
- Sakurai, H.; Era, T.; Jakt, L.M.; Okada, M.; Nakai, S.; Nishikawa, S.; Nishikawa, S. In Vitro Modeling of Paraxial and Lateral Mesoderm Differentiation Reveals Early Reversibility. Stem Cells 2006, 24, 575–586. [Google Scholar] [CrossRef]
- Ding, G.; Tanaka, Y.; Hayashi, M.; Nishikawa, S.-I.; Kataoka, H. PDGF Receptor Alpha+ Mesoderm Contributes to Endothelial and Hematopoietic Cells in Mice. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2013, 242, 254–268. [Google Scholar] [CrossRef] [Green Version]
- Siegbahn, A.; Hammacher, A.; Westermark, B.; Heldin, C.H. Differential Effects of the Various Isoforms of Platelet-Derived Growth Factor on Chemotaxis of Fibroblasts, Monocytes, and Granulocytes. J. Clin. Investig. 1990, 85, 916–920. [Google Scholar] [CrossRef]
- Fuhrman, B.; Gantman, A.; Khateeb, J.; Volkova, N.; Horke, S.; Kiyan, J.; Dumler, I.; Aviram, M. Urokinase Activates Macrophage PON2 Gene Transcription via the PI3K/ROS/MEK/SREBP-2 Signalling Cascade Mediated by the PDGFR-Beta. Cardiovasc. Res. 2009, 84, 145–154. [Google Scholar] [CrossRef] [Green Version]
- Son, D.; Na, Y.R.; Hwang, E.-S.; Seok, S.H. Platelet-Derived Growth Factor-C (PDGF-C) Induces Anti-Apoptotic Effects on Macrophages through Akt and Bad Phosphorylation. J. Biol. Chem. 2014, 289, 6225–6235. [Google Scholar] [CrossRef] [Green Version]
- Wågsäter, D.; Zhu, C.; Björck, H.M.; Eriksson, P. Effects of PDGF-C and PDGF-D on Monocyte Migration and MMP-2 and MMP-9 Expression. Atherosclerosis 2009, 202, 415–423. [Google Scholar] [CrossRef]
- Chen, C.-F.; Feng, X.; Liao, H.-Y.; Jin, W.-J.; Zhang, J.; Wang, Y.; Gong, L.-L.; Liu, J.-J.; Yuan, X.-H.; Zhao, B.-B.; et al. Regulation of T Cell Proliferation by JMJD6 and PDGF-BB during Chronic Hepatitis B Infection. Sci. Rep. 2014, 4, 6359. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, S.; Ganguly, S.; Hajian, P.; Cao, J.-N.; Agrawal, A. PDGF Upregulates CLEC-2 to Induce T Regulatory Cells. Oncotarget 2015, 6, 28621–28632. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.; Tang, H.; Lin, R.; Carr, S.G.; Wang, Z.; Babicheva, A.; Ayon, R.J.; Jain, P.P.; Xiong, M.; Rodriguez, M.; et al. Endothelial Platelet-derived Growth Factor-mediated Activation of Smooth Muscle Platelet-derived Growth Factor Receptors in Pulmonary Arterial Hypertension. Pulm. Circ. 2020, 10, 2045894020948470. [Google Scholar] [CrossRef]
- Saygin, D.; Tabib, T.; Bittar, H.E.T.; Valenzi, E.; Sembrat, J.; Chan, S.Y.; Rojas, M.; Lafyatis, R. Transcriptional Profiling of Lung Cell Populations in Idiopathic Pulmonary Arterial Hypertension. Pulm. Circ. 2020, 10, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Ntokou, A.; Dave, J.M.; Kauffman, A.C.; Sauler, M.; Ryu, C.; Hwa, J.; Herzog, E.L.; Singh, I.; Saltzman, W.M.; Greif, D.M. Macrophage-Derived PDGF-B Induces Muscularization in Murine and Human Pulmonary Hypertension. JCI Insight 2021, 6, e139067. [Google Scholar] [CrossRef]
- Selimovic, N.; Bergh, C.-H.; Andersson, B.; Sakiniene, E.; Carlsten, H.; Rundqvist, B. Growth Factors and Interleukin-6 across the Lung Circulation in Pulmonary Hypertension. Eur. Respir. J. 2009, 34, 662–668. [Google Scholar] [CrossRef] [Green Version]
- Jones, R.; Capen, D.; Jacobson, M.; Munn, L. PDGF and Microvessel Wall Remodeling in Adult Rat Lung: Imaging PDGF-AA and PDGF-Rα Molecules in Progenitor Smooth Muscle Cells Developing in Experimental Pulmonary Hypertension. Cell Tissue Res. 2006, 326, 759–769. [Google Scholar] [CrossRef]
- Overbeek, M.J.; Boonstra, A.; Voskuyl, A.E.; Vonk, M.C.; Vonk-Noordegraaf, A.; van Berkel, M.P.; Mooi, W.J.; Dijkmans, B.A.; Hondema, L.S.; Smit, E.F.; et al. Platelet-Derived Growth Factor Receptor-β and Epidermal Growth Factor Receptor in Pulmonary Vasculature of Systemic Sclerosis-Associated Pulmonary Arterial Hypertension versus Idiopathic Pulmonary Arterial Hypertension and Pulmonary Veno-Occlusive Disease: A Case-Control Study. Arthritis Res. Ther. 2011, 13, R61. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Li, L.-S.; Yang, D.-L.; Gong, Q.-H.; Deng, J.; Huang, X.-N. Inhibitory Effect of Ginsenoside Rg1 on Vascular Smooth Muscle Cell Proliferation Induced by PDGF-BB Is Involved in Nitric Oxide Formation. Evid.-Based Complement. Altern. Med. ECAM 2012, 2012, 314395. [Google Scholar] [CrossRef]
- Callera, G.E.; Yogi, A.; Briones, A.M.; Montezano, A.C.I.; He, Y.; Tostes, R.C.A.; Schiffrin, E.L.; Touyz, R.M. Vascular Proinflammatory Responses by Aldosterone Are Mediated via C-Src Trafficking to Cholesterol-Rich Microdomains: Role of PDGFR. Cardiovasc. Res. 2011, 91, 720–731. [Google Scholar] [CrossRef] [Green Version]
- Preston, I.R.; Sagliani, K.D.; Warburton, R.R.; Hill, N.S.; Fanburg, B.L.; Jaffe, I.Z. Mineralocorticoid Receptor Antagonism Attenuates Experimental Pulmonary Hypertension. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L678–L688. [Google Scholar] [CrossRef] [Green Version]
- Nissen, L.J.; Cao, R.; Hedlund, E.-M.; Wang, Z.; Zhao, X.; Wetterskog, D.; Funa, K.; Bråkenhielm, E.; Cao, Y. Angiogenic Factors FGF2 and PDGF-BB Synergistically Promote Murine Tumor Neovascularization and Metastasis. J. Clin. Investig. 2007, 117, 2766–2777. [Google Scholar] [CrossRef] [Green Version]
- ten Freyhaus, H.; Berghausen, E.M.; Janssen, W.; Leuchs, M.; Zierden, M.; Murmann, K.; Klinke, A.; Vantler, M.; Caglayan, E.; Kramer, T.; et al. Genetic Ablation of PDGF-Dependent Signaling Pathways Abolishes Vascular Remodeling and Experimental Pulmonary Hypertension. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1236–1245. [Google Scholar] [CrossRef] [Green Version]
- Dahal, B.K.; Heuchel, R.; Pullamsetti, S.S.; Wilhelm, J.; Ghofrani, H.A.; Weissmann, N.; Seeger, W.; Grimminger, F.; Schermuly, R.T. Hypoxic Pulmonary Hypertension in Mice with Constitutively Active Platelet-Derived Growth Factor Receptor-β. Pulm. Circ. 2011, 1, 259–268. [Google Scholar] [CrossRef] [Green Version]
- Yamamura, A.; Guo, Q.; Yamamura, H.; Zimnicka, A.M.; Pohl, N.M.; Smith, K.A.; Fernandez, R.A.; Zeifman, A.; Makino, A.; Dong, H.; et al. Enhanced Ca2+-Sensing Receptor Function in Idiopathic Pulmonary Arterial Hypertension. Circ. Res. 2012, 111, 469–481. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Li, M.; Warburton, R.R.; Hill, N.S.; Fanburg, B.L. The 5-HT Transporter Transactivates the PDGFβ Receptor in Pulmonary Artery Smooth Muscle Cells. FASEB J. 2007, 21, 2725–2734. [Google Scholar] [CrossRef]
- Ciuclan, L.; Hussey, M.J.; Burton, V.; Good, R.; Duggan, N.; Beach, S.; Jones, P.; Fox, R.; Clay, I.; Bonneau, O.; et al. Imatinib Attenuates Hypoxia-Induced Pulmonary Arterial Hypertension Pathology via Reduction in 5-Hydroxytryptamine through Inhibition of Tryptophan Hydroxylase 1 Expression. Am. J. Respir. Crit. Care Med. 2013, 187, 78–89. [Google Scholar] [CrossRef]
- Sheikh, A.Q.; Saddouk, F.Z.; Ntokou, A.; Mazurek, R.; Greif, D.M. Cell Autonomous and Non-Cell Autonomous Regulation of SMC Progenitors in Pulmonary Hypertension. Cell Rep. 2018, 23, 1152–1165. [Google Scholar] [CrossRef]
- Ricard, N.; Tu, L.; Le Hiress, M.; Huertas, A.; Phan, C.; Thuillet, R.; Sattler, C.; Fadel, E.; Seferian, A.; Montani, D.; et al. Increased Pericyte Coverage Mediated by Endothelial-Derived Fibroblast Growth Factor-2 and Interleukin-6 Is a Source of Smooth Muscle–Like Cells in Pulmonary Hypertension. Circulation 2014, 129, 1586–1597. [Google Scholar] [CrossRef] [Green Version]
- Lythgoe, M.P.; Rhodes, C.J.; Ghataorhe, P.; Attard, M.; Wharton, J.; Wilkins, M.R. Why Drugs Fail in Clinical Trials in Pulmonary Arterial Hypertension, and Strategies to Succeed in the Future. Pharmacol. Ther. 2016, 164, 195–203. [Google Scholar] [CrossRef]
- Rol, N.; de Raaf, M.A.; Sun, X.Q.; Kuiper, V.P.; da Silva Gonçalves Bos, D.; Happé, C.; Kurakula, K.; Dickhoff, C.; Thuillet, R.; Tu, L.; et al. Nintedanib Improves Cardiac Fibrosis but Leaves Pulmonary Vascular Remodelling Unaltered in Experimental Pulmonary Hypertension. Cardiovasc. Res. 2019, 115, 432–439. [Google Scholar] [CrossRef]
- Tsutsumi, T.; Nagaoka, T.; Yoshida, T.; Wang, L.; Kuriyama, S.; Suzuki, Y.; Nagata, Y.; Harada, N.; Kodama, Y.; Takahashi, F.; et al. Nintedanib Ameliorates Experimental Pulmonary Arterial Hypertension via Inhibition of Endothelial Mesenchymal Transition and Smooth Muscle Cell Proliferation. PLoS ONE 2019, 14, e0214697. [Google Scholar] [CrossRef] [Green Version]
- Richter, M.J.; Ewert, J.; Grimminger, F.; Ghofrani, H.A.; Kojonazarov, B.; Petrovic, A.; Seeger, W.; Schermuly, R.T.; Tello, K.; Gall, H. Nintedanib in Severe Pulmonary Arterial Hypertension. Am. J. Respir. Crit. Care Med. 2018, 198, 808–810. [Google Scholar] [CrossRef]
- Klein, M.; Schermuly, R.T.; Ellinghaus, P.; Milting, H.; Riedl, B.; Nikolova, S.; Pullamsetti, S.S.; Weissmann, N.; Dony, E.; Savai, R.; et al. Combined Tyrosine and Serine/Threonine Kinase Inhibition by Sorafenib Prevents Progression of Experimental Pulmonary Hypertension and Myocardial Remodeling. Circulation 2008, 118, 2081–2090. [Google Scholar] [CrossRef]
- Leong, Z.P.; Hikasa, Y. Effects of Toceranib Compared with Sorafenib on Monocrotaline-Induced Pulmonary Arterial Hypertension and Cardiopulmonary Remodeling in Rats. Vascul. Pharmacol. 2018, 110, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Gomberg-Maitland, M.; Maitland, M.L.; Barst, R.J.; Sugeng, L.; Coslet, S.; Perrino, T.J.; Bond, L.; LaCouture, M.E.; Archer, S.L.; Ratain, M.J. A Dosing/Cross-Development Study of the Multikinase Inhibitor Sorafenib in Patients with Pulmonary Arterial Hypertension. Clin. Pharmacol. Ther. 2010, 87, 303–310. [Google Scholar] [CrossRef] [Green Version]
- Kimura, G.; Kataoka, M.; Inami, T.; Fukuda, K.; Yoshino, H.; Satoh, T. Sorafenib as a Potential Strategy for Refractory Pulmonary Arterial Hypertension. Pulm. Pharmacol. Ther. 2017, 44, 46–49. [Google Scholar] [CrossRef]
- Guignabert, C.; Phan, C.; Seferian, A.; Huertas, A.; Tu, L.; Thuillet, R.; Sattler, C.; Le Hiress, M.; Tamura, Y.; Jutant, E.-M.; et al. Dasatinib Induces Lung Vascular Toxicity and Predisposes to Pulmonary Hypertension. J. Clin. Investig. 2016, 126, 3207–3218. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, A.; Miyaji, K.; Matsubara, H. Efficacy and Safety of Long-Term Imatinib Therapy for Patients with Pulmonary Veno-Occlusive Disease and Pulmonary Capillary Hemangiomatosis. Respir. Med. 2017, 131, 215–219. [Google Scholar] [CrossRef] [Green Version]
- Sato, H.; Sugimura, K.; Miura, M.; Konno, R.; Kozu, K.; Yaoita, N.; Shimizu, T.; Yamamoto, S.; Aoki, T.; Tatebe, S.; et al. Beneficial Effects of Imatinib in a Patient with Suspected Pulmonary Veno-Occlusive Disease. Tohoku J. Exp. Med. 2019, 247, 69–73. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Matsubara, H.; Akagi, S.; Sarashina, T.; Ejiri, K.; Kawakita, N.; Yoshida, M.; Miyoshi, T.; Watanabe, A.; Nishii, N.; et al. Nanoparticle-Mediated Drug Delivery System for Pulmonary Arterial Hypertension. J. Clin. Med. 2017, 6, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akagi, S.; Nakamura, K.; Miura, D.; Saito, Y.; Matsubara, H.; Ogawa, A.; Matoba, T.; Egashira, K.; Ito, H. Delivery of Imatinib-Incorporated Nanoparticles into Lungs Suppresses the Development of Monocrotaline-Induced Pulmonary Arterial Hypertension. Int. Heart J. 2015, 56, 354–359. [Google Scholar] [CrossRef] [Green Version]
- Wilkins, M.R.; Mckie, M.A.; Law, M.; Roussakis, A.A.; Harbaum, L.; Church, C.; Coghlan, J.G.; Condliffe, R.; Howard, L.S.; Kiely, D.G.; et al. Positioning Imatinib for Pulmonary Arterial Hypertension: A Phase I/II Design Comprising Dose Finding and Single-Arm Efficacy. Pulm. Circ. 2021, 11, 20458940211052824. [Google Scholar] [CrossRef]
- Leong, Z.P.; Okida, A.; Higuchi, M.; Yamano, Y.; Hikasa, Y. Reversal Effects of Low-Dose Imatinib Compared with Sunitinib on Monocrotaline-Induced Pulmonary and Right Ventricular Remodeling in Rats. Vascul. Pharmacol. 2018, 100, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Hatano, M.; Yao, A.; Shiga, T.; Kinugawa, K.; Hirata, Y.; Nagai, R. Imatinib Mesylate Has the Potential to Exert Its Efficacy by Down-Regulating the Plasma Concentration of Platelet-Derived Growth Factor in Patients with Pulmonary Arterial Hypertension. Int. Heart J. 2010, 51, 272–276. [Google Scholar] [CrossRef] [Green Version]
- Galkin, A.; Clemons, B.; Garcia, E.; Brooks, J.; Slee, D.; Salter-Cid, L.; Zisman, L. Abstract 11102: Gb002, A Novel Inhaled Pdgfr Kinase Inhibitor, Demonstrates Efficacy in the Su5416 Hypoxia Rat Model of Pulmonary Arterial Hypertension (Pah). Circulation 2019, 140, A11102. [Google Scholar] [CrossRef]
- Sitapara, R.; Slee, D.; Salter-Cid, L.; Zisman, L. Abstract 12947: In Vivo Efficacy of a Novel, Inhaled Pdgfra/b Inhibitor, Gb002, in The Rat Monocrotaline and Pneumonectomy Model of Pulmonary Arterial Hypertension. Circulation 2019, 140, A12947. [Google Scholar] [CrossRef]
- Frantz, R.P.; Benza, R.L.; Channick, R.N.; Chin, K.; Howard, L.S.; McLaughlin, V.V.; Sitbon, O.; Zamanian, R.T.; Hemnes, A.R.; Cravets, M.; et al. TORREY, a Phase 2 Study to Evaluate the Efficacy and Safety of Inhaled Seralutinib for the Treatment of Pulmonary Arterial Hypertension. Pulm. Circ. 2021, 11, 20458940211057071. [Google Scholar] [CrossRef]
- Leong, Z.P.; Hikasa, Y. Effects of Masitinib Compared with Tadalafil for the Treatment of Monocrotaline-Induced Pulmonary Arterial Hypertension in Rats. Vascul. Pharmacol. 2019, 122–123, 106599. [Google Scholar] [CrossRef]
- Ambade, A.S.; Jung, B.; Lee, D.; Doods, H.; Wu, D. Triple-Tyrosine Kinase Inhibition Attenuates Pulmonary Arterial Hypertension and Neointimal Formation. Transl. Res. 2019, 203, 15–30. [Google Scholar] [CrossRef] [PubMed]
- Scuri, P.; Iacovoni, A.; Abete, R.; Cereda, A.; Grosu, A.; Senni, M. An Unexpected Recovery of Patients with Pulmonary Arterial Hypertension and SARS-CoV-2 Pneumonia: A Case Series. Pulm. Circ. 2020, 10, 2045894020956581. [Google Scholar] [CrossRef] [PubMed]
- Ibarra-Sánchez, L.Á.; Gámez-Méndez, A.; Martínez-Ruiz, M.; Nájera-Martínez, E.F.; Morales-Flores, B.A.; Melchor-Martínez, E.M.; Sosa-Hernández, J.E.; Parra-Saldívar, R.; Iqbal, H.M.N. Nanostructures for Drug Delivery in Respiratory Diseases Therapeutics: Revision of Current Trends and Its Comparative Analysis. J. Drug Deliv. Sci. Technol. 2022, 70, 103219. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Gu, C.; Zhao, T.; Jia, Y.; Bao, C.; Luo, A.; Guo, Q.; Han, Y.; Wang, J.; Black, S.M.; et al. Combination Therapy with Rapamycin and Low Dose Imatinib in Pulmonary Hypertension. Front. Pharmacol. 2021, 12, 758763. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.L.; Mo, G.; Baldwin, J.R.; Peterson, P.M.; Ilaria, R.L.; Conti, I.; Cronier, D.M.; Tap, W.D. Exposure-Response Relationship of Olaratumab for Survival Outcomes and Safety When Combined with Doxorubicin in Patients with Soft Tissue Sarcoma. Cancer Chemother. Pharmacol. 2019, 83, 191–199. [Google Scholar] [CrossRef] [Green Version]
- Tap, W.D.; Wagner, A.J.; Schöffski, P.; Martin-Broto, J.; Krarup-Hansen, A.; Ganjoo, K.N.; Yen, C.-C.; Abdul Razak, A.R.; Spira, A.; Kawai, A.; et al. Effect of Doxorubicin Plus Olaratumab vs Doxorubicin Plus Placebo on Survival in Patients with Advanced Soft Tissue Sarcomas: The ANNOUNCE Randomized Clinical Trial. JAMA 2020, 323, 1266–1276. [Google Scholar] [CrossRef]
- Shen, J.; Vil, M.D.; Prewett, M.; Damoci, C.; Zhang, H.; Li, H.; Jimenez, X.; Deevi, D.S.; Iacolina, M.; Kayas, A.; et al. Development of a Fully Human Anti-PDGFRbeta Antibody That Suppresses Growth of Human Tumor Xenografts and Enhances Antitumor Activity of an Anti-VEGFR2 Antibody. Neoplasia 2009, 11, 594–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Zeitelhofer, M.; Nilsson, I.; Liu, X.; Allan, L.; Gloria, B.; Perani, A.; Murone, C.; Catimel, B.; Neville, A.M.; et al. Development of Monoclonal Anti-PDGF-CC Antibodies as Tools for Investigating Human Tissue Expression and for Blocking PDGF-CC Induced PDGFRα Signalling in Vivo. PLoS ONE 2018, 13, e0201089. [Google Scholar] [CrossRef] [PubMed]
- Duan, D.S.; Pazin, M.J.; Fretto, L.J.; Williams, L.T. A Functional Soluble Extracellular Region of the Platelet-Derived Growth Factor (PDGF) Beta-Receptor Antagonizes PDGF-Stimulated Responses. J. Biol. Chem. 1991, 266, 413–418. [Google Scholar] [CrossRef]
- Balasubramaniam, V.; Le Cras, T.D.; Ivy, D.D.; Grover, T.R.; Kinsella, J.P.; Abman, S.H. Role of Platelet-Derived Growth Factor in Vascular Remodeling during Pulmonary Hypertension in the Ovine Fetus. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 284, L826–L833. [Google Scholar] [CrossRef] [Green Version]
- Crooke, S.T.; Liang, X.-H.; Baker, B.F.; Crooke, R.M. Antisense Technology: A Review. J. Biol. Chem. 2021, 296, 100416. [Google Scholar] [CrossRef]
- Hu, C.-J.; Poth, J.M.; Zhang, H.; Flockton, A.; Laux, A.; Kumar, S.; McKeon, B.; Mouradian, G.; Li, M.; Riddle, S.; et al. Suppression of HIF2 Signalling Attenuates the Initiation of Hypoxia-Induced Pulmonary Hypertension. Eur. Respir. J. 2019, 54, 1900378. [Google Scholar] [CrossRef]
- Ogorodnikova, N.; Arenz, C. MicroRNA-145-Targeted Drug and Its Preventive Effect on Pulmonary Arterial Hypertension (Patent WO2012153135 A1). Expert Opin. Ther. Pat. 2015, 25, 723–727. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Solinc, J.; Ribot, J.; Soubrier, F.; Pavoine, C.; Dierick, F.; Nadaud, S. The Platelet-Derived Growth Factor Pathway in Pulmonary Arterial Hypertension: Still an Interesting Target? Life 2022, 12, 658. https://doi.org/10.3390/life12050658
Solinc J, Ribot J, Soubrier F, Pavoine C, Dierick F, Nadaud S. The Platelet-Derived Growth Factor Pathway in Pulmonary Arterial Hypertension: Still an Interesting Target? Life. 2022; 12(5):658. https://doi.org/10.3390/life12050658
Chicago/Turabian StyleSolinc, Julien, Jonathan Ribot, Florent Soubrier, Catherine Pavoine, France Dierick, and Sophie Nadaud. 2022. "The Platelet-Derived Growth Factor Pathway in Pulmonary Arterial Hypertension: Still an Interesting Target?" Life 12, no. 5: 658. https://doi.org/10.3390/life12050658
APA StyleSolinc, J., Ribot, J., Soubrier, F., Pavoine, C., Dierick, F., & Nadaud, S. (2022). The Platelet-Derived Growth Factor Pathway in Pulmonary Arterial Hypertension: Still an Interesting Target? Life, 12(5), 658. https://doi.org/10.3390/life12050658