
Fetal Hippocampal Connectivity Shows Dissociable Associations with Maternal Cortisol and Self-Reported Distress during Pregnancy

Abstract
:1. Introduction
2. Materials and Methods
2.1. Participants and Procedures
2.2. Measures
2.2.1. Prenatal Distress
2.2.2. Prenatal Cortisol
2.2.3. Fetal fMRI
2.2.4. fMRI Preprocessing
2.3. Analyses
2.3.1. Seed Connectivity Analyses
2.3.2. Statistical Analyses
3. Results
3.1. Associations between Self-Reported Distress and Salivary Cortisol Levels
3.2. Associations with Fetal Hippocampal Connectivity
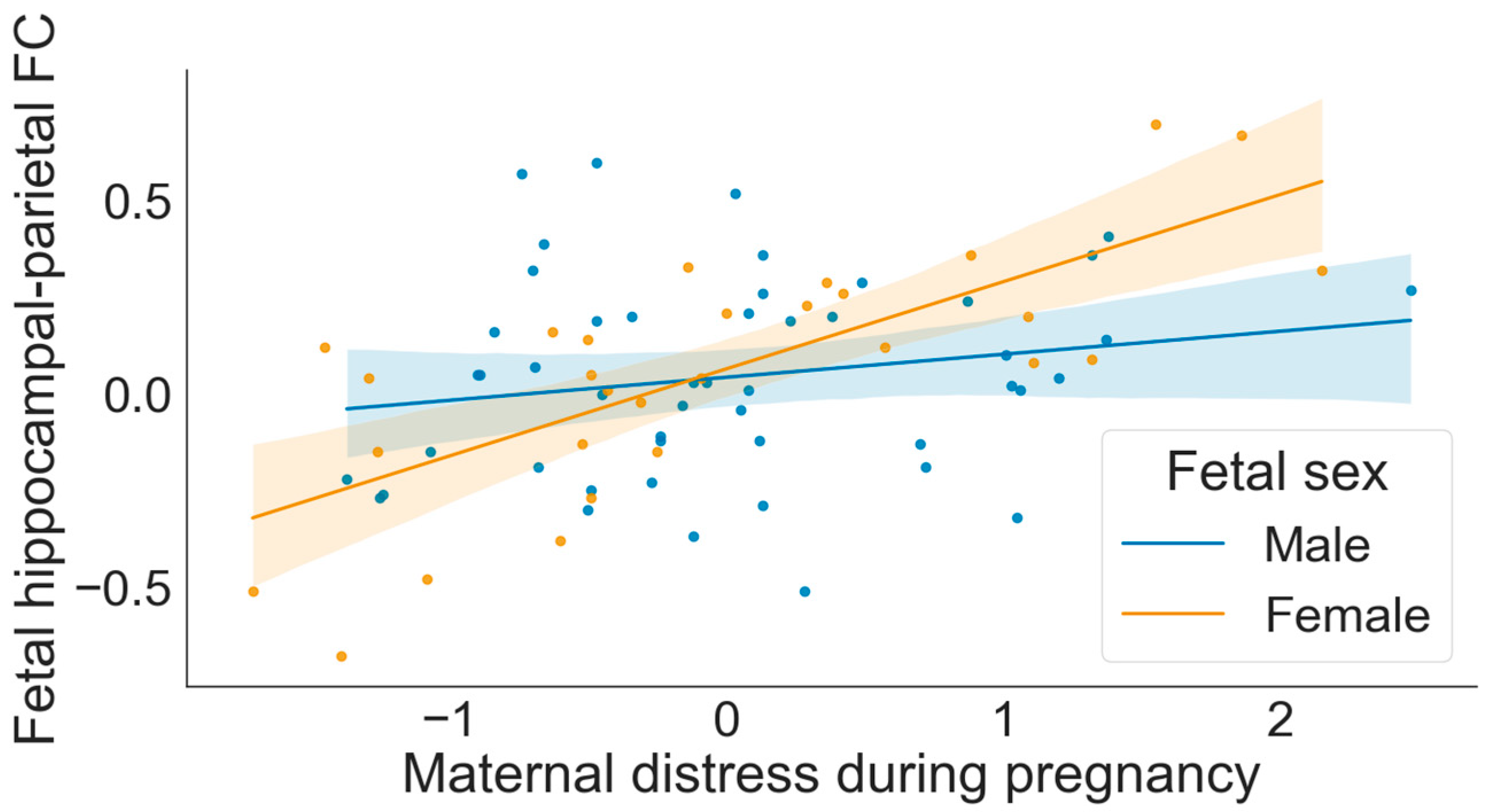
3.3. Moderation by Fetal Sex
3.4. Sensitivity Analyses
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Murphy, V.E.; Smith, R.; Giles, W.B.; Clifton, V.L. Endocrine regulation of human fetal growth: The role of the mother, placenta, and fetus. Endocr. Rev. 2006, 27, 141–169. [Google Scholar] [CrossRef] [PubMed]
- Barker, D.J. Fetal origins of coronary heart disease. BMJ 1995, 311, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Conradt, E.; Adkins, D.E.; Crowell, S.E.; Raby, K.L.; Diamond, L.M.; Ellis, B. Incorporating epigenetic mechanisms to advance fetal programming theories. Dev. Psychopathol. 2018, 30, 807–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bleker, L.S.; De Rooij, S.R.; Roseboom, T.J. Prenatal psychological stress exposure and neurodevelopment and health of children. Int. J. Environ. Res. Public Health 2019, 16, 3657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilmore, J.H.; Knickmeyer, R.C.; Gao, W. Imaging structural and functional brain development in early childhood. Nat. Rev. Neurosci. 2018, 19, 123–137. [Google Scholar] [CrossRef]
- Varcin, K.J.; Alvares, G.A.; Uljarević, M.; Whitehouse, A.J.O. Prenatal maternal stress events and phenotypic outcomes in Autism Spectrum Disorder. Autism Res. 2017, 10, 1866–1877. [Google Scholar] [CrossRef]
- Smith, S.M.; Vale, W.W. The role of the hypothalamic-pituitary-adrenal axis in neuroendocrine responses to stress. Dialogues Clin. Neurosci. 2006, 8, 383–395. [Google Scholar] [CrossRef]
- Boland, R.; Joyce, B.J.; Wallace, M.J.; Stanton, H.; Fosang, A.J.; Pierce, R.A.; Harding, R.; Hooper, S.B. Cortisol enhances structural maturation of the hypoplastic fetal lung in sheep. J. Physiol. 2004, 554, 505–517. [Google Scholar] [CrossRef] [Green Version]
- Welberg, L.A.M.; Thrivikraman, K.V.; Plotsky, P.M. Chronic maternal stress inhibits the capacity to up-regulate placental 11β-hydroxysteroid dehydrogenase type 2 activity. J. Endocrinol. 2005, 186, R7–R12. [Google Scholar] [CrossRef] [Green Version]
- Phillips, L.J.; McGorry, P.D.; Garner, B.; Thompson, K.N.; Pantelis, C.; Wood, S.J.; Berger, G. Stress, the hippocampus and the hypothalamic-pituitary-adrenal axis: Implications for the development of psychotic disorders. Aust. N. Z. J. Psychiatry 2006, 40, 725–741. [Google Scholar] [CrossRef]
- Gray, J.D.; Milner, T.A.; McEwen, B.S. Dynamic plasticity: The role of glucocorticoids, brain-derived neurotrophic factor and other trophic factors. Neuroscience 2013, 239, 214–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.J.; Diamond, D.M. The stressed hippocampus, synaptic plasticity and lost memories. Nat. Rev. Neurosci. 2002, 3, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Buss, C.; Davis, E.P.; Shahbaba, B.; Pruessner, J.C.; Head, K.; Sandman, C.A. Maternal cortisol over the course of pregnancy and subsequent child amygdala and hippocampus volumes and affective problems. Proc. Natl. Acad. Sci. USA 2012, 109, E1312–E1319. [Google Scholar] [CrossRef] [Green Version]
- Qiu, A.; Rifkin-Graboi, A.; Chen, H.; Chong, Y.-S.; Kwek, K.; Gluckman, P.D.; Fortier, M.V.; Meaney , M.J. Maternal anxiety and infants’ hippocampal development: Timing matters. Transl. Psychiatry 2013, 3, e306. [Google Scholar] [CrossRef] [Green Version]
- Scheinost, D.; Spann, M.N.; McDonough, L.; Peterson, B.S.; Monk, C. Associations between different dimensions of prenatal distress, neonatal hippocampal connectivity, and infant memory. Neuropsychopharmacology 2020, 45, 1272–1279. [Google Scholar] [CrossRef] [Green Version]
- Bale, T.L. The placenta and neurodevelopment: Sex differences in prenatal vulnerability. Dialogues Clin. Neurosci. 2022, 18, 459–464. [Google Scholar] [CrossRef]
- Sutherland, S.; Brunwasser, S.M. Sex Differences in Vulnerability to Prenatal Stress: A Review of the Recent Literature. Curr. Psychiatry Rep. 2018, 20, 1–12. [Google Scholar] [CrossRef]
- Cohen, S.; Kamarck, T.; Mermelstein, R. A global measure of perceived stress. J. Health Soc. Behav. 1983, 24, 385–396. [Google Scholar] [CrossRef]
- Radloff, L.S. The CES-D Scale: A Self-Report Depression Scale for Research in the General Population. Appl. Psychol. Meas. 1977, 1, 385–401. [Google Scholar] [CrossRef]
- Spielberger, C.D.; Gorsuch, R.L.; Lushene, R.E. Manual for the State-Trait. Anxiety Inventory. Consult. Psychol. 1970. [Google Scholar]
- Hendrix, C.L.; Brown, A.L.; McKenna, B.G.; Dunlop, A.L.; Corwin, E.J.; Brennan, P.A. Prenatal distress links maternal early life adversity to infant stress functioning in the next generation. J. Psychopathol. Clin. Sci. 2022, 131, 117. [Google Scholar] [CrossRef]
- Thomason, M.E.; Hect, J.L.; Waller, R.; Curtin, P. Interactive relations between maternal prenatal stress, fetal brain connectivity, and gestational age at delivery. Neuropsychopharmacology 2021, 46, 1839–1847. [Google Scholar] [CrossRef]
- Pruessner, J.C.; Kirschbaum, C.; Meinlschmid, G.; Hellhammer, D.H. Two formulas for computation of the area under the curve represent measures of total hormone concentration versus time-dependent change. Psychoneuroendocrinology 2003, 28, 916–931. [Google Scholar] [CrossRef]
- Shattuck, D.W.; Leahy, R.M. BrainSuite: An automated cortical surface identification tool. Med. Image Anal. 2002, 6, 129–142. [Google Scholar] [CrossRef]
- Serag, A.; Kyriakopoulou, V.; Rutherford, M.A.; Edwards, A.D.; Hajnal, J.V.; Aljabar, P.; Counsell, S.J.; Boardman, J.; Rueckert, D. A Multi-channel 4D Probabilistic Atlas of the Developing Brain: Application to Fetuses and Neonates. Ann. BMVA 2012, 2012, 1–14. [Google Scholar]
- Ji, L.; Hendrix, C.L.; Thomason, M.E. Empirical optimization of human fetal fMRI preprocessing steps. Netw. Neurosci. 2022, 1–37. [Google Scholar] [CrossRef]
- Whitfield-Gabrieli, S.; Nieto-Castanon, A. Conn: A Functional Connectivity Toolbox for Correlated and Anticorrelated Brain Networks. Brain Connect. 2012, 2, 125–141. [Google Scholar] [CrossRef] [Green Version]
- Spisák, T.; Spisák, Z.; Zunhammer, M.; Bingel, U.; Smith, S.; Nichols, T.; Kincses, T. Probabilistic TFCE: A generalized combination of cluster size and voxel intensity to increase statistical power. Neuroimage 2019, 185, 12–26. [Google Scholar] [CrossRef]
- Flournoy, J.C.; Vijayakumar, N.; Cheng, T.W.; Cosme, D.; Flannery, J.E.; Pfeifer, J.H. Improving practices and inferences in developmental cognitive neuroscience. Dev. Cogn. Neurosci. 2020, 45, 100807. [Google Scholar] [CrossRef]
- Van Den Heuvel, M.I.; Hect, J.L.; Smarr, B.L.; Qawasmeh, T.; Kriegsfeld, L.J.; Barcelona, J.; Hijazi, K.E.; Thomason, M.E. Maternal stress during pregnancy alters fetal cortico-cerebellar connectivity in utero and increases child sleep problems after birth. Sci. Rep. 2021, 11, 2228. [Google Scholar] [CrossRef] [PubMed]
- Duff, E.P.; Cunnington, R.; Egan, G.F. REX: Response exploration for neuroimaging datasets. Neuroinformatics 2007, 5, 223–234. [Google Scholar] [CrossRef]
- Belleau, E.L.; Treadway, M.T.; Pizzagalli, D.A. The Impact of Stress and Major Depressive Disorder on Hippocampal and Medial Prefrontal Cortex Morphology. Biol. Psychiatry 2019, 85, 443–453. [Google Scholar] [CrossRef]
- Ulrich-Lai, Y.; Herman, J. Neural Regulation of Endocrine and Autonomic Stress Responses. Nat. Rev. Neurosci. 2009, 10, 397–409. [Google Scholar] [CrossRef] [Green Version]
- Diorio, D.; Viau, V.; Meaney, M.J. The role of the medial prefrontal cortex (cingulate gyrus) in the regulation of hypothalamic-pituitary-adrenal responses to stress. J. Neurosci. 1993, 13, 3839–3847. [Google Scholar] [CrossRef] [Green Version]
- Bush, G.; Vogt, B.A.; Holmes, J.; Dale, A.M.; Greve, D.; Jenike, M.A.; Rosen, B.R. Dorsal anterior cingulate cortex: A role in reward-based decision making. Proc. Natl. Acad. Sci. USA 2002, 99, 523–528. [Google Scholar] [CrossRef] [Green Version]
- Rolls, E.T. The cingulate cortex and limbic systems for emotion, action, and memory. Brain Struct. Funct. 2019, 224, 3001–3018. [Google Scholar] [CrossRef] [Green Version]
- Bian, X.L.; Qin, C.; Cai, C.Y.; Zhou, Y.; Tao, Y.; Lin, Y.H.; Wu, H.Y.; Chang, L.; Luo, C.X.; Zhu, D.Y. Anterior Cingulate Cortex to Ventral Hippocampus Circuit Mediates Contextual Fear Generalization. J. Neurosci. 2019, 39, 5728–5739. [Google Scholar] [CrossRef] [Green Version]
- Weible, A.P. Remembering to attend: The anterior cingulate cortex and remote memory. Behav. Brain Res. 2013, 245, 63–75. [Google Scholar] [CrossRef]
- Bremner, J.D. Traumatic stress: Effects on the brain. Dialogues Clin. Neurosci. 2006, 8, 445–461. [Google Scholar] [CrossRef]
- Preston, A.R.; Eichenbaum, H. Interplay of hippocampus and prefrontal cortex in memory. Curr. Biol. 2013, 23, R764–R773. [Google Scholar] [CrossRef] [Green Version]
- Scaccianoce, S.; Catalani, A.; Lombardo, K.; Consoli, C.; Angelucci, L. Maternal glucocorticoid hormone influences nerve growth factor expression in the developing rat brain. Neuroreport 2001, 12, 2881–2884. [Google Scholar] [CrossRef] [PubMed]
- Mastorakos, G.; Ilias, I. Maternal and fetal hypothalamic-pituitary-adrenal axes during pregnancy and postpartum. Ann. N. Y. Acad. Sci. 2003, 997, 136–149. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.; Ho, J.T.; Torpy, D.J.; Rogers, A.; Doogue, M.; Lewis, J.G.; Czajko, R.J.; Inder, W.J. A Longitudinal Study of Plasma and Urinary Cortisol in Pregnancy and Postpartum. J. Clin. Endocrinol. Metab. 2011, 96, 1533–1540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sigurdsson, T.; Duvarci, S. Hippocampal-prefrontal interactions in cognition, behavior and psychiatric disease. Front. Syst. Neurosci. 2016, 9, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbas, H.; Blatt, G.J. Topographically specific hippocampal projections target functionally distinct prefrontal areas in the rhesus monkey. Hippocampus 1995, 5, 511–533. [Google Scholar] [CrossRef] [PubMed]
- Meyer, T.; Smeets, T.; Giesbrecht, T.; Quaedflieg, C.W.E.M.; Merckelbach, H. Acute stress differentially affects spatial configuration learning in high and low cortisol-responding healthy adults. Eur. J. Psychotraumatol. 2013, 4, 19854. [Google Scholar] [CrossRef]
- Callaghan, B.L.; Tottenham, N. The Stress Acceleration Hypothesis: Effects of early-life adversity on emotion circuits and behavior. Curr. Opin. Behav. Sci. 2016, 7, 76–81. [Google Scholar] [CrossRef] [Green Version]
- Pluess, M.; Belsky, J. Prenatal programming of postnatal plasticity? Dev. Psychopathol. 2011, 23, 29–38. [Google Scholar] [CrossRef] [Green Version]
- Schulz, A.J.; Williams, D.R.; Israel, B.A.; Lempert, L.B. Racial and Spatial Relations as Fundamental Determinants of Health in Detroit. Milbank Q. 2002, 80, 677–707. [Google Scholar] [CrossRef] [Green Version]
- Dismukes, A.; Shirtcliff, E.; Jones, C.W.; Zeanah, C.; Theall, K.; Drury, S. The development of the cortisol response to dyadic stressors in Black and White infants. Dev. Psychopathol. 2018, 30, 1995–2008. [Google Scholar] [CrossRef]
- Simon, C.D.; Adam, E.K.; Holl, J.L.; Wolfe, K.A.; Grobman, W.A.; Borders, A.E.B. Prenatal Stress and the Cortisol Awakening Response in African-American and Caucasian Women in the Third Trimester of Pregnancy. Matern. Child. Health J. 2016, 20, 2142–2149. [Google Scholar] [CrossRef] [PubMed]
- Pluess, M.; Bolten, M.; Pirke, K.M.; Hellhammer, D. Maternal trait anxiety, emotional distress, and salivary cortisol in pregnancy. Biol. Psychol. 2010, 83, 169–175. [Google Scholar] [CrossRef]
- Urizar, G.G.; Yim, I.S.; Rodriguez, A.; Schetter, C.D. The SMART Moms Program: A Randomized Trial of the Impact of Stress Management on Perceived Stress and Cortisol in Low-Income Pregnant Women. Psychoneuroendocrinology 2019, 104, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Lazarides, C.; Ward, E.B.; Buss, C.; Chen, W.P.; Voelkle, M.C.; Gillen, D.L.; Wadhwa, P.D.; Entringer, S. Psychological stress and cortisol during pregnancy: An ecological momentary assessment (EMA)-Based within- and between-person analysis. Psychoneuroendocrinology 2020, 121, 104848. [Google Scholar] [CrossRef]
- Glover, V. Prenatal stress and its effects on the fetus and the child: Possible Underlying biological mechanisms. Adv. Neurobiol. 2015, 10, 269–283. [Google Scholar] [PubMed]
- Harris, A.; Seckl, J. Glucocorticoids, prenatal stress and the programming of disease. Horm. Behav. 2011, 59, 279–289. [Google Scholar] [CrossRef]
- Cerritelli, F.; Frasch, M.G.; Antonelli, M.C.; Viglione, C.; Vecchi, S.; Chiera, M.; Manzotti, A. Review on the Vagus Nerve and Autonomic Nervous System During Fetal Development: Searching for Critical Windows. Front. Neurosci. 2021, 15, 1184. [Google Scholar] [CrossRef]
- Glover, V.; O’Donnell, K.J.; O’Connor, T.G.; Fisher, J. Prenatal maternal stress, fetal programming, and mechanisms underlying later psychopathology—A global perspective. Dev. Psychopathol. 2018, 30, 843–854. [Google Scholar] [CrossRef]
- Monk, C.; Lugo-Candelas, C.; Trumpff, C. Prenatal Developmental Origins of Future Psychopathology: Mechanisms and Pathways. Annu. Rev. Clin. Psychol. 2019, 15, 317–344. [Google Scholar] [CrossRef]
- Thomason, M.E.; Palopoli, A.C.; Jariwala, N.N.; Werchan, D.M.; Chen, A.; Adhikari, S.; Espinoza-Heredia, C.; Brito, N.H.; Trentacosta, C.J. Miswiring the brain: Human prenatal Δ9-tetrahydrocannabinol use associated with altered fetal hippocampal brain network connectivity. Dev. Cogn. Neurosci. 2021, 51, 101000. [Google Scholar] [CrossRef]
- LeWinn, K.Z.; Sheridan, M.A.; Keyes, K.M.; Hamilton, A.; McLaughlin, K.A. Sample composition alters associations between age and brain structure. Nat. Commun. 2017, 8, 874. [Google Scholar] [CrossRef] [Green Version]
- Azhari, A.; Truzzi, A.; Neoh, M.J.; Balagtas, J.P.; Tan, H.H.; Goh, P.P.; Ang, X.A.; Setoh, P.; Rigo, P.; Bornstein, M.H.; et al. A decade of infant neuroimaging research: What have we learned and where are we going? Infant Behav. Dev. 2020, 58, 101389. [Google Scholar] [CrossRef]
- Falk, E.B.; Hyde, L.W.; Mitchell, C.; Faul, J.; Gonzalez, R.; Heitzeg, M.M.; Keating, D.P.; Langa, K.M.; Martz, M.E.; Maslowsky, J.; et al. What is a representative brain? Neuroscience meets population science. Proc. Natl. Acad. Sci. USA 2013, 110, 17615–17622. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Final Sample (n = 77) | Excluded (n = 88) | Differences by Group | Final Sample | |||
|---|---|---|---|---|---|---|
| Male (n = 48) | Female (n = 29) | Differences by Fetal Sex | ||||
| M (SD) or N (%) | Stats. | M (SD) or N (%) | Stats. | |||
| Sociodemographics | ||||||
| Maternal age | 25.38 (4.42) years | 25.29 (4.83) | t = 0.12, p = 0.91 | 26.03 (4.81) | 24.32 (3.51) | t = 1.80, p = 0.08 |
| GA at fetal MRI | 32.82 (3.86) weeks | 33.17 (3.60) | t = −0.49, p = 0.62 | 33.00 (3.87) | 32.53 (3.90) | t = 0.52, p = 0.61 |
| Maternal race | X2 = 4.87, p = 0.30 | X2 = 4.54, p = 0.21 | ||||
| Black | 60 (82%) | 74 (87%) | 36 (78%) | 24 (89%) | ||
| White | 9 (12%) | 5 (6%) | 7 (15%) | 2 (7%) | ||
| Bi-racial | 3 (4%) | 4 (5%) | 3 (7%) | 0 (0%) | ||
| Asian American | 1 (1%) | 0 (0%) | 0 (0%) | 1 (4%) | ||
| Other | 0 (0%) | 2 (2%) | 0 (0%) | 0 (0%) | ||
| Latina | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | ||
| Native American | 0 (0%) | 0 (0%) | 0 (0%) | 0 (0%) | ||
| Maternal education | 37 (51%) HS diploma/GED or less | 50 (59%) | X2 = 1.27, p = 0.26 | 24 (52%) | 13 (48%) | X2 = 0.19, p = 0.66 |
| Maternal income | 43 (63%) < $20,000 | 52 (69%) | X2 = 0.36, p = 0.55 | 25 (60%) | 18 (69%) | X2 = 0.73, p = 0.39 |
| Maternal marital status | 42 (58%) single | 48 (56%) | X2 = 0.02, p = 0.90 | 28 (62%) | 14 (52%) | X2 = 1.02, p = 0.31 |
| GA at birth | 38.97 (1.47) weeks | 37.85 (3.28) | t = 2.76, p = 0.007 | 39.25 (1.56) | 38.51 (1.19) | t = 2.20, p = 0.03 |
| Birth weight | 3179.97 (536.95) g | 2999.31 (780.99) | t = 1.68, p = 0.09 | 3356.25 (540.85) | 2888.20 (387.42) | t = 4.07, p < 0.001 |
| Fetal sex | 29 (38%) female | 43 (49%) | X2 = 2.10, p = 0.15 | -- | -- | -- |
| rsfMRI characteristics | ||||||
| # low-motion volumes | 168.14 (51.84) | 163.56 (55.46) | t = 0.45, p = 0.65 | 167.79 (56.57) | 168.72 (43.82) | t = −0.08, p = 0.94 |
| Mean XYZ translation | 0.24 (0.10) mm | 0.23 (0.08) | t = 0.46, p = 0.65 | 0.22 (0.09) | 0.26 (0.11) | t = −1.49, p = 0.14 |
| Mean PYR rotation | 0.40 (0.16) mm | 0.39 (0.17) | t = 0.46, p = 0.64 | 0.39 (0.15) | 0.43 (0.18) | t = −0.90, p = 0.37 |
| Maternal prenatal distress and cortisol | ||||||
| PSS | 15.77 (6.65) | 17.13 (6.86) | t = −1.24, p = 0.22 | 16.35 (5.83) | 14.86 (7.79) | t = 0.94, p = 0.35 |
| CES-D | 15.00 (9.89) | 13.77 (9.73) | t = 0.80, p = 0.43 | 14.64 (9.65) | 15.61 (10.43) | t = −0.41, p = 0.68 |
| STAI | 36.01(8.23) | 35.96 (8.87) | t = 0.04, p = 0.97 | 36.37 (7.55) | 35.45 (9.33) | t = 0.47, p = 0.64 |
| Cortisol AUCg | 191.42 (146.30) | -- | -- | 195.80 (163.15) | 184.16 (115.45) | t = 0.34, p = 0.74 |
| Cortisol AUCi | −15.25 (105.53) | -- | -- | −14.35 (106.88) | −16.73 (105.10) | t = 0.10, p = 0.92 |
| Component 1 | |
|---|---|
| PSS | 0.89 |
| CES-D | 0.88 |
| STAI | 0.88 |
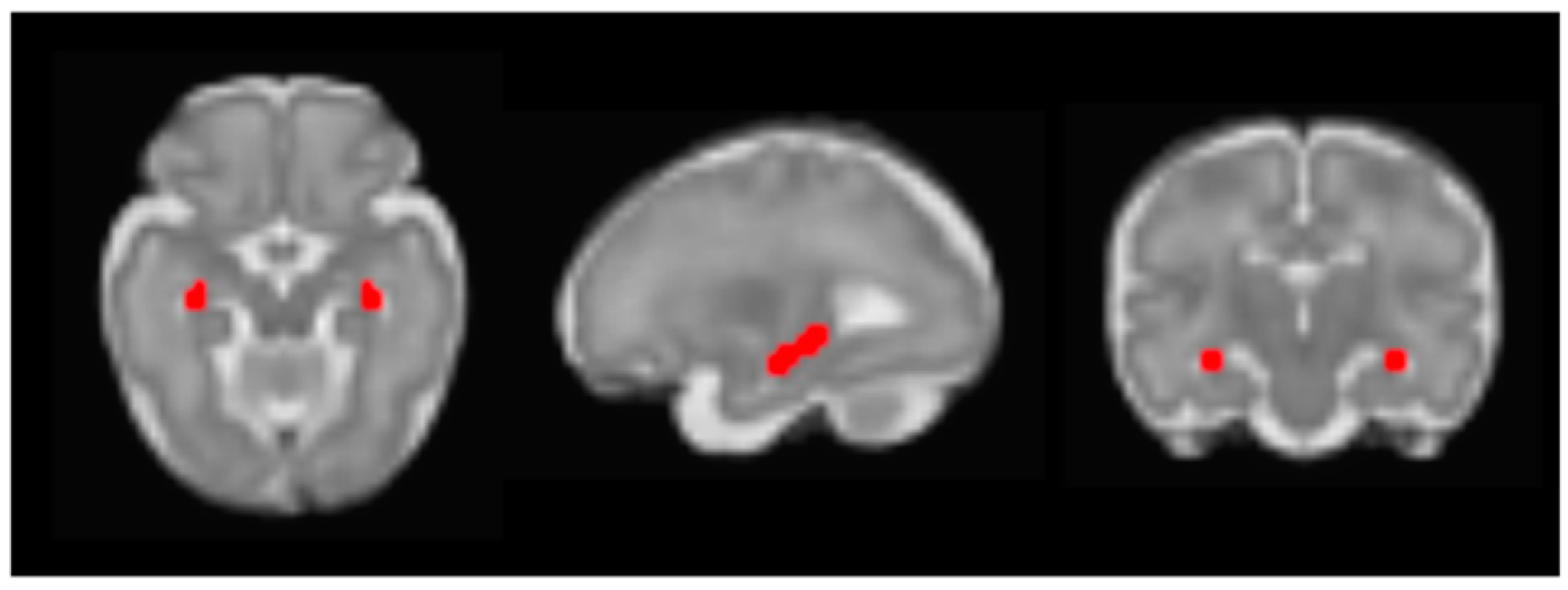
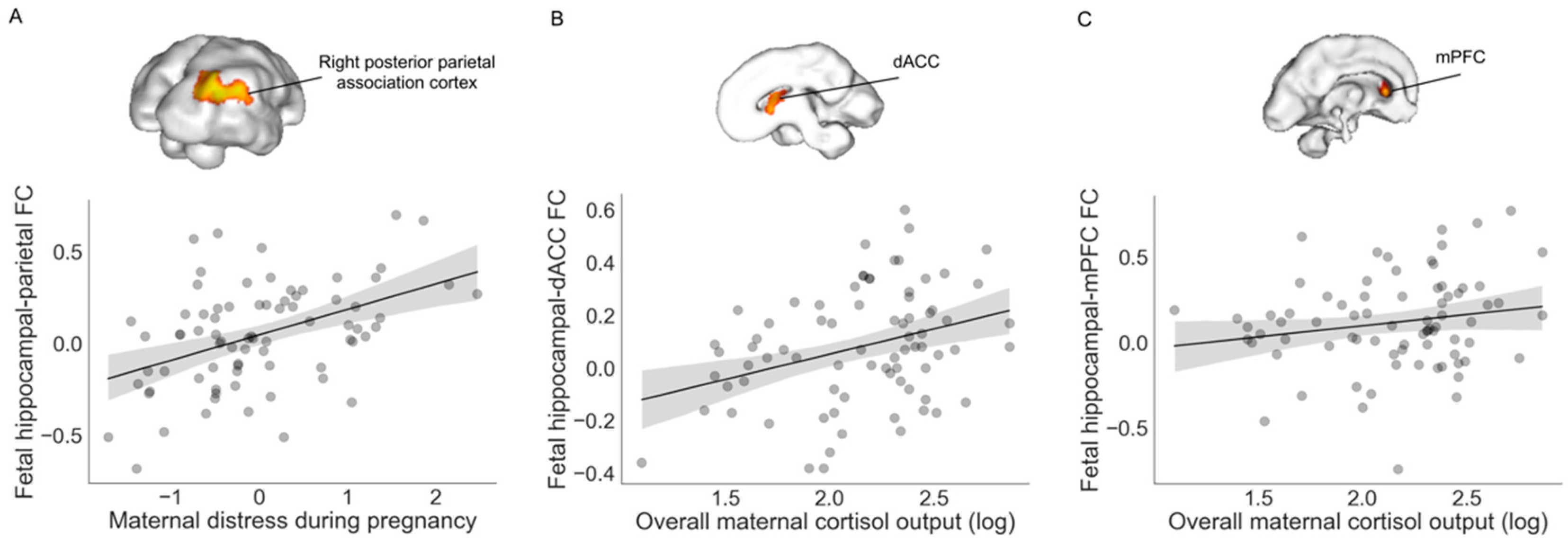
| X | Y | Z | Intensity (Fisher Z) | Direction of Effect | |
|---|---|---|---|---|---|
| Prenatal Distress | |||||
| Right posterior parietal association cortex | 30 | −28 | 6 | 3.83 | Positive |
| Prenatal Cortisol AUCg | |||||
| dACC | 2 | 16 | −2 | 2.83 | Positive |
| Left mPFC | −12 | 24 | −6 | 2.64 | Positive |
| Number Frames Included in Analysis | Mean XYZ Translation | Mean PYR Rotation | |
|---|---|---|---|
| Maternal predictors | |||
| Prenatal distress | r = 0.23, p = 0.04 | r = −0.02, p = 0.83 | r = 0.04, p = 0.73 |
| Cortisol AUCi | r = −0.15, p = 0.20 | r = −0.09, p = 0.43 | r = −0.11, p = 0.34 |
| Cortisol AUCg | r = 0.14, p = 0.21 | r = 0.25, p = 0.03 | r = 0.21, p = 0.07 |
| Fetal RSFC metrics | |||
| Hippocampal–dACC FC | r = 0.02, p = 0.86 | r = 0.07, p = 0.58 | r = 0.09, p = 0.43 |
| Hippocampal–mPFC FC | r = 0.04, p = 0.73 | r = −0.09, p = 0.44 | r = −0.17, p = 0.14 |
| Hippocampal–parietal FC | r = 0.13, p = 0.26 | r = −0.10, p = 0.38 | r = −0.04, p = 0.73 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hendrix, C.L.; Srinivasan, H.; Feliciano, I.; Carré, J.M.; Thomason, M.E. Fetal Hippocampal Connectivity Shows Dissociable Associations with Maternal Cortisol and Self-Reported Distress during Pregnancy. Life 2022, 12, 943. https://doi.org/10.3390/life12070943
Hendrix CL, Srinivasan H, Feliciano I, Carré JM, Thomason ME. Fetal Hippocampal Connectivity Shows Dissociable Associations with Maternal Cortisol and Self-Reported Distress during Pregnancy. Life. 2022; 12(7):943. https://doi.org/10.3390/life12070943
Chicago/Turabian StyleHendrix, Cassandra L., Harini Srinivasan, Integra Feliciano, Justin M. Carré, and Moriah E. Thomason. 2022. "Fetal Hippocampal Connectivity Shows Dissociable Associations with Maternal Cortisol and Self-Reported Distress during Pregnancy" Life 12, no. 7: 943. https://doi.org/10.3390/life12070943
APA StyleHendrix, C. L., Srinivasan, H., Feliciano, I., Carré, J. M., & Thomason, M. E. (2022). Fetal Hippocampal Connectivity Shows Dissociable Associations with Maternal Cortisol and Self-Reported Distress during Pregnancy. Life, 12(7), 943. https://doi.org/10.3390/life12070943