
Clinical and Molecular Aspects of Iron Metabolism in Failing Myocytes

 , , , ,
, , , ,  ,
,
Abstract
:1. Introduction
2. Iron Homeostasis in the Heart
2.1. Iron Transport
2.1.1. Transferrin Receptor 1
2.1.2. Transferrin Receptor 2
2.1.3. Divalent Metal Transporter 1
2.1.4. Zinc Transporters
2.1.5. Calcium Channels
2.1.6. Ferroportin
2.2. Iron Storage
2.3. Iron Regulation
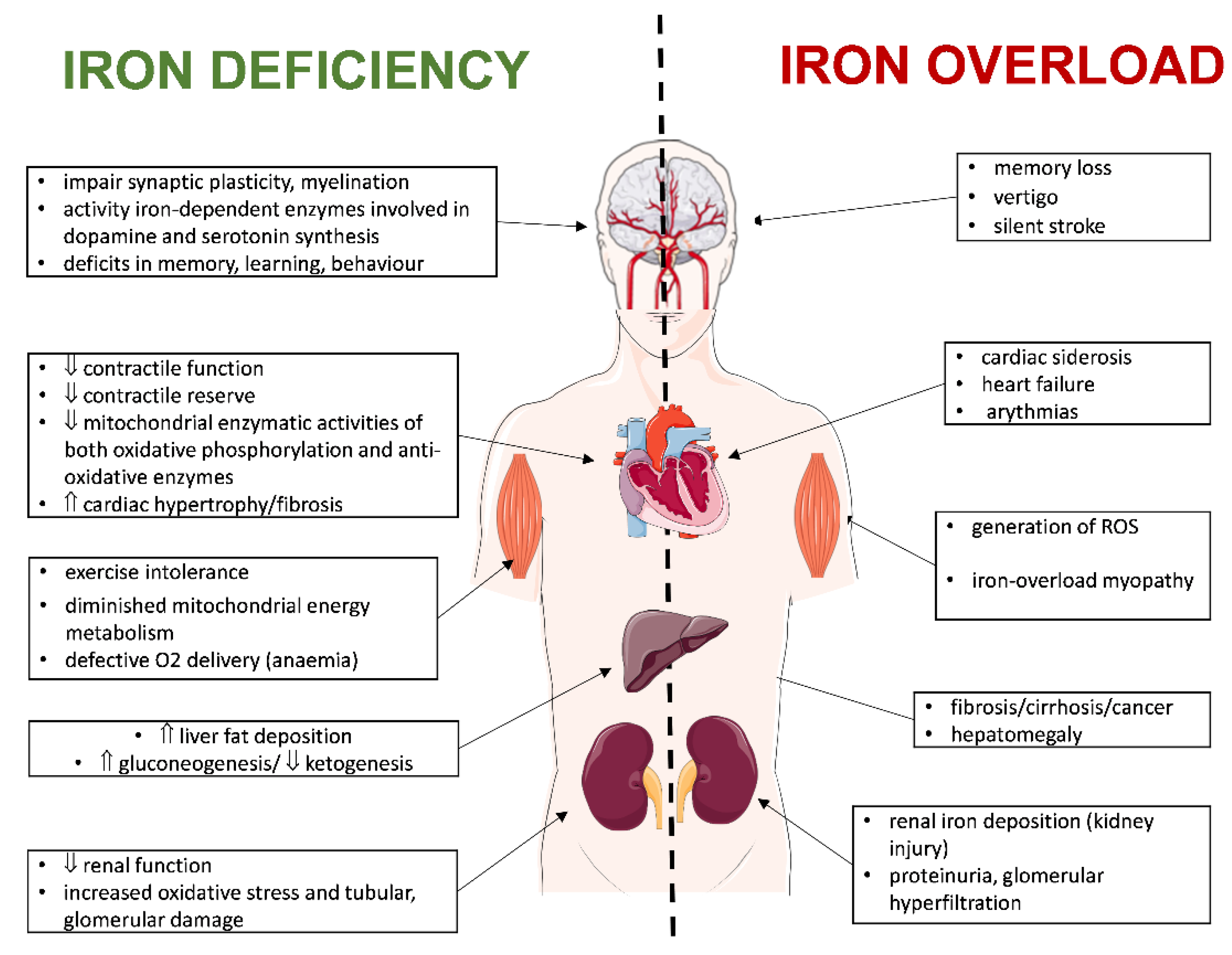
3. Dysregulation of Iron Metabolism in the Failing Heart
3.1. Iron Overload
3.1.1. Labile Iron Pool
3.1.2. Iron-Overload Consequences
3.2. Iron Deficiency
3.2.1. Reduced Iron Intake and Bioavailability
3.2.2. Reduced Iron Absorption
3.2.3. Increased Iron Loss
3.2.4. Iron-Deficiency Consequences
4. Iron Metabolism in the Failing Human Heart
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Paterek, A.; Mackiewicz, U.; Mączewski, M. Iron and the Heart: A Paradigm Shift from Systemic to Cardiomyocyte Abnormalities. J. Cell. Physiol. 2019, 234, 21613–21629. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.-L.; Ghosh, M.C.; Rouault, T.A. The Physiological Functions of Iron Regulatory Proteins in Iron Homeostasis—An Update. Front. Pharmacol. 2014, 5, 124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghafourian, K.; Shapiro, J.S.; Goodman, L.; Ardehali, H. Iron and Heart Failure. JACC Basic Transl. Sci. 2020, 5, 300–313. [Google Scholar] [CrossRef] [PubMed]
- Ponikowski, P.; Kirwan, B.-A.; Anker, S.D.; McDonagh, T.; Dorobantu, M.; Drozdz, J.; Fabien, V.; Filippatos, G.; Göhring, U.M.; Keren, A.; et al. Ferric Carboxymaltose for Iron Deficiency at Discharge after Acute Heart Failure: A Multicentre, Double-Blind, Randomised, Controlled Trial. Lancet 2020, 396, 1895–1904. [Google Scholar] [CrossRef]
- Saito, H. Metabolism of Iron Stores. Nagoya J Med Sci. 2014, 76, 235–254. [Google Scholar] [PubMed]
- Wienbergen, H.; Pfister, O.; Hochadel, M.; Michel, S.; Bruder, O.; Remppis, B.A.; Maeder, M.T.; Strasser, R.; von Scheidt, W.; Pauschinger, M.; et al. Usefulness of Iron Deficiency Correction in Management of Patients With Heart Failure [from the Registry Analysis of Iron Deficiency-Heart Failure (RAID-HF) Registry]. Am. J. Cardiol. 2016, 118, 1875–1880. [Google Scholar] [CrossRef] [PubMed]
- Jankowska, E.A.; Rozentryt, P.; Witkowska, A.; Nowak, J.; Hartmann, O.; Ponikowska, B.; Borodulin-Nadzieja, L.; von Haehling, S.; Doehner, W.; Banasiak, W.; et al. Iron Deficiency Predicts Impaired Exercise Capacity in Patients with Systolic Chronic Heart Failure. J. Card. Fail. 2011, 17, 899–906. [Google Scholar] [CrossRef]
- Kruszewski, M. Labile Iron Pool: The Main Determinant of Cellular Response to Oxidative Stress. Mutat. Res. Fundam. Mol. Mech. Mutagenesis 2003, 531, 81–92. [Google Scholar] [CrossRef]
- Lewandowska, H.; Kalinowska, M.; Brzóska, K.; Wójciuk, K.; Wójciuk, G.; Kruszewski, M. Nitrosyl Iron Complexes—Synthesis, Structure and Biology. Dalton Trans. 2011, 40, 8273–8289. [Google Scholar] [CrossRef]
- Kirchhausen, T.; Owen, D.; Harrison, S.C. Molecular Structure, Function, and Dynamics of Clathrin-Mediated Membrane Traffic. Cold Spring Harb. Perspect. Biol. 2014, 6, a016725. [Google Scholar] [CrossRef] [Green Version]
- Kawabata, H. Transferrin and Transferrin Receptors Update. Free Radic. Biol. Med. 2019, 133, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Barrientos, T.; Mao, L.; Rockman, H.A.; Sauve, A.A.; Andrews, N.C. Lethal Cardiomyopathy in Mice Lacking Transferrin Receptor in the Heart. Cell Rep. 2015, 13, 533–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, J.; Zhang, Z.; Wang, S.; Chen, Y.; Liu, C.; Xu, S.; Wang, D.; Su, J.; Ni, M.; Yu, J.; et al. Transferrin Receptor Functionally Marks Thermogenic Adipocytes. Front. Cell. Dev. Biol. 2020, 8, 572459. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Pan, X.; Pan, G.; Song, Z.; He, Y.; Zhang, S.; Ye, X.; Yang, X.; Xie, E.; Wang, X.; et al. Transferrin Receptor 1 Regulates Thermogenic Capacity and Cell Fate in Brown/Beige Adipocytes. Adv. Sci. 2020, 7, 1903366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campisi, A.; Bonfanti, R.; Raciti, G.; Bonaventura, G.; Legnani, L.; Magro, G.; Pennisi, M.; Russo, G.; Chiacchio, M.A.; Pappalardo, F.; et al. Gene Silencing of Transferrin-1 Receptor as a Potential Therapeutic Target for Human Follicular and Anaplastic Thyroid Cancer. Mol. Ther. Oncolytics 2020, 16, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Shah, Y.M.; Matsubara, T.; Ito, S.; Yim, S.-H.; Gonzalez, F.J. Intestinal Hypoxia-Inducible Transcription Factors Are Essential for Iron Absorption Following Iron Deficiency. Cell Metab. 2009, 9, 152–164. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Zhang, D.; Li, Y. LncRNAs in Cardiac Hypertrophy: From Basic Science to Clinical Application. J. Cell. Mol. Med. 2020, 24, 11638–11645. [Google Scholar] [CrossRef]
- Roetto, A.; Mezzanotte, M.; Pellegrino, R.M. The Functional Versatility of Transferrin Receptor 2 and Its Therapeutic Value. Pharmaceuticals 2018, 11, 115. [Google Scholar] [CrossRef] [Green Version]
- Kawabata, H.; Germain, R.S.; Ikezoe, T.; Tong, X.; Green, E.M.; Gombart, A.F.; Koeffler, H.P. Regulation of Expression of Murine Transferrin Receptor 2. Blood 2001, 98, 1949–1954. [Google Scholar] [CrossRef] [Green Version]
- Boero, M.; Pagliaro, P.; Tullio, F.; Pellegrino, R.M.; Palmieri, A.; Ferbo, L.; Saglio, G.; De Gobbi, M.; Penna, C.; Roetto, A. A Comparative Study of Myocardial Molecular Phenotypes of Two Tfr2β Null Mice: Role in Ischemia/Reperfusion. Biofactors 2015, 41, 360–371. [Google Scholar] [CrossRef]
- Kawabata, H.; Yang, R.; Hirama, T.; Vuong, P.T.; Kawano, S.; Gombart, A.F.; Koeffler, H.P. Molecular Cloning of Transferrin Receptor 2. A New Member of the Transferrin Receptor-like Family. J. Biol. Chem. 1999, 274, 20826–20832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nam, H.; Wang, C.-Y.; Zhang, L.; Zhang, W.; Hojyo, S.; Fukada, T.; Knutson, M.D. ZIP14 and DMT1 in the Liver, Pancreas, and Heart Are Differentially Regulated by Iron Deficiency and Overload: Implications for Tissue Iron Uptake in Iron-Related Disorders. Haematologica 2013, 98, 1049–1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumfu, S.; Chattipakorn, S.; Srichairatanakool, S.; Settakorn, J.; Fucharoen, S.; Chattipakorn, N. T-Type Calcium Channel as a Portal of Iron Uptake into Cardiomyocytes of Beta-Thalassemic Mice. Eur. J. Haematol. 2011, 86, 156–166. [Google Scholar] [CrossRef]
- Ke, Y.; Chen, Y.Y.; Chang, Y.Z.; Duan, X.L.; Ho, K.P.; Jiang, D.H.; Wang, K.; Qian, Z.M. Post-Transcriptional Expression of DMT1 in the Heart of Rat. J. Cell. Physiol. 2003, 196, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Qatato, M.; Bonadonna, M.; Palais, G.; Ertl, A.; Schmidt, G.; Polycarpou-Schwarz, M.; Karim, Z.; Galy, B. IRE-Dependent Regulation of Intestinal Dmt1 Prevails during Chronic Dietary Iron Deficiency but Is Dispensable in Conditions of Acute Erythropoietic Stress. HemaSphere 2022, 6, e693. [Google Scholar] [CrossRef]
- Tybl, E.; Gunshin, H.; Gupta, S.; Barrientos, T.; Bonadonna, M.; Celma Nos, F.; Palais, G.; Karim, Z.; Sanchez, M.; Andrews, N.C.; et al. Control of Systemic Iron Homeostasis by the 3′ Iron-Responsive Element of Divalent Metal Transporter 1 in Mice. Hemasphere 2020, 4, e459. [Google Scholar] [CrossRef]
- Ingrassia, R.; Garavaglia, B.; Memo, M. DMT1 Expression and Iron Levels at the Crossroads between Aging and Neurodegeneration. Front. Neurosci. 2019, 13, 575. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.-Y.; Jenkitkasemwong, S.; Duarte, S.; Sparkman, B.K.; Shawki, A.; Mackenzie, B.; Knutson, M.D. ZIP8 Is an Iron and Zinc Transporter Whose Cell-Surface Expression Is Upregulated by Cellular Iron Loading. J. Biol. Chem. 2012, 287, 34032–34043. [Google Scholar] [CrossRef] [Green Version]
- Gálvez-Peralta, M.; He, L.; Jorge-Nebert, L.F.; Wang, B.; Miller, M.L.; Eppert, B.L.; Afton, S.; Nebert, D.W. ZIP8 Zinc Transporter: Indispensable Role for Both Multiple-Organ Organogenesis and Hematopoiesis In Utero. PLoS ONE 2012, 7, e36055. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Gálvez-Peralta, M.; Zhang, X.; Deng, J.; Liu, Z.; Nebert, D.W. In Utero Gene Expression in the Slc39a8(Neo/Neo) Knockdown Mouse. Sci. Rep. 2018, 8, 10703. [Google Scholar] [CrossRef] [Green Version]
- Jenkitkasemwong, S.; Wang, C.-Y.; Coffey, R.; Zhang, W.; Chan, A.; Biel, T.; Kim, J.-S.; Hojyo, S.; Fukada, T.; Knutson, M.D. SLC39A14 Is Required for the Development of Hepatocellular Iron Overload in Murine Models of Hereditary Hemochromatosis. Cell Metab. 2015, 22, 138–150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopin, K.V.; Gray, I.P.; Obejero-Paz, C.A.; Thévenod, F.; Jones, S.W. Fe2+ Block and Permeation of CaV3.1 (A1G) T-Type Calcium Channels: Candidate Mechanism for Non–Transferrin-Mediated Fe2+ Influx. Mol. Pharmacol. 2012, 82, 1194–1204. [Google Scholar] [CrossRef] [Green Version]
- Oudit, G.Y.; Sun, H.; Trivieri, M.G.; Koch, S.E.; Dawood, F.; Ackerley, C.; Yazdanpanah, M.; Wilson, G.J.; Schwartz, A.; Liu, P.P.; et al. L-Type Ca2+ Channels Provide a Major Pathway for Iron Entry into Cardiomyocytes in Iron-Overload Cardiomyopathy. Nat. Med. 2003, 9, 1187–1194. [Google Scholar] [CrossRef] [PubMed]
- Kumfu, S.; Chattipakorn, S.C.; Fucharoen, S.; Chattipakorn, N. Dual T-Type and L-Type Calcium Channel Blocker Exerts Beneficial Effects in Attenuating Cardiovascular Dysfunction in Iron-Overloaded Thalassaemic Mice. Exp. Physiol. 2016, 101, 521–539. [Google Scholar] [CrossRef] [Green Version]
- Syamsunarno, M.R.A.A.; Rakhimullah, A.B.; Gamayani, U.; Kurabayasi, M.; Iso, T.; Safitri, R.; Panigoro, R. Iron Administration Affects Cardiac Calcium Channel Expression in Mice: The Role of Cardiac Calcium Channel Expression in The Heart of Iron Overload Mice Model. Indones. Biomed. J. 2020, 12, 261–266. [Google Scholar] [CrossRef]
- Rhee, J.-W.; Yi, H.; Thomas, D.; Lam, C.K.; Belbachir, N.; Tian, L.; Qin, X.; Malisa, J.; Lau, E.; Paik, D.T.; et al. Modeling Secondary Iron Overload Cardiomyopathy with Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Cell Rep. 2020, 32, 107886. [Google Scholar] [CrossRef]
- Guan, W.; Xia, M.; Ji, M.; Chen, B.; Li, S.; Zhang, M.; Liang, S.; Chen, B.; Gong, W.; Dong, C.; et al. Iron Induces Two Distinct Ca2+ Signalling Cascades in Astrocytes. Commun. Biol. 2021, 4, 525. [Google Scholar] [CrossRef] [PubMed]
- Sadaf, A.; Hasan, B.; Das, J.K.; Colan, S.; Alvi, N. Calcium Channel Blockers for Preventing Cardiomyopathy due to Iron Overload in People with Transfusion-Dependent Beta Thalassaemia. Cochrane Database Syst. Rev. 2018, 7, CD011626. [Google Scholar] [CrossRef]
- Subramaniam, V.N.; Wallace, D.F.; Dixon, J.L.; Fletcher, L.M.; Crawford, D.H. Ferroportin Disease due to the A77D Mutation in Australia. Gut 2005, 54, 1048–1049. [Google Scholar] [CrossRef] [Green Version]
- Altamura, S.; Kessler, R.; Gröne, H.-J.; Gretz, N.; Hentze, M.W.; Galy, B.; Muckenthaler, M.U. Resistance of Ferroportin to Hepcidin Binding Causes Exocrine Pancreatic Failure and Fatal Iron Overload. Cell Metab. 2014, 20, 359–367. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.-L.; Ghosh, M.C.; Ollivierre, H.; Li, Y.; Rouault, T.A. Ferroportin Deficiency in Erythroid Cells Causes Serum Iron Deficiency and Promotes Hemolysis Due to Oxidative Stress. Blood 2018, 132, 2078–2087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Garringer, H.J.; Goodwin, C.B.; Richine, B.; Acton, A.; VanDuyn, N.; Muhoberac, B.B.; Irimia-Dominguez, J.; Chan, R.J.; Peacock, M.; et al. Systemic and Cerebral Iron Homeostasis in Ferritin Knock-Out Mice. PLoS ONE 2015, 10, e0117435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferreira, C.; Santambrogio, P.; Martin, M.-E.; Andrieu, V.; Feldmann, G.; Hénin, D.; Beaumont, C. H Ferritin Knockout Mice: A Model of Hyperferritinemia in the Absence of Iron Overload. Blood 2001, 98, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Fang, X.; Cai, Z.; Wang, H.; Han, D.; Cheng, Q.; Zhang, P.; Gao, F.; Yu, Y.; Song, Z.; Wu, Q.; et al. Loss of Cardiac Ferritin H Facilitates Cardiomyopathy via Slc7a11-Mediated Ferroptosis. Circ. Res. 2020, 127, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Vanoaica, L.; Richman, L.; Jaworski, M.; Darshan, D.; Luther, S.A.; Kühn, L.C. Conditional Deletion of Ferritin H in Mice Reduces B and T Lymphocyte Populations. PLoS ONE 2014, 9, e89270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkinson, J.; Di, X.; Schönig, K.; Buss, J.L.; Kock, N.D.; Cline, J.M.; Saunders, T.L.; Bujard, H.; Torti, S.V.; Torti, F.M. Tissue-Specific Expression of Ferritin H Regulates Cellular Iron Homoeostasis in Vivo. Biochem. J. 2006, 395, 501–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mesquita, G.; Silva, T.; Gomes, A.C.; Oliveira, P.F.; Alves, M.G.; Fernandes, R.; Almeida, A.A.; Moreira, A.C.; Gomes, M.S. H-Ferritin Is Essential for Macrophages’ Capacity to Store or Detoxify Exogenously Added Iron. Sci. Rep. 2020, 10, 3061. [Google Scholar] [CrossRef] [Green Version]
- Vidal, R.; Miravalle, L.; Gao, X.; Barbeito, A.G.; Baraibar, M.A.; Hekmatyar, S.K.; Widel, M.; Bansal, N.; Delisle, M.B.; Ghetti, B. Expression of a Mutant form of the Ferritin Light Chain Gene Induces Neurodegeneration and Iron Overload in Transgenic Mice. J. Neurosci. 2008, 28, 60–67. [Google Scholar] [CrossRef] [Green Version]
- Garringer, H.J.; Irimia, J.M.; Li, W.; Goodwin, C.B.; Richine, B.; Acton, A.; Chan, R.J.; Peacock, M.; Muhoberac, B.B.; Ghetti, B.; et al. Effect of Systemic Iron Overload and a Chelation Therapy in a Mouse Model of the Neurodegenerative Disease Hereditary Ferritinopathy. PLoS ONE 2016, 11, e0161341. [Google Scholar] [CrossRef] [Green Version]
- Kosmidis, S.; Botella, J.A.; Mandilaras, K.; Schneuwly, S.; Skoulakis, E.M.C.; Rouault, T.A.; Missirlis, F. Ferritin Overexpression in Drosophila Glia Leads to Iron Deposition in the Optic Lobes and Late-Onset Behavioural Defects. Neurobiol. Dis. 2011, 43, 213–219. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Li, Y.; Liu, B.; Zhang, S.; Wu, L.; Zhu, X.; Chen, Q. Expression of Ferritin Light Chain (FTL) Is Elevated in Glioblastoma, and FTL Silencing Inhibits Glioblastoma Cell Proliferation via the GADD45/JNK Pathway. PLoS ONE 2016, 11, e0149361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levi, S.; Ripamonti, M.; Dardi, M.; Cozzi, A.; Santambrogio, P. Mitochondrial Ferritin: Its Role in Physiological and Pathological Conditions. Cells 2021, 10, 1969. [Google Scholar] [CrossRef] [PubMed]
- Bartnikas, T.B.; Campagna, D.R.; Antiochos, B.; Mulhern, H.; Pondarré, C.; Fleming, M.D. Characterization of Mitochondrial Ferritin-Deficient Mice. Am. J. Hematol. 2010, 85, 958–960. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Wang, P.; Wu, Q.; Xie, L.; Cui, Y.; Li, H.; Yu, P.; Chang, Y.-Z. The Construction and Characterization of Mitochondrial Ferritin Overexpressing Mice. Int. J. Mol. Sci. 2017, 18, 1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koeppen, A.H.; Ramirez, R.L.; Becker, A.B.; Bjork, S.T.; Levi, S.; Santambrogio, P.; Parsons, P.J.; Kruger, P.C.; Yang, K.X.; Feustel, P.J.; et al. The Pathogenesis of Cardiomyopathy in Friedreich Ataxia. PLoS ONE 2015, 10, e0116396. [Google Scholar] [CrossRef]
- Roetto, A.; Papanikolaou, G.; Politou, M.; Alberti, F.; Girelli, D.; Christakis, J.; Loukopoulos, D.; Camaschella, C. Mutant Antimicrobial Peptide Hepcidin Is Associated with Severe Juvenile Hemochromatosis. Nat. Genet. 2003, 33, 21–22. [Google Scholar] [CrossRef]
- Finberg, K.E. Regulation of Systemic Iron Homeostasis. Curr. Opin. Hematol. 2013, 20, 208–214. [Google Scholar] [CrossRef]
- Massagué, J.; Seoane, J.; Wotton, D. Smad Transcription Factors. Genes Dev. 2005, 19, 2783–2810. [Google Scholar] [CrossRef] [Green Version]
- Ramos, E.; Kautz, L.; Rodriguez, R.; Hansen, M.; Gabayan, V.; Ginzburg, Y.; Roth, M.-P.; Nemeth, E.; Ganz, T. Evidence for Distinct Pathways of Hepcidin Regulation by Acute and Chronic Iron Loading in Mice. Hepatology 2011, 53, 1333–1341. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.F.; Summerville, L.; Crampton, E.M.; Frazer, D.M.; Anderson, G.J.; Subramaniam, V.N. Combined Deletion of Hfe and Transferrin Receptor 2 in Mice Leads to Marked Dysregulation of Hepcidin and Iron Overload. Hepatology 2009, 50, 1992–2000. [Google Scholar] [CrossRef]
- Poli, M.; Luscieti, S.; Gandini, V.; Maccarinelli, F.; Finazzi, D.; Silvestri, L.; Roetto, A.; Arosio, P. Transferrin Receptor 2 and HFE Regulate Furin Expression via Mitogen-Activated Protein Kinase/Extracellular Signal-Regulated Kinase (MAPK/Erk) Signaling. Implications for Transferrin-Dependent Hepcidin Regulation. Haematologica 2010, 95, 1832–1840. [Google Scholar] [CrossRef] [PubMed]
- Pantopoulos, K. Iron Metabolism and the IRE/IRP Regulatory System: An Update. Ann. N. Y. Acad. Sci. 2004, 1012, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Thompson, M.S.; Adkins, K.M. Characteristics of the Iron-Responsive Element (IRE) Stems in the Untranslated Regions of Animal MRNAs. Open Biochem. J. 2021, 15, 26–37. [Google Scholar] [CrossRef]
- Haddad, S.; Wang, Y.; Galy, B.; Korf-Klingebiel, M.; Hirsch, V.; Baru, A.M.; Rostami, F.; Reboll, M.R.; Heineke, J.; Flögel, U.; et al. Iron-Regulatory Proteins Secure Iron Availability in Cardiomyocytes to Prevent Heart Failure. Eur. Heart J. 2017, 38, 362–372. [Google Scholar] [CrossRef] [Green Version]
- Mancardi, D.; Mezzanotte, M.; Arrigo, E.; Barinotti, A.; Roetto, A. Iron Overload, Oxidative Stress, and Ferroptosis in the Failing Heart and Liver. Antioxidants 2021, 10, 1864. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, A.J.; Das, N.K.; Ramakrishnan, S.K.; Jain, C.; Jurkovic, M.T.; Wu, J.; Nemeth, E.; Lakhal-Littleton, S.; Colacino, J.A.; Shah, Y.M. Hepatic Hepcidin/Intestinal HIF-2α Axis Maintains Iron Absorption during Iron Deficiency and Overload. J. Clin. Investig. 2019, 129, 336–348. [Google Scholar] [CrossRef]
- Camaschella, C.; Nai, A.; Silvestri, L. Iron Metabolism and Iron Disorders Revisited in the Hepcidin Era. Haematologica 2020, 105, 260–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, M.C.; Zhang, D.-L.; Jeong, S.Y.; Kovtunovych, G.; Ollivierre-Wilson, H.; Noguchi, A.; Tu, T.; Senecal, T.; Robinson, G.; Crooks, D.R.; et al. Deletion of Iron Regulatory Protein 1 Causes Polycythemia and Pulmonary Hypertension in Mice through Translational Derepression of HIF2α. Cell Metab. 2013, 17, 271–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, S.A.; Nizzi, C.P.; Chang, Y.-I.; Deck, K.M.; Schmidt, P.J.; Galy, B.; Damnernsawad, A.; Broman, A.T.; Kendziorski, C.; Hentze, M.W.; et al. The IRP1-HIF-2α Axis Coordinates Iron and Oxygen Sensing with Erythropoiesis and Iron Absorption. Cell Metab. 2013, 17, 282–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, C.Q.; Griffen, L.M.; Goldgar, D.; Drummond, C.; Skolnick, M.H.; Kushner, J.P. Prevalence of Hemochromatosis among 11,065 Presumably Healthy Blood Donors. N. Engl. J. Med. 1988, 318, 1355–1362. [Google Scholar] [CrossRef]
- Kowdley, K.V.; Brown, K.E.; Ahn, J.; Sundaram, V. ACG Clinical Guideline: Hereditary Hemochromatosis. Am. J. Gastroenterol. 2019, 114, 1202–1218. [Google Scholar] [CrossRef] [PubMed]
- Kumfu, S.; Chattipakorn, S.C.; Chattipakorn, N. T-Type and L-Type Calcium Channel Blockers for the Treatment of Cardiac Iron Overload: An Update. J. Cardiovasc. Pharmacol. 2017, 70, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Liuzzi, J.P.; Aydemir, F.; Nam, H.; Knutson, M.D.; Cousins, R.J. Zip14 (Slc39a14) Mediates Non-Transferrin-Bound Iron Uptake into Cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13612–13617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yanatori, I.; Kishi, F. DMT1 and Iron Transport. Free Radic. Biol. Med. 2019, 133, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhabyeyev, P.; Wang, S.; Oudit, G.Y. Role of Iron Metabolism in Heart Failure: From Iron Deficiency to Iron Overload. Biochim. Et Biophys. Acta (BBA) Mol. Basis Dis. 2019, 1865, 1925–1937. [Google Scholar] [CrossRef]
- Gammella, E.; Recalcati, S.; Rybinska, I.; Buratti, P.; Cairo, G. Iron-Induced Damage in Cardiomyopathy: Oxidative-Dependent and Independent Mechanisms. Oxidative Med. Cell. Longev. 2015, 2015, e230182. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Cao, F.; Yin, H.; Huang, Z.; Lin, Z.; Mao, N.; Sun, B.; Wang, G. Ferroptosis: Past, Present and Future. Cell Death Dis. 2020, 11, 88. [Google Scholar] [CrossRef]
- Zhao, Y.; Huang, Z.; Peng, H. Molecular Mechanisms of Ferroptosis and Its Roles in Hematologic Malignancies. Front. Oncol. 2021, 11, 743006. [Google Scholar] [CrossRef]
- Laftah, A.H.; Sharma, N.; Brookes, M.J.; McKie, A.T.; Simpson, R.J.; Iqbal, T.H.; Tselepis, C. Tumour Necrosis Factor Alpha Causes Hypoferraemia and Reduced Intestinal Iron Absorption in Mice. Biochem. J. 2006, 397, 61–67. [Google Scholar] [CrossRef] [Green Version]
- Toblli, J.E.; Cao, G.; Angerosa, M. Cardiovascular Outcomes of Intravenous Iron in Perspective of Clinical Trials and the Use of Different Iron Preparations. Int. J. Cardiol. 2015, 187, 196–197. [Google Scholar] [CrossRef] [Green Version]
- Friedman, J.R.; Nunnari, J. Mitochondrial Form and Function. Nature 2014, 505, 335–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seibler, P.; Burbulla, L.F.; Dulovic, M.; Zittel, S.; Heine, J.; Schmidt, T.; Rudolph, F.; Westenberger, A.; Rakovic, A.; Münchau, A.; et al. Iron Overload Is Accompanied by Mitochondrial and Lysosomal Dysfunction in WDR45 Mutant Cells. Brain 2018, 141, 3052–3064. [Google Scholar] [CrossRef] [PubMed]
- Merrill, J.F.; Thomson, D.M.; Hardman, S.E.; Hepworth, S.D.; Willie, S.; Hancock, C.R. Iron Deficiency Causes a Shift in AMP-Activated Protein Kinase (AMPK) Subunit Composition in Rat Skeletal Muscle. Nutr. Metab. 2012, 9, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leermakers, P.A.; Remels, A.H.V.; Zonneveld, M.I.; Rouschop, K.M.A.; Schols, A.M.W.J.; Gosker, H.R. Iron Deficiency-Induced Loss of Skeletal Muscle Mitochondrial Proteins and Respiratory Capacity; the Role of Mitophagy and Secretion of Mitochondria-Containing Vesicles. FASEB J. 2020, 34, 6703–6717. [Google Scholar] [CrossRef] [Green Version]
- Hoes, M.F.; Grote Beverborg, N.; Kijlstra, J.D.; Kuipers, J.; Swinkels, D.W.; Giepmans, B.N.G.; Rodenburg, R.J.; van Veldhuisen, D.J.; de Boer, R.A.; van der Meer, P. Iron Deficiency Impairs Contractility of Human Cardiomyocytes through Decreased Mitochondrial Function. Eur. J. Heart Fail. 2018, 20, 910–919. [Google Scholar] [CrossRef] [Green Version]
- Heather, L.C.; Carr, C.A.; Stuckey, D.J.; Pope, S.; Morten, K.J.; Carter, E.E.; Edwards, L.M.; Clarke, K. Critical Role of Complex III in the Early Metabolic Changes Following Myocardial Infarction. Cardiovasc. Res. 2010, 85, 127–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rineau, E.; Gaillard, T.; Gueguen, N.; Procaccio, V.; Henrion, D.; Prunier, F.; Lasocki, S. Iron Deficiency without Anemia Is Responsible for Decreased Left Ventricular Function and Reduced Mitochondrial Complex I Activity in a Mouse Model. Int. J. Cardiol. 2018, 266, 206–212. [Google Scholar] [CrossRef]
- Beard, J.L. Iron Biology in Immune Function, Muscle Metabolism and Neuronal Functioning. J. Nutr. 2001, 131, 568S–579S. [Google Scholar] [CrossRef]
- Van Swelm, R.P.L.; Wetzels, J.F.M.; Swinkels, D.W. The Multifaceted Role of Iron in Renal Health and Disease. Nat. Rev. Nephrol. 2020, 16, 77–98. [Google Scholar] [CrossRef]
- Hassan, R.H.; Kandil, S.M.; Zeid, M.S.; Zaki, M.E.; Fouda, A.E. Kidney Injury in Infants and Children with Iron-Deficiency Anemia before and after Iron Treatment. Hematology 2017, 22, 565–570. [Google Scholar] [CrossRef] [Green Version]
- Ponikowski, P.; Filippatos, G.; Colet, J.C.; Willenheimer, R.; Dickstein, K.; Lüscher, T.; Gaudesius, G.; von Eisenhart Rothe, B.; Mori, C.; Greenlaw, N.; et al. The Impact of Intravenous Ferric Carboxymaltose on Renal Function: An Analysis of the FAIR-HF Study. Eur. J. Heart Fail. 2015, 17, 329–339. [Google Scholar] [CrossRef] [PubMed]
- Courbon, G.; Martinez-Calle, M.; David, V. Simultaneous Management of Disordered Phosphate and Iron Homeostasis to Correct Fibroblast Growth Factor 23 and Associated Outcomes in Chronic Kidney Disease. Curr. Opin. Nephrol. Hypertens. 2020, 29, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.; Neves, P.; Gozzelino, R. Multilevel Impacts of Iron in the Brain: The Cross Talk between Neurophysiological Mechanisms, Cognition, and Social Behavior. Pharmaceuticals 2019, 12, E126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, S.; Yuan, Y.; Kuang, Y.; Li, X. Iron Metabolism and Immune Regulation. Front. Immunol. 2022, 13, 816282. [Google Scholar] [CrossRef] [PubMed]
- Wopereis, D.M.; Du Puy, R.S.; van Heemst, D.; Walsh, J.P.; Bremner, A.; Bakker, S.J.L.; Bauer, D.C.; Cappola, A.R.; Ceresini, G.; Degryse, J.; et al. The Relation Between Thyroid Function and Anemia: A Pooled Analysis of Individual Participant Data. J. Clin. Endocrinol. Metab. 2018, 103, 3658–3667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kontoghiorghes, G.J.; Kleanthous, M.; Kontoghiorghe, C.N. The History of Deferiprone (L1) and the Paradigm of the Complete Treatment of Iron Overload in Thalassaemia. Mediterr. J. Hematol. Infect. Dis. 2020, 12, e2020011. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, V.G.; Tongers, J.; Bode, J.; Berliner, D.; Widder, J.D.; Escher, F.; Mutsenko, V.; Chung, B.; Rostami, F.; Guba-Quint, A.; et al. Cardiac Iron Concentration in Relation to Systemic Iron Status and Disease Severity in Non-Ischaemic Heart Failure with Reduced Ejection Fraction. Eur. J. Heart Fail. 2020, 22, 2038–2046. [Google Scholar] [CrossRef]
- Lewis, G.D.; Malhotra, R.; Hernandez, A.F.; McNulty, S.E.; Smith, A.; Felker, G.M.; Tang, W.H.W.; LaRue, S.J.; Redfield, M.M.; Semigran, M.J.; et al. Effect of Oral Iron Repletion on Exercise Capacity in Patients with Heart Failure with Reduced Ejection Fraction and Iron Deficiency: The IRONOUT HF Randomized Clinical Trial. JAMA 2017, 317, 1958–1966. [Google Scholar] [CrossRef]
- McDonagh, T.A.; Metra, M.; Adamo, M.; Gardner, R.S.; Baumbach, A.; Böhm, M.; Burri, H.; Butler, J.; Čelutkienė, J.; Chioncel, O.; et al. 2021 ESC Guidelines for the Diagnosis and Treatment of Acute and Chronic Heart Failure. Eur. Heart J. 2021, 42, 3599–3726. [Google Scholar] [CrossRef]
- Leszek, P.; Sochanowicz, B.; Brzóska, K.; Kraj, L.; Kuśmierczyk, M.; Śmigielski, W.; Rywik, T.M.; Sobieszczańska-Małek, M.; Rozentryt, P.; Kruszewski, M. Accurate Noninvasive Assessment of Myocardial Iron Load in Advanced Heart Failure Patients. Dis. Markers 2020, 2020, 8885189. [Google Scholar] [CrossRef]
- Leszek, P.; Sochanowicz, B.; Brzóska, K.; Danko, B.; Kraj, L.; Kuśmierczyk, M.; Piotrowski, W.; Sobieszczańska-Małek, M.; Rywik, T.M.; Polkowska-Motrenko, H.; et al. Does Myocardial Iron Load Determine the Severity of Heart Insufficiency? Int. J. Cardiol. 2015, 182, 191–193. [Google Scholar] [CrossRef] [PubMed]
- Kozłowska, B.; Sochanowicz, B.; Kraj, L.; Palusińska, M.; Kołsut, P.; Szymański, Ł.; Lewicki, S.; Śmigielski, W.; Kruszewski, M.; Leszek, P. Expression of Iron Metabolism Proteins in Patients with Chronic Heart Failure. J. Clin. Med. 2022, 11, 837. [Google Scholar] [CrossRef] [PubMed]
- Tajes, M.; Díez-López, C.; Enjuanes, C.; Moliner, P.; Ferreiro, J.L.; Garay, A.; Jiménez-Marrero, S.; Yun, S.; Sosa, S.G.; Alcoberro, L.; et al. Neurohormonal Activation Induces Intracellular Iron Deficiency and Mitochondrial Dysfunction in Cardiac Cells. Cell Biosci. 2021, 11, 89. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Jamieson, K.L.; Grenier, J.; Nikhanj, A.; Tang, Z.; Wang, F.; Wang, S.; Seidman, J.G.; Seidman, C.E.; Thompson, R.; et al. Myocardial Iron Deficiency and Mitochondrial Dysfunction in Advanced Heart Failure in Humans. J. Am. Heart Assoc. 2022, 11, e022853. [Google Scholar] [CrossRef]
- Bi, Y.; Ajoolabady, A.; Demillard, L.J.; Yu, W.; Hilaire, M.L.; Zhang, Y.; Ren, J. Dysregulation of Iron Metabolism in Cardiovascular Diseases: From Iron Deficiency to Iron Overload. Biochem. Pharmacol. 2021, 190, 114661. [Google Scholar] [CrossRef]
- Zolk, O.; Solbach, T.F.; Eschenhagen, T.; Weidemann, A.; Fromm, M.F. Activation of Negative Regulators of the Hypoxia-Inducible Factor (HIF) Pathway in Human End-Stage Heart Failure. Biochem. Biophys. Res. Commun. 2008, 376, 315–320. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| RNA | Protein | Translation | Effect |
|---|---|---|---|
| FtH/L | Ferritin | ↓ | ↓ Iron-storage protein |
| DMT1 | Divalent Metal Transporter | ↑ | ↑ Iron import |
| TfR1 | Transferrin Receptor | ↑ | ↑ Iron import |
| Fpn1 | Ferroportin 1 | ↓ | ↓ Iron export |
| ACO2 | Mitochondrial Aconitase 2 | ↓ | ↓ TCA cycle |
| HIF2α | Hypoxia-Inducible Factor 2α | ↓ | ↓ Hypoxia response |
| Classification | Primary Iron Overload | Protein Defect/Mechanism |
|---|---|---|
| Type 1A | Homozygosity for the C282Y mutation in HFE—80% of hemochromatosis | Involved in hepcidin synthesis via BMP6, interaction with TFR1 |
| Type 1B | Mutation in HFE gene—H63D, S65C—2%–10% of cases | Involved in hepcidin synthesis via BMP6, interaction with TFR1 |
| Type 2A | HJV mutation | Involved in hepcidin synthesis, BMP co-receptor |
| Type 2B | HAMP mutation | Downregulation of iron efflux from erythrocytes |
| Type 3 | TFR2 mutation | Involved in hepcidin synthesis, interaction with transferrin |
| Type 4 | SLC40A1 (FPN) mutation | Duodenal iron export |
| Classification | Secondary Iron Overload | Protein Defect/Mechanism |
| Iron-loading anemias | Hemoglobin synthesis disturbances—thalassemia major, hemoglobin H The abnormal breakdown of red blood cells—chronic hemolytic anemia, sickle cell anemia, pyruvate kinase deficiency hereditary spherocytosis Red blood cells’ synthesis disturbances— aplastic anemia | Excessive release of iron from red blood cells, chronic transfusions lead to iron overload as humans can not actively remove excess iron, |
| Parenteral iron overload | RBC transfusions, iron-dextran injections, long-term hemodialysis | Patients requiring recurrent transfusions due to anemia or CKD |
| Chronic liver disease | Alcoholic liver disease, non-alcoholic fatty liver disease, viral liver disease—hepatitis C, hepatitis B, metabolic diseases—porphyria cutanea tarda dysmetabolic iron overload syndrome, | Hepcidin deficiency |
| Miscellaneous | malignancy (HCC, breast cancer, hematologic malignancies), chronic inflammatory states (systemic lupus erythematosus, rheumatoid arthritis) | Increased iron absorption (possibly from elevated growth differentiation factor 15) |
| Other | HCC, hepatocellular carcinoma, NAFLD, non-alcoholic fatty liver disease | Hepcidin dysregulation due to insulin resistance |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kozłowska, B.; Sochanowicz, B.; Kraj, L.; Palusińska, M.; Kołsut, P.; Szymański, Ł.; Lewicki, S.; Kruszewski, M.; Załęska-Kocięcka, M.; Leszek, P. Clinical and Molecular Aspects of Iron Metabolism in Failing Myocytes. Life 2022, 12, 1203. https://doi.org/10.3390/life12081203
Kozłowska B, Sochanowicz B, Kraj L, Palusińska M, Kołsut P, Szymański Ł, Lewicki S, Kruszewski M, Załęska-Kocięcka M, Leszek P. Clinical and Molecular Aspects of Iron Metabolism in Failing Myocytes. Life. 2022; 12(8):1203. https://doi.org/10.3390/life12081203
Chicago/Turabian StyleKozłowska, Bogna, Barbara Sochanowicz, Leszek Kraj, Małgorzata Palusińska, Piotr Kołsut, Łukasz Szymański, Sławomir Lewicki, Marcin Kruszewski, Marta Załęska-Kocięcka, and Przemysław Leszek. 2022. "Clinical and Molecular Aspects of Iron Metabolism in Failing Myocytes" Life 12, no. 8: 1203. https://doi.org/10.3390/life12081203
APA StyleKozłowska, B., Sochanowicz, B., Kraj, L., Palusińska, M., Kołsut, P., Szymański, Ł., Lewicki, S., Kruszewski, M., Załęska-Kocięcka, M., & Leszek, P. (2022). Clinical and Molecular Aspects of Iron Metabolism in Failing Myocytes. Life, 12(8), 1203. https://doi.org/10.3390/life12081203