
Behavioral and Molecular Effects of Thapsigargin-Induced Brain ER- Stress: Encompassing Inflammation, MAPK, and Insulin Signaling Pathway
 ,
,  and
and
Abstract
:1. Introduction
2. Results
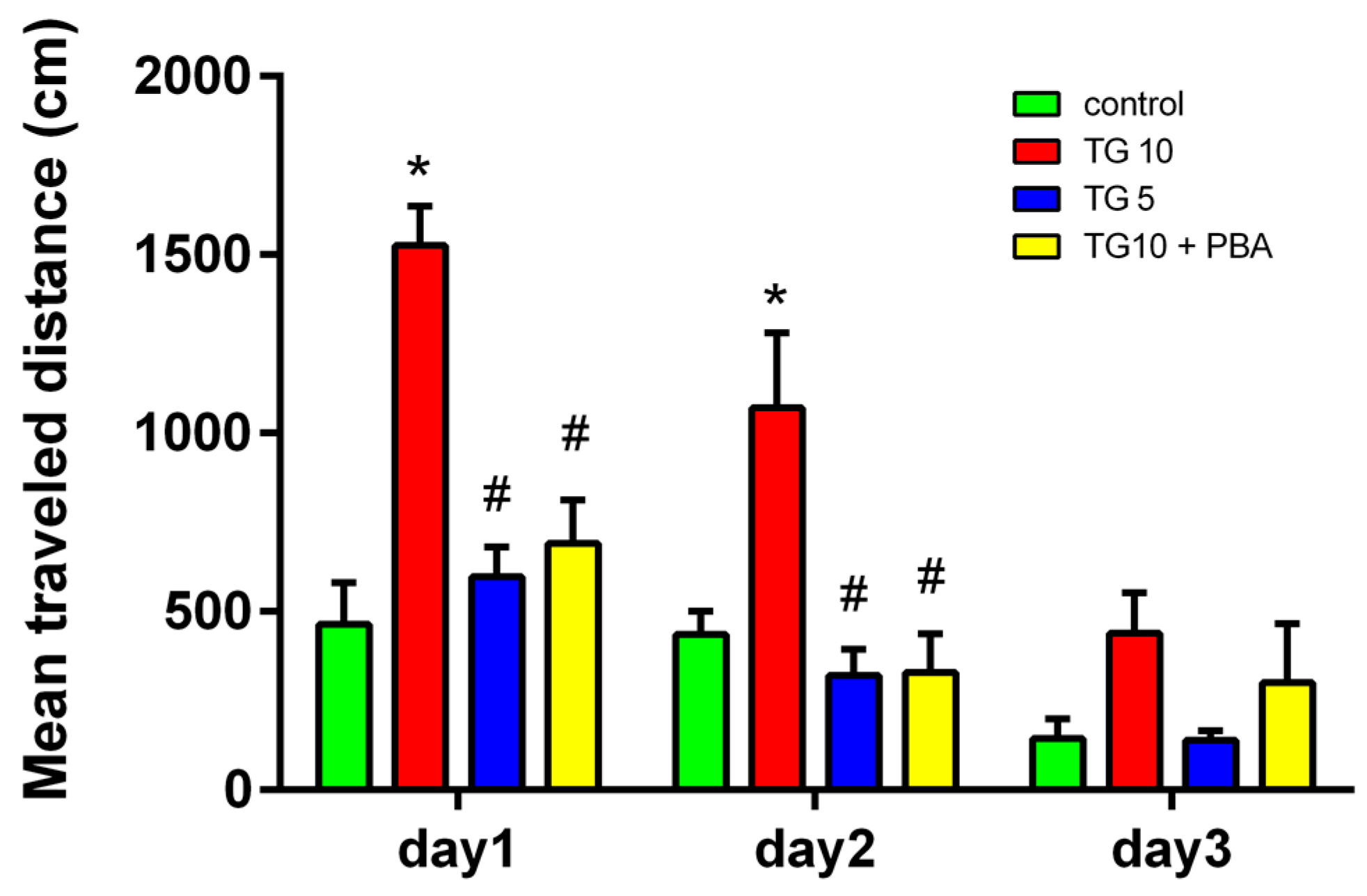
2.1. Behavioral Results
2.2. Results of PCR Assessments
2.3. Results of Western Blot Analysis
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Materials
4.3. Drugs Preparation and Administration
4.3.1. TG Microinjection
4.3.2. PBA Administration
4.4. Stereotaxic Surgery
4.5. Behavioral Test
MWM Apparatus
4.6. Procedure
4.7. Tissue Preparation
4.8. RNA Isolation and qPCR
4.9. Western Blot Analysis
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Alzheimer’s Disease International. World Alzheimer Report 2018; Alzheimer’s Disease International: London, UK, 2018. [Google Scholar]
- De Strooper, B. Proteases and Proteolysis in Alzheimer Disease: A Multifactorial View on the Disease Process. Physiol. Rev. 2010, 90, 465–494. [Google Scholar] [CrossRef]
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef]
- Caceres, A.; Kosik, K.S. Inhibition of neurite polarity by tau antisense oligonucleotides in primary cerebellar neurons. Nature 1990, 343, 461–463. [Google Scholar] [CrossRef]
- Stoothoff, W.H.; Johnson, G.V. Tau phosphorylation: Physiological and pathological consequences. Biochim. Biophys. Acta 2005, 1739, 280–297. [Google Scholar] [CrossRef]
- Gong, C.-X. Hyperphosphorylation of Microtubule-Associated Protein Tau: A Promising Therapeutic Target for Alzheimer Disease. Curr. Med. Chem. 2008, 15, 2321–2328. [Google Scholar] [CrossRef]
- McDonald, M.P.; Dahl, E.E.; Overmier, J.B.; Mantyh, P.; Cleary, J. Effects of an exogenous β-amyloid peptide on retention for spatial learning. Behav. Neural Biol. 1994, 62, 60–67. [Google Scholar] [CrossRef]
- Chen, S.; Frederickson, R.C.; Brunden, K.R. Neuroglial-mediated immunoinflammatory responses in Alzheimer’s disease: Complement activation and therapeutic approaches. Neurobiol. Aging 1996, 17, 781–787. [Google Scholar] [CrossRef]
- Maurice, T.; Lockhart, B.P.; Privat, A. Amnesia induced in mice by centrally administered β-amyloid peptides involves cholinergic dysfunction. Brain Res. 1996, 706, 181–193. [Google Scholar] [CrossRef]
- Pepeu, G.; Giovannelli, L.; Casamenti, F.; Cali, C.; Bartolini, L. Chapter 27 Amyloid β-peptides injection into the cholinergic nuclei: Morphological, neurochemical and behavioral effects. Prog. Brain Res. 1996, 109, 273–282. [Google Scholar] [CrossRef]
- A Sweeney, W.; Luedtke, J.; McDonald, M.P.; Overmier, J. Intrahippocampal Injections of Exogenous β-Amyloid Induce Postdelay Errors in an Eight-Arm Radial Maze. Neurobiol. Learn. Mem. 1997, 68, 97–101. [Google Scholar] [CrossRef] [Green Version]
- Harkany, T.; O’Mahony, S.; Kelly, J.; Soós, K.; Törõ, I.; Penke, B.; Luiten, P.; Nyakas, C.; Gulya, K.; Leonard, B. β-Amyloid(Phe(SO3H)24)25–35 in rat nucleus basalis induces behavioral dysfunctions, impairs learning and memory and disrupts cortical cholinergic innervation. Behav. Brain Res. 1998, 90, 133–145. [Google Scholar] [CrossRef]
- Oka, J.-I.; Suzuki, E.; Goto, N.; Kameyama, T. Endogenous GLP-1 modulates hippocampal activity in β-amyloid protein-treated rats. Neuroreport 1999, 10, 2961–2964. [Google Scholar] [CrossRef]
- Games, D.; Adams, D.; Alessandrini, R.; Barbour, R.; Borthelette, P.; Blackwell, C.; Carr, T.; Clemens, J.; Donaldson, T.; Gillespie, F.; et al. Alzheimer-type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nature 1995, 373, 523–527. [Google Scholar] [CrossRef]
- Lee, J.W.; Lee, Y.K.; Yuk, D.Y.; Choi, D.Y.; Ban, S.B.; Oh, K.W.; Hong, J.T. Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J. Neuroinflamm. 2008, 5, 37. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Liu, Y.; Jiang, Y.; Ding, J.; Li, L. Geniposide Ameliorates Learning Memory Deficits, Reduces Tau Phosphorylation and Decreases Apoptosis via GSK3β Pathway in Streptozotocin-Induced Alzheimer Rat Model. Brain Pathol. 2014, 24, 261–269. [Google Scholar] [CrossRef] [PubMed]
- Beatty, W.W.; Butters, N.; Janowsky, D.S. Patterns of memory failure after scopolamine treatment: Implications for cholinergic hypotheses of dementia. Behav. Neural Biol. 1986, 45, 196–211. [Google Scholar] [CrossRef]
- Ebert, U.; Kirch, W. Scopolamine model of dementia: Electroencephalogram findings and cognitive performance. Eur. J. Clin. Investig. 1998, 28, 944–949. [Google Scholar] [CrossRef]
- Kopelman, M.D.; Corn, T.H. Cholinergic ‘blockade’ as a model for cholinergic depletion. A comparison of the memory deficits with those of Alzheimer-type dementia and the alcoholic Korsakoff syndrome. Brain 1988, 111, 1079–1110. [Google Scholar] [CrossRef]
- Baleriola, J.; Walker, C.A.; Jean, Y.Y.; Crary, J.F.; Troy, C.M.; Nagy, P.L.; Hengst, U. Axonally Synthesized ATF4 Transmits a Neurodegenerative Signal across Brain Regions. Cell 2014, 158, 1159–1172. [Google Scholar] [CrossRef]
- Cornejo, V.H.; Hetz, C. The unfolded protein response in Alzheimer’s disease. Semin. Immunopathol. 2013, 35, 277–292. [Google Scholar] [CrossRef]
- Endres, K.; Reinhardt, S. ER-stress in Alzheimer’s disease: Turning the scale? Am. J. Neurodegener. Dis. 2013, 2, 247–265. [Google Scholar] [PubMed]
- Barbero-Camps, E.; Fernández, A.; Baulies, A.; Martinez, L.; Fernández-Checa, J.C.; Colell, A. Endoplasmic Reticulum Stress Mediates Amyloid β Neurotoxicity via Mitochondrial Cholesterol Trafficking. Am. J. Pathol. 2014, 184, 2066–2081. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, T.; Sadleir, K.R.; Maus, E.; Velliquette, R.A.; Zhao, J.; Cole, S.L.; Eimer, W.A.; Hitt, B.; Bembinster, L.A.; Lammich, S.; et al. Phosphorylation of the Translation Initiation Factor eIF2α Increases BACE1 Levels and Promotes Amyloidogenesis. Neuron 2008, 60, 988–1009. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, S.; Schuck, F.; Grösgen, S.; Riemenschneider, M.; Hartmann, T.; Postina, R.; Grimm, M.; Endres, K. Unfolded protein response signaling by transcription factor XBP-1 regulates ADAM10 and is affected in Alzheimer’s disease. FASEB J. 2014, 28, 978–997. [Google Scholar] [CrossRef] [PubMed]
- Pintado, C.; Macías, S.; Domínguez-Martín, H.; Castaño, A.; Ruano, D. Neuroinflammation alters cellular proteostasis by producing endoplasmic reticulum stress, autophagy activation and disrupting ERAD activation. Sci. Rep. 2017, 7, 8100. [Google Scholar] [CrossRef] [PubMed]
- Biswas, J.; Gupta, S.; Verma, D.K.; Singh, S. Streptozotocin alters glucose transport, connexin expression and endoplasmic reticulum functions in neurons and astrocytes. Neuroscience 2017, 356, 151–166. [Google Scholar] [CrossRef]
- Hoozemans, J.J.M.; Veerhuis, R.; Van Haastert, E.S.; Rozemuller, J.M.; Baas, F.; Eikelenboom, P.; Scheper, W. The unfolded protein response is activated in Alzheimer’s disease. Acta Neuropathol. 2005, 110, 165–172. [Google Scholar] [CrossRef]
- Scheper, W.; Hoozemans, J.J.M. The unfolded protein response in neurodegenerative diseases: A neuropathological perspective. Acta Neuropathol. 2015, 130, 315–331. [Google Scholar] [CrossRef]
- Hoozemans, J.J.; van Haastert, E.S.; Nijholt, D.A.; Rozemuller, A.J.; Eikelenboom, P.; Scheper, W. The Unfolded Protein Response Is Activated in Pretangle Neurons in Alzheimer’s Disease Hippocampus. Am. J. Pathol. 2009, 174, 1241–1251. [Google Scholar] [CrossRef]
- Lee, J.H.; Won, S.M.; Suh, J.; Son, S.J.; Moon, G.J.; Park, U.-J.; Gwag, B.J. Induction of the unfolded protein response and cell death pathway in Alzheimer’s disease, but not in aged Tg2576 mice. Exp. Mol. Med. 2010, 42, 386–394. [Google Scholar] [CrossRef] [Green Version]
- Westrate, L.M.; Lee, J.E.; Prinz, W.A.; Voeltz, G.K. Form Follows Function: The Importance of Endoplasmic Reticulum Shape. Annu. Rev. Biochem. 2015, 84, 791–811. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.; Ron, D. The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Science 2011, 334, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Ozcan, U. Unfolded Protein Response Signaling and Metabolic Diseases. J. Biol. Chem. 2014, 289, 1203–1211. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Novoa, I.; Zhang, Y.; Zeng, H.; Wek, R.; Schapira, M.; Ron, D. Regulated Translation Initiation Controls Stress-Induced Gene Expression in Mammalian Cells. Mol. Cell 2000, 6, 1099–1108. [Google Scholar] [CrossRef]
- Han, J.; Back, S.H.; Hur, J.; Lin, Y.-H.; Gildersleeve, R.; Shan, J.; Yuan, C.L.; Krokowski, D.; Wang, S.; Hatzoglou, M.; et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol. 2013, 15, 481–490. [Google Scholar] [CrossRef]
- Cao, S.S.; Kaufman, R.J. Unfolded protein response. Curr. Biol. 2012, 22, R622–R626. [Google Scholar] [CrossRef] [PubMed]
- Glimcher, L.H. XBP1: The last two decades. Ann. Rheum. Dis. 2009, 69 (Suppl. 1), i67–i71. [Google Scholar] [CrossRef]
- Yoshida, H.; Matsui, T.; Yamamoto, A.; Okada, T.; Mori, K. XBP1 mRNA Is Induced by ATF6 and Spliced by IRE1 in Response to ER Stress to Produce a Highly Active Transcription Factor. Cell 2001, 107, 881–891. [Google Scholar] [CrossRef]
- Calfon, M.; Zeng, H.; Urano, F.; Till, J.H.; Hubbard, S.R.; Harding, H.P.; Clark, S.G.; Ron, D. IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 2002, 415, 92–96. [Google Scholar] [CrossRef]
- Rasmussen, U.; Christensen, S.B.; Sandberg, F. Thapsigargine and thapsigargicine, two new histamine liberators from Thapsia garganica L. Acta Pharm. Suec. 1978, 15, 133–140. [Google Scholar]
- Rogers, T.B.; Inesi, G.; Wade, R.; Lederer, W.J. Use of thapsigargin to study Ca2+ homeostasis in cardiac cells. Biosci. Rep. 1995, 15, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, J.D.; Kaufman, R.J. The endoplasmic reticulum and the unfolded protein response. Semin. Cell Dev. Biol. 2007, 18, 716–731. [Google Scholar] [CrossRef] [PubMed]
- Kolb, P.S.; Ayaub, E.A.; Zhou, W.; Yum, V.; Dickhout, J.G.; Ask, K. The therapeutic effects of 4-phenylbutyric acid in maintaining proteostasis. Int. J. Biochem. Cell Biol. 2015, 61, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Özcan, U.; Yilmaz, E.; Özcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Görgün, C.Z.; Hotamisligil, G.S. Chemical Chaperones Reduce ER Stress and Restore Glucose Homeostasis in a Mouse Model of Type 2 Diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef]
- Gong, B.; Zhang, L.Y.; Lam, D.S.C.; Pang, C.P.; Yam, G.H.F. Sodium 4-phenylbutyrate ameliorates the effects of cataract-causing mutant gammaD-crystallin in cultured cells. Mol. Vis. 2010, 16, 997–1003. [Google Scholar]
- Li, C.-Y.; Lee, J.-S.; Ko, Y.-G.; Kim, J.-I.; Seo, J.-S. Heat Shock Protein 70 Inhibits Apoptosis Downstream of Cytochrome c Release and Upstream of Caspase-3 Activation. J. Biol. Chem. 2000, 275, 25665–25671. [Google Scholar] [CrossRef]
- Lourenco, M.V.; Clarke, J.R.; Frozza, R.L.; Bomfim, T.R.; Forny-Germano, L.; Batista, A.F.; Sathler, L.B.; Brito-Moreira, J.; Amaral, O.B.; Silva, C.A.; et al. TNF-α Mediates PKR-Dependent Memory Impairment and Brain IRS-1 Inhibition Induced by Alzheimer’s β-Amyloid Oligomers in Mice and Monkeys. Cell Metab. 2013, 18, 831–843. [Google Scholar] [CrossRef]
- Chen, C.-M.; Wu, C.-T.; Chiang, C.-K.; Liao, B.-W.; Liu, S.-H. C/EBP Homologous Protein (CHOP) Deficiency Aggravates Hippocampal Cell Apoptosis and Impairs Memory Performance. PLoS ONE 2012, 7, e40801. [Google Scholar] [CrossRef]
- Wang, Y.-W.; Zhou, Q.; Zhang, X.; Qian, Q.-Q.; Xu, J.-W.; Ni, P.-F.; Qian, Y.-N. Mild endoplasmic reticulum stress ameliorates lipopolysaccharide-induced neuroinflammation and cognitive impairment via regulation of microglial polarization. J. Neuroinflamm. 2017, 14, 233. [Google Scholar] [CrossRef]
- Lin, L.; Cao, J.; Yang, S.; Fu, Z.; Zeng, P.; Chu, J.; Ning, L.; Zhang, T.; Shi, Y.; Tian, Q.; et al. Endoplasmic reticulum stress induces spatial memory deficits by activatingGSK-3. J. Cell. Mol. Med. 2018, 22, 3489–3502. [Google Scholar] [CrossRef]
- Obulesu, M.; Lakshmi, M.J. Apoptosis in Alzheimer’s Disease: An Understanding of the Physiology, Pathology and Therapeutic Avenues. Neurochem. Res. 2014, 39, 2301–2312. [Google Scholar] [CrossRef] [PubMed]
- He, P.; Zhong, Z.; Lindholm, K.; Berning, L.; Lee, W.; Lemere, C.; Staufenbiel, M.; Li, R.; Shen, Y. Deletion of tumor necrosis factor death receptor inhibits amyloid β generation and prevents learning and memory deficits in Alzheimer’s mice. J. Cell Biol. 2007, 178, 829–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clark, I.; Atwood, C.; Bowen, R.; Paz-Filho, G.; Vissel, B. Tumor Necrosis Factor-Induced Cerebral Insulin Resistance in Alzheimer’s Disease Links Numerous Treatment Rationales. Pharmacol. Rev. 2012, 64, 1004–1026. [Google Scholar] [CrossRef] [PubMed]
- Moloney, A.M.; Griffin, R.J.; Timmons, S.; O’Connor, R.; Ravid, R.; O’Neill, C. Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer’s disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol. Aging 2010, 31, 224–243. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Sprenkle, N.T.; Sims, S.G.; Sánchez, C.L.; Meares, G.P. Endoplasmic reticulum stress and inflammation in the central nervous system. Mol. Neurodegener. 2017, 12, 42. [Google Scholar] [CrossRef]
- Sims-Robinson, C.; Bakeman, A.; Glasser, R.; Boggs, J.; Pacut, C.; Feldman, E. The role of endoplasmic reticulum stress in hippocampal insulin resistance. Exp. Neurol. 2016, 277, 261–267. [Google Scholar] [CrossRef]
- de la Monte, S.M. Triangulated Mal-Signaling in Alzheimer’s Disease: Roles of Neurotoxic Ceramides, ER Stress, and Insulin Resistance Reviewed. J. Alzheimer’s Dis. 2012, 30 (Suppl. 2), S231–S249. [Google Scholar] [CrossRef]
- Areias, M.F.C.; Prada, P.O. Mechanisms of insulin resistance in the amygdala: Influences on food intake. Behav. Brain Res. 2015, 282, 209–217. [Google Scholar] [CrossRef]
- Yue, J.; López, J.M. Understanding MAPK Signaling Pathways in Apoptosis. Int. J. Mol. Sci. 2020, 21, 2346. [Google Scholar] [CrossRef]
- Lee, J.K.; Kim, N.-J. Recent Advances in the Inhibition of p38 MAPK as a Potential Strategy for the Treatment of Alzheimer’s Disease. Molecules 2017, 22, 1287. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.K.; Choi, E.-J. Compromised MAPK signaling in human diseases: An update. Arch. Toxicol. 2015, 89, 867–882. [Google Scholar] [CrossRef] [PubMed]
- Ghasemi, R.; Haeri, A.; Dargahi, L.; Mohamed, Z.; Ahmadiani, A. Insulin in the Brain: Sources, Localization and Functions. Mol. Neurobiol. 2013, 47, 145–171. [Google Scholar] [CrossRef] [PubMed]
- Iloun, P.; Abbasnejad, Z.; Janahmadi, M.; Ahmadiani, A.; Ghasemi, R. Investigating the role of P38, JNK and ERK in LPS induced hippocampal insulin resistance and spatial memory impairment: Effects of insulin treatment. EXCLI J. 2018, 17, 825–839. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.J.; Kim, S.-K.; Jin, S.M.; Hwang, E.-M.; Kim, Y.S.; Huh, K.; Mook-Jung, I. IFN-γ-induced BACE1 expression is mediated by activation of JAK2 and ERK1/2 signaling pathways and direct binding of STAT1 to BACE1 promoter in astrocytes. Glia 2006, 55, 253–262. [Google Scholar] [CrossRef]
- Colombo, A.; Bastone, A.; Ploia, C.; Sclip, A.; Salmona, M.; Forloni, G.; Borsello, T. JNK regulates APP cleavage and degradation in a model of Alzheimer’s disease. Neurobiol. Dis. 2009, 33, 518–525. [Google Scholar] [CrossRef]
- Kirouac, L.; Rajic, A.J.; Cribbs, D.H.; Padmanabhan, J. Activation of Ras-ERK Signaling and GSK-3 by Amyloid Precursor Protein and Amyloid Beta Facilitates Neurodegeneration in Alzheimer’s Disease. eNeuro 2017, 4, 1–21. [Google Scholar] [CrossRef]
- Darling, N.J.; Cook, S.J. The role of MAPK signalling pathways in the response to endoplasmic reticulum stress. Biochim. Biophys. Acta 2014, 1843, 2150–2163. [Google Scholar] [CrossRef]
- Xue, L.-X.; Liu, H.-Y.; Cui, Y.; Dong, Y.; Wang, J.-Q.; Ji, Q.-Y.; He, J.-T.; Yao, M.; Wang, Y.Y.; Shao, Y.K.; et al. Neuroprotective effects of Activin A on endoplasmic reticulum stress-mediated apoptotic and autophagic PC12 cell death. Neural Regen. Res. 2017, 12, 779–786. [Google Scholar] [CrossRef]
- Sekine, Y.; Takeda, K.; Ichijo, H. The ASK1-MAP Kinase Signaling in ER Stress and Neurodegenerative Diseases. Curr. Mol. Med. 2006, 6, 87–97. [Google Scholar] [CrossRef]
- Liang, L.; Chen, J.; Zhan, L.; Lu, X.; Sun, X.; Sui, H.; Zheng, L.; Xiang, H.; Zhang, F. Endoplasmic Reticulum Stress Impairs Insulin Receptor Signaling in the Brains of Obese Rats. PLoS ONE 2015, 10, e0126384. [Google Scholar] [CrossRef]
- Spinelli, M.; Fusco, S.; Grassi, C. Brain insulin resistance impairs hippocampal plasticity. Vitam. Horm. 2020, 114, 281–306. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-S.; Li, B.; Rhew, K.Y.; Oh, H.-W.; Lim, H.-D.; Lee, W.; Chae, H.-J.; Kim, H.-R. The regulatory mechanism of 4-phenylbutyric acid against ER stress-induced autophagy in human gingival fibroblasts. Arch. Pharmacal Res. 2012, 35, 1269–1278. [Google Scholar] [CrossRef]
- Woltjer, R.L.; McMahan, W.; Milatovic, D.; Kjerulf, J.D.; Shie, F.-S.; Rung, L.G.; Montine, K.S.; Montine, T.J. Effects of chemical chaperones on oxidative stress and detergent-insoluble species formation following conditional expression of amyloid precursor protein carboxy-terminal fragment. Neurobiol. Dis. 2007, 25, 427–437. [Google Scholar] [CrossRef]
- Costa-Mattioli, M.; Gobert, D.; Harding, H.; Herdy, B.; Azzi, M.; Bruno, M.; Bidinosti, M.; Ben Mamou, C.; Marcinkiewicz, E.; Yoshida, M.; et al. Translational control of hippocampal synaptic plasticity and memory by the eIF2α kinase GCN2. Nature 2005, 436, 1166–1170. [Google Scholar] [CrossRef] [PubMed]
- Costa-Mattioli, M.; Gobert, D.; Stern, E.; Gamache, K.; Colina, R.; Cuello, C.; Sossin, W.; Kaufman, R.; Pelletier, J.; Rosenblum, K.; et al. eIF2α Phosphorylation Bidirectionally Regulates the Switch from Short- to Long-Term Synaptic Plasticity and Memory. Cell 2007, 129, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Belforte, J.E.; Lu, Y.; Yabe, Y.; Pickel, J.; Smith, C.B.; Je, H.; Lu, B.; Nakazawa, K. eIF2 Phosphorylation-Dependent Translation in CA1 Pyramidal Cells Impairs Hippocampal Memory Consolidation without Affecting General Translation. J. Neurosci. 2010, 30, 2582–2594. [Google Scholar] [CrossRef]
- Ma, T.; Trinh, M.A.; Wexler, A.J.; Bourbon, C.; Gatti, E.; Pierre, P.; Cavener, D.R.; Klann, E. Suppression of eIF2α kinases alleviates Alzheimer’s disease–related plasticity and memory deficits. Nat. Neurosci. 2013, 16, 1299–1305. [Google Scholar] [CrossRef]
- Zhang, X.; Xu, L.; He, D.; Ling, S. Endoplasmic Reticulum Stress-Mediated Hippocampal Neuron Apoptosis Involved in Diabetic Cognitive Impairment. BioMed Res. Int. 2013, 2013, 924327. [Google Scholar] [CrossRef]
- Samali, A.; Fitzgerald, U.; Deegan, S.; Gupta, S. Methods for Monitoring Endoplasmic Reticulum Stress and the Unfolded Protein Response. Int. J. Cell Biol. 2010, 2010, 830307. [Google Scholar] [CrossRef]
- Kokame, K.; Kato, H.; Miyata, T. Identification of ERSE-II, a New cis-Acting Element Responsible for the ATF6-dependent Mammalian Unfolded Protein Response. J. Biol. Chem. 2001, 276, 9199–9205. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Z.; Lawson, B.; Brewer, J.W.; Zinszner, H.; Sanjay, A.; Mi, L.J.; Boorstein, R.; Kreibich, G.; Hendershot, L.M.; Ron, D. Signals from the stressed endoplasmic reticulum induce C/EBP-homologous protein (CHOP/GADD153). Mol. Cell. Biol. 1996, 16, 4273–4280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Ron, D. Stress-Induced Phosphorylation and Activation of the Transcription Factor CHOP (GADD153) by p38 MAP Kinase. Science 1996, 272, 1347–1349. [Google Scholar] [CrossRef] [PubMed]
- Limjindaporn, T.; Wongwiwat, W.; Noisakran, S.; Srisawat, C.; Netsawang, J.; Puttikhunt, C.; Kasinrerk, W.; Avirutnan, P.; Thiemmeca, S.; Sriburi, R.; et al. Interaction of dengue virus envelope protein with endoplasmic reticulum-resident chaperones facilitates dengue virus production. Biochem. Biophys. Res. Commun. 2009, 379, 196–200. [Google Scholar] [CrossRef]
- Chen, T.-H.; Chiang, Y.-H.; Hou, J.-N.; Cheng, C.-C.; Sofiyatun, E.; Chiu, C.-H.; Chen, W.-J. XBP1-Mediated BiP/GRP78 Upregulation Copes with Oxidative Stress in Mosquito Cells during Dengue 2 Virus Infection. BioMed Res. Int. 2017, 2017, 3519158. [Google Scholar] [CrossRef]
- Ghanbari-Maman, A.; Ghasemian-Roudsari, F.; Aliakbari, S.; Gholamipour-Badie, H.; Khodagholi, F.; Shaerzadeh, F.; Daftari, M. Calcium Channel Blockade Ameliorates Endoplasmic Reticulum Stress in the Hippocampus Induced by Amyloidopathy in the Entorhinal Cortex. Iran. J. Pharm. Res. 2019, 18, 1466–1476. [Google Scholar] [CrossRef]
- Moreno, J.A.; Tiffany-Castiglioni, E. The Chaperone Grp78 in Protein Folding Disorders of the Nervous System. Neurochem. Res. 2015, 40, 329–335. [Google Scholar] [CrossRef]
- Fu, Z.-Q.; Yang, Y.; Song, J.; Jiang, Q.; Liu, Z.-C.; Wang, Q.; Zhu, L.-Q.; Wang, J.-Z.; Tian, Q. LiCl Attenuates Thapsigargin-Induced Tau Hyperphosphorylation by Inhibiting GSK-3β In Vivo and In Vitro. J. Alzheimer’s Dis. 2010, 21, 1107–1117. [Google Scholar] [CrossRef]
- Liu, Z.-C.; Fu, Z.-Q.; Song, J.; Zhang, J.-Y.; Wei, Y.-P.; Chu, J.; Han, L.; Na Qu, N.; Wang, J.-Z.; Tian, Q. Bip Enhanced the Association of GSK-3β with Tau During ER Stress Both in vivo and in vitro. J. Alzheimer’s Dis. 2012, 29, 727–740. [Google Scholar] [CrossRef]
- Endo, M.; Mori, M.; Akira, S.; Gotoh, T. C/EBP Homologous Protein (CHOP) Is Crucial for the Induction of Caspase-11 and the Pathogenesis of Lipopolysaccharide-Induced Inflammatio. J. Immunol. 2006, 176, 6245–6253. [Google Scholar] [CrossRef]
- Tsai, Y.C.; Weissman, A.M. The Unfolded Protein Response, Degradation from the Endoplasmic Reticulum, and Cancer. Genes Cancer 2010, 1, 764–778. [Google Scholar] [CrossRef] [PubMed]
- Onaka, Y.; Wada, S.; Yamaguchi, T.; Yoneyama, M.; Ogita, K. Preventive effect of olanzapine on trimethyltin neurotoxicity in mice: Evaluation of hippocampal neuronal loss, microglial activation, and cognitive dysfunction. Glob. Drugs Ther. 2018, 3, 5. [Google Scholar] [CrossRef] [Green Version]
- Donev, R.; Kolev, M.; Millet, B.; Thome, J. Neuronal death in Alzheimer’s disease and therapeutic opportunities. J. Cell. Mol. Med. 2009, 13, 4329–4348. [Google Scholar] [CrossRef] [PubMed]
- Galehdar, Z.; Swan, P.; Fuerth, B.; Callaghan, S.M.; Park, D.S.; Cregan, S.P. Neuronal Apoptosis Induced by Endoplasmic Reticulum Stress Is Regulated by ATF4-CHOP-Mediated Induction of the Bcl-2 homology 3-only Member PUMA. J. Neurosci. 2010, 30, 16938–16948. [Google Scholar] [CrossRef] [PubMed]
- Crowley, L.; Waterhouse, N.J. Detecting Cleaved Caspase-3 in Apoptotic Cells by Flow Cytometry. Cold Spring Harb. Protoc. 2016, 2016, pdb-rot087312. [Google Scholar] [CrossRef] [PubMed]
- Snigdha, S.; Smith, E.; Prieto, G.A.; Cotman, C.W. Caspase-3 activation as a bifurcation point between plasticity and cell death. Neurosci. Bull. 2012, 28, 14–24. [Google Scholar] [CrossRef]
- Matsunaga, D.; Sreekumar, P.G.; Ishikawa, K.; Terasaki, H.; Barron, E.; Cohen, P.; Kannan, R.; Hinton, D.R. Humanin Protects RPE Cells from Endoplasmic Reticulum Stress-Induced Apoptosis by Upregulation of Mitochondrial Glutathione. PLoS ONE 2016, 11, e0165150. [Google Scholar] [CrossRef]
- Sun, H.; Yang, Y.; Shao, H.; Sun, W.; Gu, M.; Wang, H.; Jiang, L.; Qu, L.; Sun, D.; Gao, Y. Sodium Arsenite-Induced Learning and Memory Impairment Is Associated with Endoplasmic Reticulum Stress-Mediated Apoptosis in Rat Hippocampus. Front. Mol. Neurosci. 2017, 10, 286. [Google Scholar] [CrossRef]
- Blomgren, K.; Leist, M.; Groc, L. Pathological apoptosis in the developing brain. Apoptosis 2007, 12, 993–1010. [Google Scholar] [CrossRef]
- Li, C.; Wang, Q.; Li, L.; Liu, Y.; Diao, H. Arachidonic acid attenuates learning and memory dysfunction induced by repeated isoflurane anesthesia in rats. Int. J. Clin. Exp. Med. 2015, 8, 12365–12373. [Google Scholar]
- Scorrano, L.; Oakes, S.A.; Opferman, J.T.; Cheng, E.H.; Sorcinelli, M.D.; Pozzan, T.; Korsmeyer, S.J. BAX and BAK Regulation of Endoplasmic Reticulum Ca 2+: A Control Point for Apoptosis. Science 2003, 300, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Ferri, K.F.; Kroemer, G. Organelle-specific initiation of cell death pathways. Nat. Cell Biol. 2001, 3, E255–E263. [Google Scholar] [CrossRef]
- McCullough, K.D.; Martindale, J.L.; Klotz, L.-O.; Aw, T.-Y.; Holbrook, N.J. Gadd153 Sensitizes Cells to Endoplasmic Reticulum Stress by Down-Regulating Bcl2 and Perturbing the Cellular Redox State. Mol. Cell. Biol. 2001, 21, 1249–1259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deniaud, A.; el Dein, O.S.; Maillier, E.; Poncet, D.; Kroemer, G.; Lemaire, C.; Brenner, C. Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene 2007, 27, 285–299. [Google Scholar] [CrossRef]
- Kuhn, H.G.; Biebl, M.; Wilhelm, D.; Li, M.; Friedlander, R.; Winkler, J. Increased generation of granule cells in adult Bcl-2-overexpressing mice: A role for cell death during continued hippocampal neurogenesis. Eur. J. Neurosci. 2005, 22, 1907–1915. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.-Q.; Xu, Z.-P.; Zhang, S.; Cao, X.-S.; Liu, T.-S. Simulated weightlessness aggravates hypergravity-induced impairment of learning and memory and neuronal apoptosis in rats. Behav. Brain Res. 2009, 199, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yin, C.-P.; Tai, Y.-L.; Zhao, Z.-J.; Hou, Z.-Y.; Wang, Q.-J. Apoptosis inhibition is involved in improvement of sevoflurane-induced cognitive impairment following normobaric hyperoxia preconditioning in aged rats. Exp. Ther. Med. 2021, 21, 203. [Google Scholar] [CrossRef]
- He, Y.; Ruganzu, J.B.; Lin, C.; Ding, B.; Zheng, Q.; Wu, X.; Ma, R.; Liu, Q.; Wang, Y.; Jin, H.; et al. Tanshinone IIA ameliorates cognitive deficits by inhibiting endoplasmic reticulum stress-induced apoptosis in APP/PS1 transgenic mice. Neurochem. Int. 2019, 133, 104610. [Google Scholar] [CrossRef]
- Li, Y.; Schwabe, R.F.; DeVries-Seimon, T.; Yao, P.M.; Gerbod-Giannone, M.-C.; Tall, A.R.; Davis, R.J.; Flavell, R.; Brenner, D.A.; Tabas, I. Free cholesterol-loaded macrophages are an abundant source of tumor necrosis factor-alpha and interleukin-6: Model of NF-kappaB- and map kinase-dependent inflammation in advanced atherosclerosis. J. Biol. Chem. 2005, 280, 21763–21772. [Google Scholar] [CrossRef]
- Wang, Z.; Huang, Y.; Cheng, Y.; Tan, Y.; Wu, F.; Wu, J.; Shi, H.; Zhang, H.; Yu, X.; Gao, H.; et al. Endoplasmic reticulum stress-induced neuronal inflammatory response and apoptosis likely plays a key role in the development of diabetic encephalopathy. Oncotarget 2016, 7, 78455–78472. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, H.; Hiramatsu, N.; Hayakawa, K.; Tagawa, Y.; Okamura, M.; Ogata, R.; Huang, T.; Nakajima, S.; Yao, J.; Paton, A.W.; et al. Activation of the Akt-NF-κB Pathway by Subtilase Cytotoxin through the ATF6 Branch of the Unfolded Protein Response. J. Immunol. 2009, 183, 1480–1487. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Joe, Y.; Surh, Y.-J.; Chung, H.T. Differential Regulation of Toll-Like Receptor-Mediated Cytokine Production by Unfolded Protein Response. Oxidative Med. Cell. Longev. 2018, 2018, 9827312. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, K.; Nakajima, S.; Hiramatsu, N.; Okamura, M.; Huang, T.; Saito, Y.; Tagawa, Y.; Tamai, M.; Takahashi, S.; Yao, J.; et al. ER Stress Depresses NF-κB Activation in Mesangial Cells through Preferential Induction of C/EBPβ. J. Am. Soc. Nephrol. 2009, 21, 73–81. [Google Scholar] [CrossRef] [PubMed]
- Grootjans, J.; Kaser, A.; Kaufman, R.J.; Blumberg, J.G.R.S. The unfolded protein response in immunity and inflammation. Nat. Rev. Immunol. 2016, 16, 469–484. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.K.; Choi, E.-J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Biophys. Acta 2010, 1802, 396–405. [Google Scholar] [CrossRef]
- Gual, P.; Le Marchand-Brustel, Y.; Tanti, J.-F. Positive and negative regulation of insulin signaling through IRS-1 phosphorylation. Biochimie 2005, 87, 99–109. [Google Scholar] [CrossRef]
- Hemmati, F.; Ghasemi, R.; Ibrahim, N.M.; Dargahi, L.; Mohamed, Z.; Raymond, A.A.; Ahmadiani, A. Crosstalk Between Insulin and Toll-like Receptor Signaling Pathways in the Central Nervous system. Mol. Neurobiol. 2014, 50, 797–810. [Google Scholar] [CrossRef]
- Gregor, M.F.; Hotamisligil, G.S. Inflammatory Mechanisms in Obesity. Annu. Rev. Immunol. 2011, 29, 415–445. [Google Scholar] [CrossRef]
- Ozcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.-H.; Iwakoshi, N.N.; Özdelen, E.; Tuncman, G.; Görgün, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic Reticulum Stress Links Obesity, Insulin Action, and Type 2 Diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef]
- Nakatani, Y.; Kaneto, H.; Kawamori, D.; Yoshiuchi, K.; Hatazaki, M.; Matsuoka, T.-A.; Ozawa, K.; Ogawa, S.; Hori, M.; Yamasaki, Y.; et al. Involvement of Endoplasmic Reticulum Stress in Insulin Resistance and Diabetes. J. Biol. Chem. 2005, 280, 847–851. [Google Scholar] [CrossRef]
- Arnold, S.E.; Arvanitakis, Z.; Macauley-Rambach, S.L.; Koenig, A.M.; Wang, H.-Y.; Ahima, R.S.; Craft, S.; Gandy, S.; Buettner, C.; Stoeckel, L.E.; et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nat. Rev. Neurol. 2018, 14, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Iurlaro, R.; Muñoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef] [PubMed]
- Belegri, E.; Rijnsburger, M.; Eggels, L.; Unmehopa, U.; Scheper, W.; Boelen, A.; la Fleur, S.E. Effects of Fat and Sugar, Either Consumed or Infused toward the Brain, on Hypothalamic ER Stress Markers. Front. Neurosci. 2017, 11, 270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timberlake, M.; Dwivedi, Y. Altered Expression of Endoplasmic Reticulum Stress Associated Genes in Hippocampus of Learned Helpless Rats: Relevance to Depression Pathophysiology. Front. Pharmacol. 2015, 6, 319. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sequence | Amplicon (bp) | Reference |
|---|---|---|---|
| s-XBP1 | F-CTGAGTCCGAATCAGGTCAG | 59 | [124] |
| R-ATCCATGGGAGATGTTCTGG | |||
| us-XBP1 | F-GTCCGCAGCACTCAGACTAC | 268 | [124] |
| R-ATGAGGTCCCCATGACAGA | |||
| ATF6 | F-CGAGGGGAGGTGTCTGTTTC | 97 | [125] |
| R-GTCTTCACTGGTCCATGAGG | |||
| ATF4 | F-CTGAACAGCGAAGTGTTGGC | 224 | [124] |
| R-TCTGTCCCGGAAAAGGCATC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Askari, S.; Javadpour, P.; Rashidi, F.S.; Dargahi, L.; Kashfi, K.; Ghasemi, R. Behavioral and Molecular Effects of Thapsigargin-Induced Brain ER- Stress: Encompassing Inflammation, MAPK, and Insulin Signaling Pathway. Life 2022, 12, 1374. https://doi.org/10.3390/life12091374
Askari S, Javadpour P, Rashidi FS, Dargahi L, Kashfi K, Ghasemi R. Behavioral and Molecular Effects of Thapsigargin-Induced Brain ER- Stress: Encompassing Inflammation, MAPK, and Insulin Signaling Pathway. Life. 2022; 12(9):1374. https://doi.org/10.3390/life12091374
Chicago/Turabian StyleAskari, Sahar, Pegah Javadpour, Fatemeh Sadat Rashidi, Leila Dargahi, Khosrow Kashfi, and Rasoul Ghasemi. 2022. "Behavioral and Molecular Effects of Thapsigargin-Induced Brain ER- Stress: Encompassing Inflammation, MAPK, and Insulin Signaling Pathway" Life 12, no. 9: 1374. https://doi.org/10.3390/life12091374