
The IgCAM CAR Regulates Gap Junction-Mediated Coupling on Embryonic Cardiomyocytes and Affects Their Beating Frequency

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Embryonic Cardiomyocyte and Heart Organ Cultures
2.3. Preparation of the Fiber Knob of the Adenovirus
2.4. Calcium Imaging of Cultured Cardiomyocytes
2.5. Calcium Concentration Estimation
2.6. Analysis of Cell–Cell Coupling by Lucifer Yellow
2.7. Whole-Cell PATCH-Clamp Recordings of Cultured Cardiomyocytes
2.8. Microarray Analysis of mRNAs in the Embryonic Heart
2.9. qPCR to Quantify the Level of mRNA of Selected Genes
2.10. Biochemical Methods
2.11. Immunoprecipitation
2.12. Immunocytochemistry and Immunohistochemistry
2.13. Statistical Analysis
3. Results
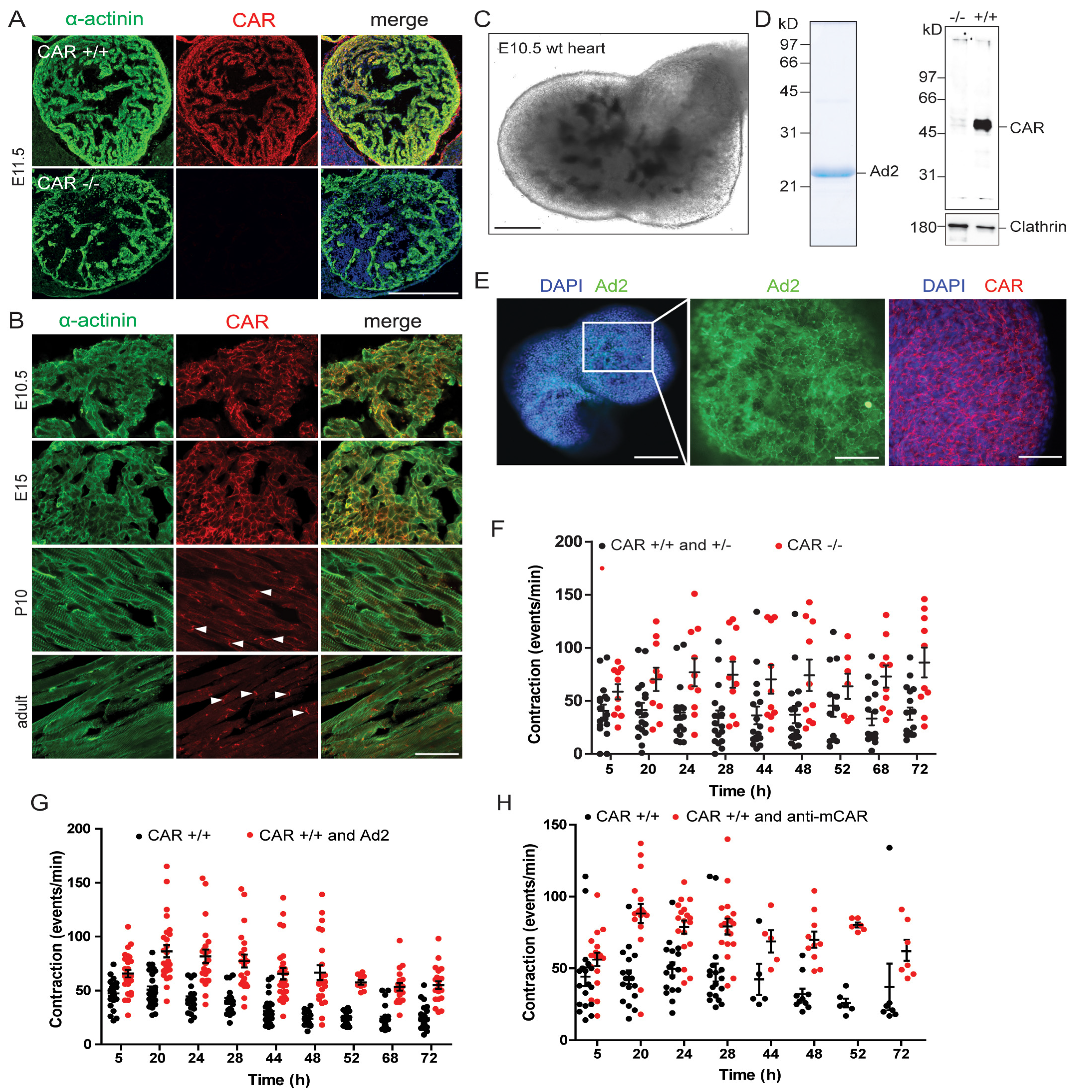
3.1. The Absence of the CAR Resulted in Increased Spontaneous Beating Frequencies of Cultured Embryonic Cardiomyocytes
3.1.1. Mechanisms of Calcium Cycling Were Not Impaired in CAR-Deficient Cardiomyocytes
3.1.2. Electrophysiological Properties Were Not Changed in CAR-Deficient Cardiomyocytes
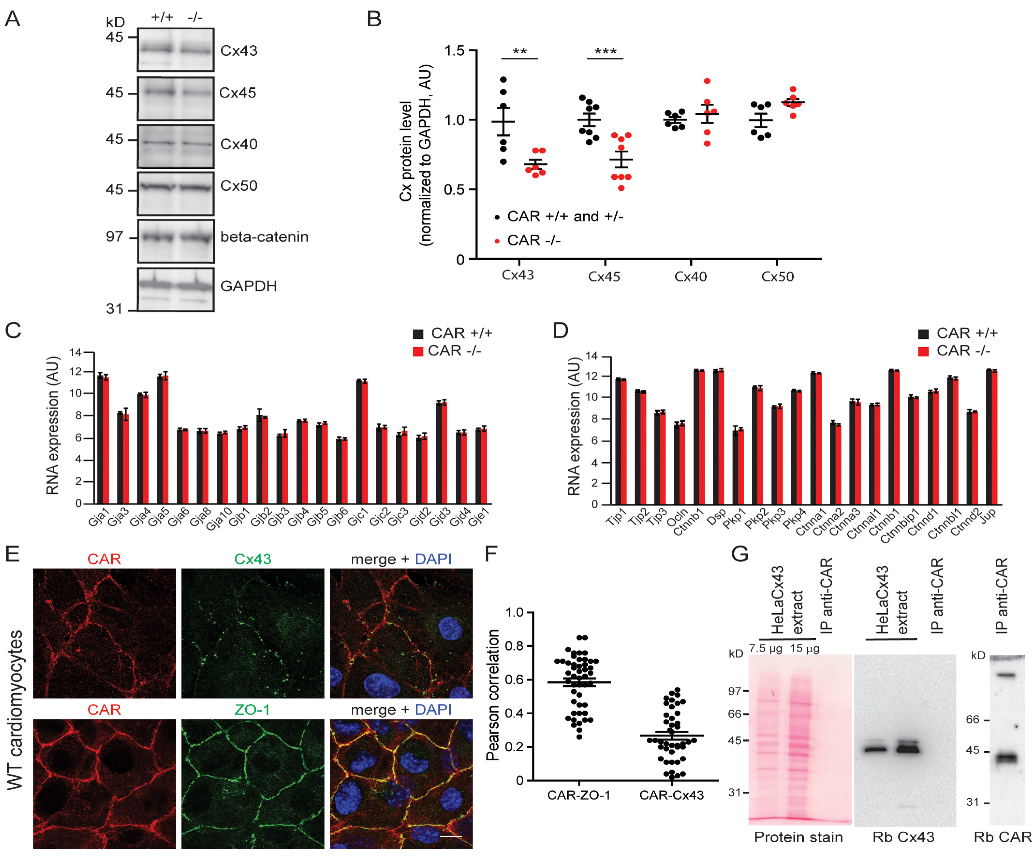
3.2. Gap Junction-Mediated Coupling Is Increased in CAR-Deficient Cardiomyocytes
3.3. Increased Beating of CAR-Deficient Embryonic Hearts in Organ Cultures
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rathjen, F.G. The CAR group of Ig cell adhesion proteins—Regulators of gap junctions ? Bioessays 2020, 42, 2000031. [Google Scholar] [CrossRef] [PubMed]
- Matthäus, C.; Schreiber, J.; Jüttner, R.; Rathjen, F.G. The Ig CAM CAR is Implicated in Cardiac Development and Modulates Electrical Conduction in the Mature Heart. J. Cardiovasc. Dev. Dis. 2014, 1, 111–120. [Google Scholar] [CrossRef] [Green Version]
- Patzke, C.; Max, K.E.A.; Behlke, J.; Schreiber, J.; Schmidt, H.; Dorner, A.A.; Kröger, S.; Henning, M.; Otto, A.; Heinemann, U.; et al. The coxsackievirus-adenovirus receptor reveals complex homophilic and heterophilic interactions on neural cells. J. Neurosci. 2010, 30, 2897–2910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, C.J.; Shieh, J.T.C.; Pickles, R.J.; Okegawa, T.; Hsieh, J.-T.T.; Bergelson, J.M. The coxsackievirus and adenovirus receptor is a transmembrane component of the tight junction. Proc. Natl. Acad. Sci. USA 2001, 98, 15191–15196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coyne, C.B.; Bergelson, J.M. CAR: A virus receptor within the tight junctions. Adv. Drug Deliv. Rev. 2005, 57, 869–882. [Google Scholar] [CrossRef] [PubMed]
- Raschperger, E.; Thyberg, J.; Pettersson, S.; Philipson, L.; Fuxe, J.; Pettersson, R.F. The coxsackie- and adenovirus receptor (CAR) is an in vivo marker for epithelial tight junctions, with a potential role in regulating permeability and tissue homeostasis. Exp.Cell Res. 2006, 312, 1566–1580. [Google Scholar] [CrossRef]
- Tomko, R.P.; Xu, R.; Philipson, L. HCAR and MCAR: The human and mouse cellular receptors for subgroup C adenoviruses and group B coxsackieviruses. Proc. Natl. Acad. Sci. USA 1997, 94, 3352–3356. [Google Scholar] [CrossRef] [Green Version]
- Tomko, R.P.; Johansson, C.B.; Totrov, M.; Abagyan, R.; Frisén, J.; Philipson, L.; Frisen, J.; Philipson, L.; Frisén, J.; Philipson, L. Expression of the adenovirus receptor and its interaction with the fiber knob. Exp. Cell Res. 2000, 255, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Dorner, A.A.A.; Wegmann, F.; Butz, S.; Wolburg-Buchholz, K.; Wolburg, H.; Mack, A.; Nasdala, I.; August, B.; Westermann, J.; Rathjen, F.G.; et al. Coxsackievirus-adenovirus receptor (CAR) is essential for early embryonic cardiac development. J. Cell Sci. 2005, 118, 3509–3521. [Google Scholar] [CrossRef] [Green Version]
- Honda, T.; Saitoh, H.; Masuko, M.; Katagiri-Abe, T.; Tominaga, K.; Kozakai, I.; Kobayashi, K.; Kumanishi, T.; Watanabe, Y.G.; Odani, S.; et al. The coxsackievirus-adenovirus receptor protein as a cell adhesion molecule in the developing mouse brain. Brain Res. Mol. Brain Res. 2000, 77, 19–28. [Google Scholar] [CrossRef]
- Hotta, Y.; Honda, T.; Naito, M.; Kuwano, R. Developmental distribution of coxsackie virus and adenovirus receptor localized in the nervous system. Brain Res. Dev. Brain Res. 2003, 143, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Asher, D.R.; Cerny, A.M.; Weiler, S.R.; Horner, J.W.; Keeler, M.L.; Neptune, M.A.; Jones, S.N.; Bronson, R.T.; DePinho, R.A.; Finberg, R.W. Coxsackievirus and adenovirus receptor is essential for cardiomyocyte development. Genesis 2005, 42, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Lim, B.K.; Xiong, D.; Dorner, A.; Youn, T.J.; Yung, A.; Liu, T.I.; Gu, Y.; Dalton, N.D.; Wright, A.T.; Evans, S.M.; et al. Coxsackievirus and adenovirus receptor (CAR) mediates atrioventricular-node function and connexin 45 localization in the murine heart. J. Clin. Investig. 2008, 118, 2758–2770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poller, W.; Fechner, H.; Noutsias, M.; Tschoepe, C.; Schultheiss, H.-P.P. Highly variable expression of virus receptors in the human cardiovascular system. Implications for cardiotropic viral infections and gene therapy. Z. Kardiol. 2002, 91, 978–991. [Google Scholar] [CrossRef] [PubMed]
- Shaw, C.A.; Holland, P.C.; Sinnreich, M.; Allen, C.; Sollerbrant, K.; Karpati, G.; Nalbantoglu, J. Isoform-specific expression of the Coxsackie and adenovirus receptor (CAR) in neuromuscular junction and cardiac intercalated discs. BMC Cell Biol. 2004, 5, 42. [Google Scholar] [CrossRef] [Green Version]
- Lisewski, U.; Shi, Y.; Wrackmeyer, U.; Fischer, R.; Chen, C.; Schirdewan, A.; Juttner, R.; Rathjen, F.; Poller, W.; Radke, M.H.; et al. The tight junction protein CAR regulates cardiac conduction and cell-cell communication. J. Exp. Med. 2008, 205, 2369–2379. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.-W.W.; Zhou, B.; Yu, Q.-C.C.; Shin, S.J.; Jiao, K.; Schneider, M.D.; Baldwin, H.S.; Bergelson, J.M. Cardiomyocyte-specific deletion of the coxsackievirus and adenovirus receptor results in hyperplasia of the embryonic left ventricle and abnormalities of sinuatrial valves. Circ. Res. 2006, 98, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Pazirandeh, A.; Sultana, T.; Mirza, M.; Rozell, B.; Hultenby, K.; Wallis, K.; Vennstrom, B.; Davis, B.; Arner, A.; Heuchel, R.; et al. Multiple phenotypes in adult mice following inactivation of the Coxsackievirus and Adenovirus Receptor (Car) gene. PLoS ONE 2011, 6, e20203. [Google Scholar] [CrossRef] [Green Version]
- Caruso, L.; Yuen, S.; Smith, J.; Husain, M.; Opavsky, M.A. Cardiomyocyte-targeted overexpression of the coxsackie-adenovirus receptor causes a cardiomyopathy in association with beta-catenin signaling. J. Mol. Cell Cardiol. 2010, 48, 1194–1205. [Google Scholar] [CrossRef]
- Noutsias, M.; Fechner, H.; De Jonge, H.; Wang, X.; Dekkers, D.; Pauschinger, M.; Bergelson, J.; Warraich, R.; Yacoub, M.; Lamers, J.; et al. Human coxsackie-adenovirus receptor is colocalized with integrins alpha(v)beta(3) and alpha(v)beta(5) on the cardiomyocyte sarcolemma and upregulated in dilated cardiomyopathy: Implications for cardiotropic viral infections. Circulation 2001, 104, 275–280. [Google Scholar] [CrossRef]
- Toivonen, R.; Mäyränpää, M.I.; Kovanen, P.T.; Savontaus, M.; Mayranpaa, M.I.; Kovanen, P.T.; Savontaus, M. Dilated cardiomyopathy alters the expression patterns of CAR and other adenoviral receptors in human heart. Histochem. Biol. 2010, 133, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Kodama, M.; Masuko, M.; Yamaura, M.; Fuse, K.; Uesugi, Y.; Hirono, S.; Okura, Y.; Kato, K.; Hotta, Y.; et al. Expression of coxsackievirus and adenovirus receptor in hearts of rats with experimental autoimmune myocarditis. Circ. Res. 2000, 86, 275–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuen, S.; Smith, J.; Caruso, L.; Balan, M.; Opavsky, M.A. The coxsackie-adenovirus receptor induces an inflammatory cardiomyopathy independent of viral infection. J. Mol. Cell. Cardiol. 2011, 50, 826–840. [Google Scholar] [CrossRef]
- Fechner, H.; Noutsias, M.; Tschoepe, C.; Hinze, K.; Wang, X.; Escher, F.; Pauschinger, M.; Dekkers, D.; Vetter, R.; Paul, M.; et al. Induction of Coxsackievirus-Adenovirus-Receptor Expression During Myocardial Tissue Formation and Remodeling: Identification of a Cell-to-Cell Contact-Dependent Regulatory Mechanism. Circulation 2003, 107, 876–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kashimura, T.; Kodama, M.; Hotta, Y.; Hosoya, J.; Yoshida, K.; Ozawa, T.; Watanabe, R.; Okura, Y.; Kato, K.; Hanawa, H.; et al. Spatiotemporal changes of coxsackievirus and adenovirus receptor in rat hearts during postnatal development and in cultured cardiomyocytes of neonatal rat. Virchows Arch. 2004, 444, 283–292. [Google Scholar] [CrossRef] [PubMed]
- Wrackmeyer, U.; Kaldrack, J.; Jüttner, R.; Pannasch, U.; Gimber, N.; Freiberg, F.; Purfürst, B.; Kainmueller, D.; Schmitz, D.; Haucke, V.; et al. The cell adhesion protein CAR is a negative regulator of synaptic transmission. Sci. Rep. 2019, 9, 6768. [Google Scholar] [CrossRef] [Green Version]
- Zussy, C.; Loustalot, F.; Junyent, F.; Gardoni, F.; Bories, C.; Valero, J.; Desarmenien, M.G.; Bernex, F.; Henaff, D.; Bayo-Puxan, N.; et al. Coxsackievirus Adenovirus Receptor Loss Impairs Adult Neurogenesis, Synapse Content, and Hippocampus Plasticity. J. Neurosci. 2016, 36, 9558–9571. [Google Scholar] [CrossRef] [Green Version]
- Verdino, P.; Witherden, D.A.; Havran, W.L.; Wilson, I.A. The molecular interaction of CAR and JAML recruits the central cell signal transducer PI3K. Science 2010, 329, 1210–1214. [Google Scholar] [CrossRef] [Green Version]
- Van Raaij, M.J.; Chouin, E.; van der Zandt, H.; Bergelson, J.M.; Cusack, S.; van der, Z.H.; Bergelson, J.M.; Cusack, S. Dimeric structure of the coxsackievirus and adenovirus receptor D1 domain at 1.7 A resolution. Struct. Fold. Des. 2000, 8, 1147–1155. [Google Scholar] [CrossRef] [Green Version]
- Mirza, M.; Hreinsson, J.; Strand, M.-L.L.; Hovatta, O.; Soder, O.; Philipson, L.; Pettersson, R.F.; Sollerbrant, K. Coxsackievirus and adenovirus receptor (CAR) is expressed in male germ cells and forms a complex with the differentiation factor JAM-C in mouse testis. Exp. Cell Res. 2006, 312, 817–830. [Google Scholar] [CrossRef]
- Witherden, D.A.; Verdino, P.; Rieder, S.E.; Garijo, O.; Mills, R.E.; Teyton, L.; Fischer, W.H.; Wilson, I.A.; Havran, W.L.; Robyn, E.; et al. The junctional adhesion molecule JAML is a costimulatory receptor for epithelial gammadelta T cell activation. Science 2010, 329, 1205–1210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zen, K.; Liu, Y.; Mccall, I.C.; Wu, T.; Lee, W.; Babbin, B.A.; Nusrat, A.; Parkos, C.A. Neutrophil Migration across Tight Junctions Is Mediated by Adhesive Interactions between Epithelial Coxsackie and Adenovirus Receptor and a Junctional Adhesion Molecule-like Protein on Neutrophils. Mol. Biol. Cell 2005, 16, 2694–2703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coyne, C.B.; Voelker, T.; Pichla, S.L.; Bergelson, J.M. The coxsackievirus and adenovirus receptor interacts with the multi-PDZ domain protein-1 (MUPP-1) within the tight junction. J. Biol. Chem. 2004, 279, 48079–48084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolawole, A.O.; Sharma, P.; Yan, R.; Lewis, K.J.E.; Xu, Z.; Hostetler, H.A.; Ashbourne Excoffon, K.J.D. The PDZ1 and PDZ3 domains of MAGI-1 regulate the eight-exon isoform of the coxsackievirus and adenovirus receptor. J. Virol. 2012, 86, 9244–9254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirza, M.; Raschperger, E.; Philipson, L.; Pettersson, R.F.; Sollerbrant, K. The cell surface protein coxsackie- and adenovirus receptor (CAR) directly associates with the Ligand-of-Numb Protein-X2 (LNX2). Exp. Cell Res. 2005, 309, 110–120. [Google Scholar] [CrossRef]
- Sollerbrant, K.; Raschperger, E.; Mirza, M.; Engstrom, U.; Philipson, L.; Ljungdahl, P.O.; Pettersson, R.F.; Engström, U.; Philipson, L.; Ljungdahl, P.O.; et al. The Coxsackievirus and adenovirus receptor (CAR) forms a complex with the PDZ domain-containing protein ligand-of-numb protein-X (LNX). J. Biol. Chem. 2003, 278, 7439–7444. [Google Scholar] [CrossRef] [Green Version]
- Dyer, L.A.; Patterson, C. A Novel Ex vivo Culture Method for the Embryonic Mouse Heart. J. Vis. Exp. 2013, 24, e50359. [Google Scholar] [CrossRef] [Green Version]
- Awasthi, V.; Meinken, G.; Springer, K.; Srivastava, S.C.; Freimuth, P. Biodistribution of Radioiodinated Adenovirus Fiber Protein Knob Domain after Intravenous Injection in Mice. J. Virol. 2004, 78, 6431–6438. [Google Scholar] [CrossRef] [Green Version]
- Freimuth, P.; Springer, K.; Berard, C.; Hainfeld, J.I.M.; Bewley, M.; Flanagan, J.; York, N.; Flanagan, J. Coxsackievirus and adenovirus receptor amino-terminal immunoglobulin V-related domain binds adenovirus type 2 and fiber knob from adenovirus type 12. J. Virol. 1999, 73, 1392–1398. [Google Scholar] [CrossRef] [Green Version]
- Kirby, I.A.N.; Davison, E.; Beavil, A.J.; Soh, C.P.C.; Wickham, T.J.; Roelvink, P.W.; Kovesdi, I.; Sutton, B.J.; Santis, G. Identification of contact residues and definition of the CAR-binding site of adenovirus type 5 fiber protein. J. Virol. 2000, 74, 2804–2813. [Google Scholar] [CrossRef]
- Engvall, E. Enzyme immunoassay ELISA and EMIT. Methods Enzym. 1980, 70, 419–439. [Google Scholar]
- Voigt, N.; Li, N.; Wang, Q.; Wang, W.; Trafford, A.W.; Abu-Taha, I.; Sun, Q.; Wieland, T.; Ravens, U.; Nattel, S.; et al. Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+-Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation 2012, 125, 2059–2070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doeller, J.E.; Wittenberg, B.A. Intracellular calcium and high-energy in isolated cardiac myocytes. Am. Physiol. Soc. 1990, 259, H1851–H1859. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Nolan, T. Pitfalls of Quantitative Real-time PCR. J. Biomol. Tech. 2004, 15, 155–166. [Google Scholar] [PubMed]
- Pfaffl, M.W. Quantification strategies in real-time PCR. In A-Z of Quantitative PCR; International University Line (IUL): La Jolla, CA, USA, 2004; pp. 87–112. [Google Scholar]
- Elfgang, C.; Eckert, R.; Lichtenberg-Frate, H.; Butterweck, A.; Traub, O.; Klein, R.A.; Hulser, D.F.; Willecke, K. Specific permeability and selective formation of gap junction channels in connexin-transfected HeLa cells. J. Cell Biol. 1995, 129, 805–817. [Google Scholar] [CrossRef]
- Langhorst, H.; Juttner, R.; Groneberg, D.; Mohtashamdolatshahi, A.; Pelz, L.; Purfurst, B.; Schmidt-Ott, K.M.; Friebe, A.; Rathjen, F.G. The IgCAM CLMP regulates expression of Connexin43 and Connexin45 in intestinal and ureteral smooth muscle contraction in mice. Dis. Model. Mech. 2018, 11, dmm032128. [Google Scholar] [CrossRef] [Green Version]
- Fischer, R.; Poller, W.; Schultheiss, H.; Gotthardt, M. CAR-diology—A virus receptor in the healthy and diseased heart. J. Mol. Med. 2009, 87, 879–884. [Google Scholar] [CrossRef]
- Bers, D.M. Cardiac excitation contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef]
- Frank, K.; Kranias, E.G. Phospholamban and cardiac contractility. Ann. Med. 2000, 32, 572–578. [Google Scholar] [CrossRef]
- Simmerman, H.K.; Jones, L.R. Phospholamban: Protein structure, mechanism of action, and role in cardiac function. Physiol. Rev. 1998, 78, 921–947. [Google Scholar] [CrossRef] [Green Version]
- Bers, D.M. Cardiac sarcoplasmic reticulum calcium leak: Basis and roles in cardiac dysfunction. Annu. Rev. Physiol. 2014, 76, 107–127. [Google Scholar] [CrossRef] [PubMed]
- Neef, S.; Dybkova, N.; Sossalla, S.; Ort, K.R.; Fluschnik, N.; Neumann, K.; Seipelt, R.; Schöndube, F.A.; Hasenfuss, G.; Maier, L.S. CaMKII-Dependent diastolic SR Ca2+ leak and elevated diastolic Ca2+ levels in right atrial myocardium of patients with atrial fibrillation. Circ. Res. 2010, 106, 1134–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, J.A.; van Veen, T.A.B.; de Bakker, J.M.T.; van Rijen, H.V.M. Cardiac connexins and impulse propagation. J. Mol. Cell. Cardiol. 2010, 48, 76–82. [Google Scholar] [CrossRef]
- Alcoléa, S.; Théveniau-Ruissy, M.; Jarry-Guichard, T.; Marics, I.; Tzouanacou, E.; Chauvin, J.P.; Briand, J.P.; Moorman, A.F.; Lamers, W.H.; Gros, D.B. Downregulation of connexin 45 gene products during mouse heart development. Circ. Res. 1999, 84, 1365–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solan, J.L.; Lampe, P.D. Kinase programs spatiotemporally regulate gap junction assembly and disassembly: Effects on wound repair. Semin. Dev. Biol. 2016, 50, 40–48. [Google Scholar] [CrossRef] [Green Version]
- Bewley, M.C.; Springer, K.; Zhang, Y.B.; Freimuth, P.; Flanagan, J.M. Structural analysis of the mechanism of adenovirus binding to its human cellular receptor, CAR. Science 1999, 286, 1579–1583. [Google Scholar] [CrossRef] [Green Version]
- Roelvink, P.W.; Lee, G.M.; Einfeld, D.A.; Kovesdi, I.; Wickham, T.J. Identification of a conserved receptor-binding site on the fiber proteins of CAR-recognizing adenoviridae. Science 1999, 286, 1568–1571. [Google Scholar] [CrossRef]
- Walters, R.W.; Freimuth, P.; Moninger, T.O.; Ganske, I.; Zabner, J.; Welsh, M.J.; York, N. Adenovirus Fiber Disrupts CAR-Mediated Intercellular Adhesion Allowing Virus Escape. Cell 2002, 110, 789–799. [Google Scholar] [CrossRef] [Green Version]
- Matthaus, C.; Langhorst, H.; Schutz, L.; Juttner, R.; Rathjen, F.G. Cell-cell communication mediated by the CAR subgroup of immunoglobulin cell adhesion molecules in health and disease. Mol. Cell Neurosci. 2017, 81, 32–40. [Google Scholar] [CrossRef] [Green Version]
- Kurtenbach, S.S.; Kurtenbach, S.S.; Zoidl, G. Gap junction modulation and its implications for heart function. Front. Physiol. 2014, 5, 82. [Google Scholar] [CrossRef]
- Bukauskas, F.F.; Jordan, K.; Bukauskiene, A.; Bennett, M.V.L.; Lampe, P.D.; Laird, D.W.; Verselis, V.K. Clustering of connexin 43—Enhanced green fluorescent protein gap junction channels and functional coupling in living cells. Proc. Natl. Acad. Sci. USA 2000, 97, 2556–2561. [Google Scholar] [CrossRef] [PubMed]
- Verheule, S.; Kaese, S. Connexin diversity in the heart: Insights from transgenic mouse models. Front. Pharmacol. 2013, 4, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egashira, K.; Nishii, K.; Nakamura, K.I.; Kumai, M.; Morimoto, S.; Shibata, Y. Conduction abnormality in gap junction protein connexin45-deficient embryonic stem cell-derived cardiac myocytes. Anat. Rec. Part A Discov. Mol. Cell. Evol. Biol. 2004, 280, 973–979. [Google Scholar] [CrossRef]
- Van Rijen, H.V.M.; Eckardt, D.; Degen, J.; Theis, M.; Ott, T.; Willecke, K.; Jongsma, H.J.; Opthof, T.; De Bakker, J.M.T. Slow Conduction and Enhanced Anisotropy Increase the Propensity for Ventricular Tachyarrhythmias in Adult Mice with Induced Deletion of Connexin43. Circulation 2004, 109, 1048–1055. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutstein, D.E.; Morley, G.E.; Tamaddon, H.; Vaidya, D.; Schneider, M.D.; Chen, J.; Chien, K.R.; Stuhlmann, H.; Fishman, G.I. Conduction Slowing and Sudden Arrhythmic Death in Mice with Cardiac-Restricted Inactivation of Connexin43. Circ. Res. 2001, 88, 333–339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reaume, A.; de Sousa, P.; Kulkarni, S.; Langille, B.; Zhu, D.; Davies, T.; Juneja, S.; Kidder, G.; Rossant, J. Cardiac malformation in neonatal mice lacking connexin43. Science 1995, 267, 1831–1834. [Google Scholar] [CrossRef]
- Nishii, K.; Kumai, M.; Egashira, K.; Miwa, T.; Hashizume, K.; Miyano, Y.; Shibata, Y. Mice lacking connexin45 conditionally in cardiac myocytes display embryonic lethality similar to that of germline knockout mice without endocardial cushion defect. Cell Commun. Adhes. 2003, 10, 365–369. [Google Scholar] [CrossRef]
- Gu, H.; Smith, F.C.; Taffet, S.M.; Delmar, M. High incidence of cardiac malformations in Connexin40-deficient mice. Circ. Res. 2003, 93, 201–206. [Google Scholar] [CrossRef]
- Kumai, M.; Nishii, K.; Nakamura, K.; Takeda, N.; Suzuki, M.; Shibata, Y. Loss of connexin45 causes a cushion defect in early cardiogenesis. Development 2000, 127, 3501–3512. [Google Scholar] [CrossRef]
- Ya, J.; Erdtsieck-Ernste, E.B.; de Boer, P.A.; van Kempen, M.J.; Jongsma, H.; Gros, D.; Moorman, A.F.; Lamers, W.H. Heart defects in connexin43-deficient mice. Circ. Res. 1998, 82, 360–366. [Google Scholar] [CrossRef] [Green Version]
- Pelz, L.; Purfürst, B.; Rathjen, F.G. The cell adhesion molecule BT-IgSF is essential for a functional blood-testis barrier and male fertility in mice. J. Biol. Chem. 2017, 292, 21490–21503. [Google Scholar] [CrossRef] [PubMed]
- Pelz, L.; Dossou, L.; Kompier, N.; Jüttner, R.S.G.; Meyer, N.; Lowenstein, E.D.; Lahmann, I.; Kettenmann, H.; Birchmeier, C.; Rathjen, F.G. The IgCAM BT-IgSF (IgSF11) is essential for connexin43-mediated astrocyte-astrocyte and ependymal cell-cell coupling. bioRxiv 2022. [Google Scholar] [CrossRef]
- Hunter, A.W.; Barker, R.J.; Zhu, C.; Gourdie, R.G. Zonula occludens-1 alters connexin43 gap junction size and organization by influencing channel accretion. Mol. Biol. Cell 2005, 16, 5686–5698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giepmans, B.N.G.; Moolenaar, W.H. The gap junction protein connexin43 interacts with the second PDZ domain of the zona occludens-1 protein. Curr. Biol. 1998, 8, 931–934. [Google Scholar] [CrossRef] [Green Version]
- Toyofuku, T.; Yabuki, M.; Otsu, K.; Kuzuya, T.; Hori, M.; Tada, M. Direct association of the gap junction protein connexin-43 with ZO-1 in cardiac myocytes. J. Biol. Chem. 1998, 273, 12725–12731. [Google Scholar] [CrossRef] [Green Version]
- Hunter, A.W.; Gourdie, R.G. The second PDZ domain of zonula occludens-1 is dispensable for targeting to connexin 43 gap junctions. Cell Commun. Adhes. 2008, 15, 55–63. [Google Scholar] [CrossRef]
- Rhett, J.M.; Jourdan, J.; Gourdie, R.G. Connexin 43 connexon to gap junction transition is regulated by zonula occludens-1. Mol. Biol. Cell 2011, 22, 1516–1528. [Google Scholar] [CrossRef]
- Maass, K.; Shibayama, J.; Chase, S.E.; Willecke, K.; Delmar, M. Size, and Localization of Cardiac Gap Junction Plaques. Circ. Res. 2007, 101, 1283–1291. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matthaeus, C.; Jüttner, R.; Gotthardt, M.; Rathjen, F.G. The IgCAM CAR Regulates Gap Junction-Mediated Coupling on Embryonic Cardiomyocytes and Affects Their Beating Frequency. Life 2023, 13, 14. https://doi.org/10.3390/life13010014
Matthaeus C, Jüttner R, Gotthardt M, Rathjen FG. The IgCAM CAR Regulates Gap Junction-Mediated Coupling on Embryonic Cardiomyocytes and Affects Their Beating Frequency. Life. 2023; 13(1):14. https://doi.org/10.3390/life13010014
Chicago/Turabian StyleMatthaeus, Claudia, René Jüttner, Michael Gotthardt, and Fritz G. Rathjen. 2023. "The IgCAM CAR Regulates Gap Junction-Mediated Coupling on Embryonic Cardiomyocytes and Affects Their Beating Frequency" Life 13, no. 1: 14. https://doi.org/10.3390/life13010014