Abstract
The vascular system of the prenatal brain is crucial for the development of the central nervous system. Communication between vessels and neural cells is bidirectional, and dysfunctional communication can lead to neurodevelopmental diseases. In the present review, we introduce neurodevelopmental and neuropsychiatric diseases potentially caused by disturbances in the neurovascular system and discuss candidate genes responsible for neurovascular system impairments. In contrast to diseases that can manifest during the developing stage, we have also summarized the disturbances of the neurovascular system in neurodegenerative diseases including Alzheimer’s disease and Parkinson’s disease. Furthermore, we discussed the role of abnormal vascularization and dysfunctional vessels in the development of neurovascular-related diseases.
1. Introduction
During the development of the murine central nervous system (CNS), the establishment of neural plates and the beginning of neural tube formation occur around embryonic day 7.5 (E7.5). Subsequently, dorsoventral patterning of neuroepithelial cells of the neural plate is established by E9.5 [1], with the proliferation of progenitors, differentiation into neural stem cells (NSCs), and migration of differentiated neurons and glia initiates to form the cerebral cortex in mice. CNS vascularization is initiated by the formation of the perineural vascular plexus (PNVP) at E8.5–10.5 in mice [2]. After the PNVP covers the CNS by E9, vessel ingression from the periventricular plexus into the cortex occurs at approximately E11.5 [3]. The association between embryonic NSCs and the vasculature thus appears necessary for CNS development [4,5]. According to this hypothesis, disturbances in brain vascularization during early life, including prenatal life, could impact brain formation through impaired neurogenesis. Conversely, impaired NSC characteristics reportedly influence brain vascularization, thereby suggesting that a close relationship between the nervous and vascular systems is essential for brain assembly [3,6]. In the present review, we introduce certain disorders that may be attributed to disturbances in brain vascularization.
In contrast to brain vascularization during early life, dysfunction of brain vessels may be involved in the development of cognitive decline. It has long been suggested that Alzheimer’s disease (AD), which accounts for a large proportion of dementia, is closely associated with cerebrovascular dysfunction [7,8]. In addition, cerebral amyloid angiopathy (CAA) has been detected in patients with AD [9]. Dementia with CAA alone is considered a form of vascular dementia, classified as AD when combined with significant AD pathology. Although there exists an overlap between AD and vascular dementia, and mixed dementia exists as well, this review focuses on the relationship between AD and cerebrovascular abnormalities. In addition to AD, vascular abnormalities in other neurodegenerative disorders, including Parkinson’s disease (PD), are discussed in our review.
The present review aims to shed light on the interplay between neurons and vascular system in neurodevelopmental, neuropsychiatric, and neuro-degenerative diseases.
2. Neural Stem Cells and Vascularization
During neural development, the vascular niche creates a specialized microenvironment via direct physical contact and secreted soluble factors.
In the embryonic neocortex, an avascular region without capillary vessel invasion is specifically constructed in the ventricular zone where mitotic NSCs are located, and NSCs transiently express HIF-1α, thereby attracting vascular endothelial tip cells. As a result, NSCs in contact with the pseudopodia of capillaries showed properties of undifferentiation, suggesting that direct contact with special ECs plays an important role in maintaining stemness [6]. Mitotic NSCs in the ventral telencephalon also induce vascular filopodia formation toward the ventricle in a cell cycle-dependent manner to regulate stem cell behavior [10]. In contrast, the adult subventricular zone (SVZ) is highly vascularized by a rich plexus of blood vessels [11]; however, the blood-brain barrier (BBB) in the SVZ has unique sites that have fewer glial endfeet and less pericyte (PE) coverage, allowing direct contact between NSCs and ECs [12]. In contrast, it is well established that soluble factors released from ECs regulate the behavior of NSCs, demonstrating the crucial role of the vascular niche in promoting the proliferation and differentiation of progenitors through soluble secreted cues [11,13,14,15]. Age-related changes in the vascular niche of the SVZ contribute to NSC depletion and dysfunction. For example, it has been suggested that the BBB in the SVZ is vulnerable and sensitive to age-dependent changes. Dividing NSCs are tightly juxtaposed with SVZ blood vessels during homeostasis and regeneration [12] as small circulating molecules in the blood enter the SVZ [12]. Recent evidence suggests reciprocal regulation between choroid plexuses and NSCs in a time- and region-dependent manner [16]. This is accomplished by region-specific secretion of molecules from each choroid plexus-cerebrospinal fluid (CSF) system as well as the competence of brain-region-specific NSCs to respond to the signaling molecules distributed in the CSF [16]. In addition, a recent study indicated that NSCs in the SVZ are particularly sensitive to age-related changes in the secretome of the lateral ventricle choroid plexus [17]. These findings suggest that the brain-specific capillary milieu influences NSCs expansion during development and deconstructs with age.
3. Neurodevelopmental and Neuropsychiatric Disorders Associated with Abnormal Vascularization
3.1. 22q11.2 Deletion Syndrome
The 22q11.2 deletion syndrome (22qDS; also known as DiGeorge’s syndrome) is caused by a 2.5-Mb hemizygous deletion of approximately 46 protein-coding genes on chromosome 22, including the Tbx1 gene [18]. Individuals with this syndrome are at high risk of neuropsychiatric disorders, including intellectual disability, schizophrenia, attention-deficit hyperactivity disorder, autism spectrum disorder, anxiety disorders, and seizures [19]. Based on neuroimaging studies, individuals with 22qDS exhibit reduced cortical thickness in specific brain regions [20,21], tortuous vessels, and subtle differences in cortical lamination [22], suggesting that neurogenesis and angiogenesis may be involved in the potential mechanisms underlying the neuropsychiatric phenotype of this syndrome.
Among the hemizygously deleted genes in 22qDS, TBX1 has been implicated in neurogenesis and angiogenesis in animal models. Tbx1, a transcription factor participating in organ development during prenatal life, reportedly plays a role in brain angiogenesis [23] (Figure 1). Furthermore, Tbx1 heterozygous deficiency-mediated brain vascular anomalies such as brain vessel hyperplasia and increased filopodial density were recently demonstrated to be restored by Tbx1-Cre-induced activation of the vascular endothelial growth factor receptor 3 (Vegfr3) transgene, suggesting that the brain vascular phenotype caused by Tbx1 loss of function is associated with the dysregulated expression of Vegfr3 [24]. Tbx1-deficient mice were shown to exhibit abnormalities in brain ECs, along with enhanced angiogenic sprouting, resulting in an expanded vascular network [25]. However, the expanded vascular network is not functionally inactivated [26]. Tbx1+/− mice showed the reduced proliferation of cortical progenitors and disturbed migration of glutamatergic cortical projection neurons and γ-aminobutyric acid (GABA)-mediated inhibitory neurons in the prenatal brain, with altered lamination documented in the adult cerebral cortex [25] (Figure 1).
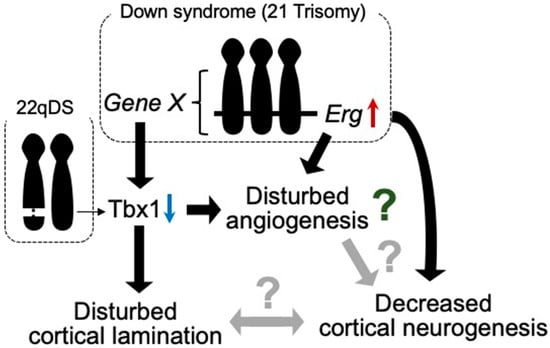
Figure 1.
Tbx1 possibly relates to vascular abnormalities in 22qDS and DS. Triplication of Erg gene reduces prenatal cortical neurogenesis. ERG is dominantly expressed in endothelial cells and is suggested to play a role in angiogenesis. In DS, increased expression of certain gene(s) in Hsa21 decreases the expression of Tbx1. In 22qDS, a 3 Mb or a nested 1.5 Mb deletion of Hsa22q11.2 includes the TBX1 gene. TBX1 is suggested to play a role in cortical lamination and prenatal cortical neurogenesis. Thus, TBX1 may be involved in the molecular mechanism of common aspects in these syndromes, such as intellectual disability.
Autism spectrum disorder (ASD) is a group of neurodevelopmental conditions characterized by early-onset dysfunctions in communication, impairments in social interaction, and repetitive and stereotyped behaviors and interests. Tbx1+/− mice reportedly exhibit ADS-related behavioral phenotypes such as impaired social interaction, ultrasonic vocalization, memory-based behavioral alternation, working memory, and thigmotaxis [26,27], along with low fractional anisotropy signals, deficits in myelinated axons in the fimbria, and selectively delayed spatial memory acquisition [28], suggesting that Tbx1 is a gene responsible for the phenotypes of 22q11.2 hemizygosity-associated ASD. In addition, these mice demonstrated peripheral lymphatic vessel development via the Tbx1-mediated regulation of Vegfr3 gene expression [29]. Based on these observations in Tbx1+/− mice, disturbances in angiogenesis, lymphangiogenesis, and neurogenesis may be responsible for the psychiatric phenotypes in 22qDS, such as ASD (Figure 1).
In addition to Tbx1, claudin-5 (CLDN5) is also encoded in the region of hemizygous deletion in 22qDS. Interestingly, the frequency of schizophrenia is significantly elevated in 22qDS [30]. Furthermore, the CLDN5 variant rs10314, which is associated with a decreased claudin-5 expression, was detected in the remaining 22q11.2 region in 9 of 15 22qDS subjects with schizophrenia but in only 8 of 44 22qDS subjects without schizophrenia [31]. Cldn5-deficient mice exhibit size-selective leakage of the BBB to molecules with a weight up to 800 Da, despite displaying a normal development and morphology of cerebral vessels [32]. Therefore, CLDN5 is thought to play a role in the formation of the BBB. Taken together, these results suggest that BBB dysfunction caused by a deficiency of CLDN5 may lead to a high incidence of schizophrenia in 22qDS.
3.2. Down Syndrome (DS)
DS is a typical aneuploidy caused by the presence of an extra copy of human chromosome 21 (Hsa21). Individuals with DS exhibit numerous clinical features, including intellectual disability, developmental delay (growth retardation), characteristic facial features, and early onset AD-like dementia [33]. In autopsy studies, embryonic neurogenesis has been suggested to be decreased in human fetuses with DS compared with the non-DS population [34,35,36]. In addition, studies with mouse models of DS revealed the presence of decreased neurogenesis in the developing prenatal brain [37,38]. Several candidate genes that are associated with decreased prenatal cortical neurogenesis have been identified [39].
Although vascular malformations of the brain have not been reported in individuals with DS or mouse models of DS, several studies have demonstrated suppressed tumor angiogenesis in mouse models of DS [40,41]. These studies revealed that Rcan1, Jam-b, Adamts1, Erg, and Pttg1lp may be associated with the inhibition of tumor angiogenesis. In contrast to tumor angiogenesis, triplication of the Erg gene reduces cortical neurogenesis in the embryonic brains of DS model mice [42] (Figure 1).
Murine ERG is pre-dominantly expressed in mesodermal tissues, including the endothelial, precartilaginous, and urogenital areas, during embryogenesis [43]. ERG has also been suggested to play a role in the regulation of endothelial homeostasis, vascular development, and angiogenesis [44]. These findings indicate that Erg is one of the genes responsible for neurovascular abnormalities in developing brains with DS. In addition, we reported that Tbx1 expression is reduced in the brain of Ts1Cje mice, as well as in other mouse models of DS, during both prenatal and postnatal life [45] (Figure 1). Accordingly, Tbx1 may play a role in brain vascularization [23]. Using an inducible-X-inactive specific transcript method, silencing of Hsa21 in induced pluripotent stem cells (iPSCs) with DS showed that triplication of Hsa21 influenced the expression of genes related to neurogenesis and angiogenesis [46] (Figure 1). These reports suggest that anomalies in vascular development may be disturbed in prenatal brains with DS.
3.3. Schizophrenia
Schizophrenia is a complex neuropsychiatric disorder with an unknown etiology and poorly defined neuropathological and neurobiological features. Current genetic and neurobiological analyses have implicated neuronal developmental and synaptic plasticity abnormalities [47], neurotransmitters [48], microglia [49], and oligodendrocyte dysfunction [50] in schizophrenia. Alterations in prenatal brain development have been implicated as major risk factors for schizophrenia [51]. Imaging studies using postmortem brains of patients with schizophrenia have revealed abnormalities in cortical cell-type composition and macroscopic tissue organization, possibly stemming from aberrant brain development, such as a reduced density of parvalbumin (PV)-expressing GABAergic neurons in the prefrontal cortex [52], reduced cortical layer thickness [53,54], and enlarged lateral ventricles [55]. Human iPSCs with a mutation in the disrupted-in-schizophrenia 1 (DISC1) gene, which is a risk factor for a wide array of psychiatric illnesses, including schizophrenia, exhibit elevated WNT signaling activity with an altered expression of neuronal fate-related genes including an increased expression of dorsal progenitor markers and decreased expression of ventral progenitor markers [56,57]. Furthermore, in an organoid model of human iPSCs with a mutation in DISC1, a disorganized ventricular structure, decreased proliferation of neural progenitors, and disturbed formation of cortical layers 2/3 were observed [58]. Since these abnormalities are improved by a WNT antagonist, they seem to be caused by WNT signaling activation [59].
In addition, studies with iPSCs derived from patients with schizophrenia also suggest a disturbance in cortical neurogenesis with alterations in WNT signaling [60]. It has been shown that disturbed Wnt/β-catenin signaling affects cortical neurogenesis and ventricular morphogenesis in rodents, similar to the results obtained using iPSC models of DISC1 mutation [60,61]. In line with these observations, decreased WNT signaling activity has also been detected in iPSC-derived brain organoids from patients with schizophrenia [59]. Therefore, reduced WNT activity enhances differentiation into cortical GABAergic neurons with a reduced proliferation of NSCs [62].
Disruption of the BBB has been documented in schizophrenia, suggesting an association between BBB hyperpermeability and the pathogenesis of schizophrenia [63,64,65,66,67]. In an in vitro study using patient-derived ECs, human iPSCs consistently revealed an intrinsic failure in brain microvascular endothelial-like cells of patients with schizophrenia, thereby affecting proper angiogenesis and the BBB function, which may contribute to altered neurovascular crosstalk during schizophrenia [68]. Furthermore, epidemiological findings suggest that the risk of certain types of cancer, such as respiratory cancer, is significantly lower in patients with schizophrenia than in those without schizophrenia [69,70,71,72,73,74]. This reduced frequency of certain cancers may be attributed to the impairment of tumor angiogenesis. Accumulating evidence therefore suggests that abnormal vascularization in the brain with schizophrenia may impact brain development.
4. Neurodegenerative Disorders Associated with Abnormal Vascularization
4.1. AD and Cerebrovascular Abnormalities
In contrast to neurodevelopmental diseases, aging is a predominant risk factor for dementia. The incidence of dementia is estimated to double every 5.0–5.5 years in the over-65 population [75]. According to an estimation by AD International, approximately 130 million people worldwide will suffer from dementia by 2050. AD accounts for 60%–80% of all dementia cases. According to the amyloid cascade hypothesis, the accumulation of amyloid-β (Aβ) in the brain is the first trigger for the development of AD, followed by the formation of neurofibrillary tangles and synaptic and neuronal loss, which are pathological alterations directly related to cognitive decline [76]. Neurofibrillary tangles are formed by the intraneuronal accumulation of hyperphosphorylated tau protein. Aβ is generated from the Aβ precursor protein (APP) by sequential processing with β- and γ-secretases, followed by deposition into the brain parenchyma to form senile plaques in AD; this accumulation in the cerebrovascular space results in CAA formation.
CAA has also been detected in the brains of patients with AD. As described above, a strong relationship between AD and cerebrovascular abnormalities has long been suggested. Although the pathogenesis of AD remains unclear, numerous accumulated reports suggest the involvement of the neurovascular unit [77], composed of cellular components such as neurons, astrocytes, microglia, pericytes, ECs, and smooth muscle cells, in the pathogenesis of AD (Figure 2).
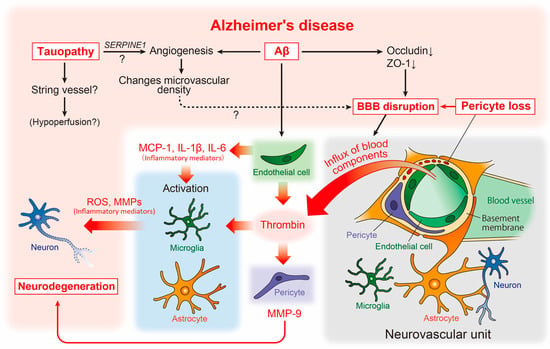
Figure 2.
Neurovascular unit and cerebrovascular abnormality-related neurodegenerative pathways in AD pathophysiology. The relationship between vascular and brain component cells in the predicted AD pathology is depicted, including key neurodegenerative molecules. Aβ, amyloid-β; AD, Alzheimer’s disease; BBB, blood-brain barrier; IL, interleukin; MCP-1, monocyte chemoattractant protein-1; MMPs, matrix metalloproteinases; ROS, reactive oxygen species.
At the macrostructural level, an altered microvascular density and numerous atrophic vessels are well established in the AD brain [78] (Figure 2). Enhanced microvascular density has been observed in the brains of an amyloid pathology mouse model (Tg2576 mice), as well as in human AD [79]. This enhancement was accompanied by increased angiogenesis and the disrupted expression of tight junction proteins such as occludin and zonula occludens-1 (ZO-1), suggesting that amyloidogenic events induce angiogenesis, which may affect the increased microvascular density and BBB disruption (Figure 2).
However, studies assessing the capillary density in human AD have shown conflicting findings, with some reporting an increase and others showing a decrease in capillary density [78]. This discrepancy may be explained by tauopathy. Recent studies in transgenic models of tauopathy (Tg4510 mice) have revealed that vascular density initially increases, followed by a decrease [80]. Moreover, the increased expression of angiogenesis-related genes such as Vegfa, Serpine1, and Plau was observed in ECs in the same mouse model, while the SERPINE1 expression was elevated in human AD (Figure 2). Thus, Aβ and tau pathologies may independently affect vascular abnormalities, and the phenotype of vascular abnormality may vary according to the stage of AD. In the same study, aged mice with tauopathy showed an increased number of small-diameter blood vessels (<4 μm) lacking red blood cells, with adhered leukocytes often restricting downstream flow [80]. Small vessels may correspond to the “string vessels”, characterized by collapsed capillaries and dying ECs in the AD brain [81]. Further analysis of the effect of tauopathy on the formation of blood vessels may provide novel insights into the molecular mechanisms underlying blood hypoperfusion in the AD brain (Figure 2).
In contrast, VEGF binds to Aβ and is deposited in plaques in the brains of patients with AD, likely resulting in deficiency of available VEGF under hypoperfusion [82]. Moreover, the overexpression of VEGF in the CNS in a transgenic mouse model of AD significantly improved the integrity of the cerebrovasculature and functionally rescued mice from memory impairments [83], suggesting that VEGF can be effective in combating neurodegeneration and vascular dysfunction that occur during the progression of AD. In addition, lymphatic vessels are regulated by signaling between VEGF-C and its receptor VEGFR3, whereas impairments in this pathway lead to a deficiency in meningeal lymphatic vessels in the brain [84,85,86].
Reportedly, vascular abnormalities correspond to pericyte degeneration [87], and studies of postmortem brains have shown a correlation between pericyte loss and BBB disruption [88] (Figure 2). BBB disruption results in an influx of blood components such as thrombin into the brain parenchyma [89]. Thrombin activates microglia and astrocytes, increasing the release of neurotoxic reactive oxygen species and matrix metalloproteinases (MMPs), respectively [90,91] (Figure 2). Interestingly, the microvessels of patients with AD release significantly more inflammatory proteins, including thrombin and MMPs, than those of non-demented individuals [92]. Subsequent studies have revealed that thrombin synthesis is highly upregulated in ECs [93], whereas pericytes react most sensitively to thrombin and markedly increase MMP-9 production [94]. Furthermore, Aβ promotes the production of inflammatory mediators including monocyte chemoattractant protein (MCP)-1, interleukin (IL)-1β, and IL-6 [95] from endothelial cells. This suggests that, along with neurons and glial cells, vascular cells themselves may actively and directly contribute to the neurodegenerative process in AD [7] (Figure 2).
A significant decrease in acetylcholine synthase (ChAT) activity [96,97] and nicotinic acetylcholine receptors [98] has been observed in autopsied brains of patients with AD. Furthermore, cholinergic neurons in the nucleus basalis of Meynert in the basal forebrain have been shown to be impaired in the very early pathological stages of AD [99,100]. Thus, the profound involvement of cholinergic signaling disruption in the pathogenesis of Alzheimer’s has been recognized. These findings gave rise to the cholinergic hypothesis [101] and led to the development of acetylcholinesterase inhibitors (AChEIs) such as donepezil, galantamine, and rivastigmine [102]. However, these drugs are symptomatic treatments for AD with limited efficacy and duration. Consequently, there is a need to develop new disease-modifying therapies (DMTs) that can intervene in the pathogenesis of AD. However, the development of DMTs has been hindered by undetectable efficacy and the emergence of side effects in clinical trials. Therefore, at present, drug treatment for AD still relies on AChEIs [103].
The profound association between basal forebrain vasculopathy and cholinergic degeneration, which is detected early in AD pathology, and its involvement in AD pathogenesis have attracted much attention. Indeed, in humans, basal forebrain atrophy due to cholinergic neuron degeneration, altered cerebral blood flow, and exacerbation of Aβ lesions has been detected in parallel [104]. In a mouse model of AD, it was demonstrated that specific neurodegeneration of cholinergic neurons induced by murine p75NTR saporin (mu p75-SAP), a highly specific cholinergic immunotoxin, is associated with increased Aβ plaque deposition [105]. However, the association between the loss of cholinergic innervation, decreased vascular reactivity, and decreased clearance of brain Aβ has not yet been elucidated. Smooth muscle cells, which regulate arterial contraction and contribute to the regulation of cerebral blood flow in the brain, are innervated by cholinergic neurons that originate from the basal forebrain. ACh, released from cholinergic neurons, induces vasodilation by stimulating nitric oxide (NO) production, primarily through activation of endothelial nitric oxide synthase (eNOS). Thus, ACh contributes to regional arteriolar dilation and increased cerebral blood flow during neurovascular communication through eNOS activation. This suggests that eNOS is involved in AD pathology. In fact, decreased eNOS expression has been reported in the occipital cortex, which is hypoperfused in AD [106]. Furthermore, eNOS-deficient mice showed increased CAA levels without increased Aβ production [107]. However, one mechanism underlying ACh activation of eNOS is suggested to occur via the insulin-receptor substrate/PI3K/Akt pathway [108]. In line with this hypothesis, stimulation of the PI3K/Akt/eNOS pathway by fasudil hydrochloride, a selective ROCK inhibitor, increases cerebral blood flow [109]. Nizari et al. recently demonstrated the role of loss of cholinergic innervation in the onset and progression of CAA in a mouse model of brain Aβ pathology treated with mu p75-SAP. The results further indicated that the intramural periarterial drainage pathway via regulation of the vascular function by eNOS may be involved in the clearance mechanism in fasudil-treated mice [110]. These findings support the importance of the interrelationship between cholinergic innervation and the vascular function in Aβ accumulation in the brain. Furthermore, the data suggest that activation of the cholinergic neuron-eNOS axis may enhance the efficiency of Aβ removal from the brain.
Early-onset familial AD (FAD) represents less than 1% of all AD cases and is caused by a single genetic mutation of either APP, PSEN1, and PSEN2 [111]. These mutations not only increase the production of Aβ but also vary the ratio of Aβ species to the aggregation-prone form (Aβ1-42) [76]. However, in the analysis of late-onset AD (sporadic AD), APOE ε4 was first identified as a risk gene. APOE ε4 is still considered the strongest genetic risk factor, including for early-onset AD. Furthermore, genome-wide association studies (GWAS) have identified more than 40 loci that are linked to AD risk and suggested that microglia are strongly implicated as the major cell type expressing GWAS genes [112]. Thus, there is a consensus that genetic factors have a strong influence on the development of AD, suggesting genetic overlap between AD and vascular pathology, primarily due to APOE [113]. However, little is known about the substantial association between genetic factors for AD and cerebrovascular abnormalities. One reason for this includes the difficulty of performing analyses due to the nature of blood vessels, which have abundant extracellular matrix around them and are rigid.
In this context, Yang et al. recently developed vessel isolation and nuclei extraction for sequencing (VINE-seq) to profile the major vascular and perivascular cell types of the human brain through 143,793 single-nucleus transcriptomes from nine individuals with AD and eight individuals with no cognitive impairment [114]. As a result, they demonstrated that at least 30 of the top 45 GWAS genes were enriched in cells of the human cerebrovascular system, again confirming the deep involvement of blood vessels and AD. They further found that GWAS genes predominantly expressed in microglia in mice, such as APOE, CASS4, INPP5D, and HLA-DRB1, were found to be strongly expressed in vascular cells in humans; this suggests the hypothesis that some AD risk genes and pathways may have been evolutionarily transferred from microglia to the vasculature from mice to humans. In addition, they also note that there is little overlap in the GWAS gene expression between mouse and human vascular cells, raising questions about the use of mice in studying cerebrovascular problems in human disease [114].
However, animal models, including mice, will be an essential tool for elucidating the mechanisms that substantially link genetic and pathological changes. Lee et al. identified FMNL2 from a GWAS with 6568 AD cases and 8101 control subjects by interaction analysis with cardio and cerebrovascular risk factors [115]. Using the Aβ-injected zebrafish and transgenic mouse (APdE9 mice) models of AD, they further experimentally found that FMNL2 is upregulated and expressed in astrocytes with Aβ burden and loosens gliovascular interactions to promote Aβ clearance in brains. Thus, by identifying a stage- and cell-specific role of FMNL2, they suggest a compensatory function for this protein in AD pathophysiology and propose it as a target molecule for new drug development [115].
Appropriate interventions to address the risk of vascular dysfunction in daily life have been beneficial in reducing the prevalence of AD [116]. Understanding the molecular mechanisms underlying the pathophysiology of AD angiopathy is expected to lead to the development of DMTs and diagnostic strategies for AD.
4.2. Other Neurodegenerative Diseases and Cerebrovascular Abnormalities
Other neurodegenerative disorders, such as blood vessel alterations, BBB disruption, cerebral blood flow abnormalities, amyotrophic lateral sclerosis (ALS) [117,118], Huntington’s disease (HD) [119], and PD [120], are known. ALS is characterized by progressive dysfunction and degeneration of motor neurons. Perturbation of the BBB and the blood-spinal cord barrier (BSCB) has been observed in a mouse model of ALS [121]. The decreased expression of tight junction-related proteins, including ZO-1 and CLDN5, has been shown in the spinal cord microvessels of ALS model mice [121], indicating that brain/spinal cord vasculature dysmorphology and dysfunction may be involved in pathogenesis or disease progression.
HD, an inherited autosomal dominant neurodegenerative disease, is caused by the expansion of cytosine–adenine–guanine (CAG) repeats in the huntingtin gene. Accumulating evidence indicates that HD is associated with cerebrovascular changes, including increased microvascular density [122,123,124] and BBB dysfunction [122,125,126]. Cerebrovascular alterations may be involved in HD pathogenesis.
Vascular abnormalities in PD have been investigated and summarized previously. It has been suggested that increased angiogenic vessels in the brain regions are affected not only in patients with PD but also in rodent models of PD [127,128,129,130]. Newly generated vessels are immature and prone to BBB leakage, particularly when pericyte recruitment is impaired. Indeed, a dysfunctional BBB, resulting in high vascular permeability, has been demonstrated in a number of PD models [129,131,132,133] and individuals with PD [134,135,136]. A study using rubidium-82-PET consistently failed to detect BBB leakage in patients [136]. Thus, abnormalities in the vascular system may be a common pathogenic mechanism in a number of neurodegenerative disorders, such as AD, ALS, HD, and PD, and may represent a possible therapeutic target.
5. Conclusions
Anomalies in vascularization seem to be common in certain neurodevelopmental and neuropsychiatric diseases and neurodegenerative disorders. Interestingly, the expression of Tbx1, which is suggested to play a role in abnormal prenatal vascularization, is known to be disturbed in two developmental diseases, 22qDS and DS. Shared aspects of these syndromes, such as intellectual disability, may be associated with an abnormal Tbx1 function. An increased frequency of schizophrenia is observed in individuals with 22qDS [19]. There is a report examining whether or not the TBX1 gene is associated with schizophrenia, and results suggest that a couple of rare mutations in the TBX1 gene may contribute to the patho-genesis of schizophrenia in some patients [137]. In contrast to developmental diseases, BBB dysfunction and abnormal vascularization have also been implicated in AD. A high prevalence of early-onset AD has been detected in individuals with DS, with symptoms appearing before the age of 65 years old [138]. Furthermore, approximately 75% of individuals with DS over 60 years old exhibit clinical evidence of dementia [139]. The abnormal development of vessels in the brains with DS may contribute to the pathogenesis of early-onset AD. Thus, clarifying the role of abnormal brain vessels in neurodevelopmental disorders and neurodegenerative diseases may afford a new target for pharmacotherapy for these neurological diseases. Other factors, such as sex differences, should be considered in the vascular abnormality-based neurological diseases. Since the cerebral vasculature expressing several sex steroid receptors is a direct target of androgen, androgen therefore affects the cerebral vasculature, and it may be the foundation of sex differences in neurological diseases [140].
Author Contributions
Conceptualization: K.I. and K.-i.M.; writing—original draft preparation: K.I. and K.T.; writing—review and editing: K.-i.M.; visualization, K.I. and K.T.; supervision, K.I. and K.-i.M.; project administration, K.I.; funding acquisition, K.I. All authors have read and agreed to the published version of the manuscript.
Funding
This work was in part supported by JSPS KAKENHI (Grant Number 22H04822 and 22K07033).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Dessaud, E.; McMahon, A.P.; Briscoe, J. Pattern formation in the vertebrate neural tube: A sonic hedgehog morphogen-regulated transcriptional network. Development 2008, 135, 2489–2503. [Google Scholar] [CrossRef]
- Hogan, K.A.; Ambler, C.A.; Chapman, D.L.; Bautch, V.L. The neural tube patterns vessels developmentally using the VEGF signaling pathway. Development 2004, 131, 1503–1513. [Google Scholar] [CrossRef]
- Vasudevan, A.; Long, J.E.; Crandall, J.E.; Rubenstein, J.L.; Bhide, P.G. Compartment-specific transcription factors orchestrate angiogenesis gradients in the embryonic brain. Nat. Neurosci. 2008, 11, 429–439. [Google Scholar] [CrossRef]
- Bjornsson, C.S.; Apostolopoulou, M.; Tian, Y.; Temple, S. It takes a village: Constructing the neurogenic niche. Dev. Cell 2015, 32, 435–446. [Google Scholar] [CrossRef]
- Takashima, S.; Watanabe, C.; Ema, M.; Mizutani, K.I. Interaction of the nervous system and vascular system is required for the proper assembly of the neocortex. Neurochem. Int. 2019, 129, 104481. [Google Scholar] [CrossRef]
- Komabayashi-Suzuki, M.; Yamanishi, E.; Watanabe, C.; Okamura, M.; Tabata, H.; Iwai, R.; Ajioka, I.; Matsushita, J.; Kidoya, H.; Takakura, N.; et al. Spatiotemporally Dependent Vascularization Is Differently Utilized among Neural Progenitor Subtypes during Neocortical Development. Cell Rep. 2019, 29, 1113–1129. [Google Scholar] [CrossRef]
- Toledo, J.B.; Arnold, S.E.; Raible, K.; Brettschneider, J.; Xie, S.X.; Grossman, M.; Monsell, S.E.; Kukull, W.A.; Trojanowski, J.Q. Contribution of cerebrovascular disease in autopsy confirmed neurodegenerative disease cases in the National Alzheimer’s Coordinating Centre. Brain 2013, 136, 2697–2706. [Google Scholar] [CrossRef]
- De la Torre, J.C.; Mussivand, T. Can disturbed brain microcirculation cause Alzheimer’s disease? Neurol. Res. 1993, 15, 146–153. [Google Scholar] [CrossRef]
- Viswanathan, A.; Greenberg, S.M. Cerebral amyloid angiopathy in the elderly. Ann. Neurol. 2011, 70, 871–880. [Google Scholar] [CrossRef]
- Di Marco, B.; Crouch, E.E.; Shah, B.; Duman, C.; Paredes, M.F.; Ruiz de Almodovar, C.; Huang, E.J.; Alfonso, J. Reciprocal Interaction between Vascular Filopodia and Neural Stem Cells Shapes Neurogenesis in the Ventral Telencephalon. Cell Rep. 2020, 33, 108256. [Google Scholar] [CrossRef]
- Ihrie, R.A.; Alvarez-Buylla, A. Lake-front property: A unique germinal niche by the lateral ventricles of the adult brain. Neuron 2011, 70, 674–686. [Google Scholar] [CrossRef]
- Tavazoie, M.; Van der Veken, L.; Silva-Vargas, V.; Louissaint, M.; Colonna, L.; Zaidi, B.; Garcia-Verdugo, J.M.; Doetsch, F. A specialized vascular niche for adult neural stem cells. Cell Stem Cell 2008, 3, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Goderie, S.K.; Jin, L.; Karanth, N.; Sun, Y.; Abramova, N.; Vincent, P.; Pumiglia, K.; Temple, S. Endothelial cells stimulate self-renewal and expand neurogenesis of neural stem cells. Science 2004, 304, 1338–1340. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Wang, Y.; Kokovay, E.; Lin, G.; Chuang, S.M.; Goderie, S.K.; Roysam, B.; Temple, S. Adult SVZ stem cells lie in a vascular niche: A quantitative analysis of niche cell-cell interactions. Cell Stem Cell 2008, 3, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Kokovay, E.; Goderie, S.; Wang, Y.; Lotz, S.; Lin, G.; Sun, Y.; Roysam, B.; Shen, Q.; Temple, S. Adult SVZ lineage cells home to and leave the vascular niche via differential responses to SDF1/CXCR4 signaling. Cell Stem Cell 2010, 7, 163–173. [Google Scholar] [CrossRef]
- Johansson, P.A. The choroid plexuses and their impact on developmental neurogenesis. Front. Neurosci. 2014, 8, 340. [Google Scholar] [CrossRef]
- Silva-Vargas, V.; Maldonado-Soto, A.R.; Mizrak, D.; Codega, P.; Doetsch, F. Age-Dependent Niche Signals from the Choroid Plexus Regulate Adult Neural Stem Cells. Cell Stem Cell 2016, 19, 643–652. [Google Scholar] [CrossRef]
- Gur, R.E.; Bassett, A.S.; McDonald-McGinn, D.M.; Bearden, C.E.; Chow, E.; Emanuel, B.S.; Owen, M.; Swillen, A.; Van den Bree, M.; Vermeesch, J.; et al. A neurogenetic model for the study of schizophrenia spectrum disorders: The International 22q11.2 Deletion Syndrome Brain Behavior Consortium. Mol. Psychiatry 2017, 22, 1664–1672. [Google Scholar] [CrossRef]
- Zinkstok, J.R.; Boot, E.; Bassett, A.S.; Hiroi, N.; Butcher, N.J.; Vingerhoets, C.; Vorstman, J.A.S.; van Amelsvoort, T.A.M.J. Neurobiological perspective of 22q11.2 deletion syndrome. Lancet Psychiatry 2019, 6, 951–960. [Google Scholar] [CrossRef]
- Bearden, C.E.; van Erp, T.G.; Dutton, R.A.; Tran, H.; Zimmermann, L.; Sun, D.; Geaga, J.A.; Simon, T.J.; Glahn, D.C.; Cannon, T.D.; et al. Mapping cortical thickness in children with 22q11.2 deletions. Cereb. Cortex 2007, 17, 1889–1898. [Google Scholar] [CrossRef]
- Sun, D.; Ching, C.R.K.; Lin, A.; Forsyth, J.K.; Kushan, L.; Vajdi, A.; Jalbrzikowski, M.; Hansen, L.; Villalon-Reina, J.E.; Qu, X.; et al. Large-scale mapping of cortical alterations in 22q11.2 deletion syndrome: Convergence with idiopathic psychosis and effects of deletion size. Mol. Psychiatry 2020, 25, 1822–1834. [Google Scholar] [CrossRef] [PubMed]
- Wu, P.; Teot, L.; Murdoch, G.; Monaghan-Nichols, A.P.; McFadden, K. Neuropathology of 22q11 deletion syndrome in an infant. Pediatr. Dev. Pathol. 2014, 17, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, S.; Martucciello, S.; Fulcoli, F.G.; Bilio, M.; Ferrentino, R.; Nusco, E.; Illingworth, E. Tbx1 regulates brain vascularization. Hum. Mol. Genet. 2014, 23, 78–89. [Google Scholar] [CrossRef] [PubMed]
- Cioffi, S.; Flore, G.; Martucciello, S.; Bilio, M.; Turturo, M.G.; Illingworth, E. VEGFR3 modulates brain microvessel branching in a mouse model of 22q11.2 deletion syndrome. Life Sci. Alliance 2022, 5, e202101308. [Google Scholar] [CrossRef] [PubMed]
- Flore, G.; Cioffi, S.; Bilio, M.; Illingworth, E. Cortical Development Requires Mesodermal Expression of Tbx1, a Gene Haploinsufficient in 22q11.2 Deletion Syndrome. Cereb. Cortex 2017, 27, 2210–2225. [Google Scholar]
- Hiramoto, T.; Kang, G.; Suzuki, G.; Satoh, Y.; Kucherlapati, R.; Watanabe, Y.; Hiroi, N. Tbx1: Identification of a 22q11.2 gene as a risk factor for autism spectrum disorder in a mouse model. Hum. Mol. Genet. 2011, 20, 4775–4785. [Google Scholar] [CrossRef]
- Takahashi, T.; Okabe, S.; Broin, P.Ó.; Nishi, A.; Ye, K.; Beckert, M.V.; Izumi, T.; Machida, A.; Kang, G.; Abe, S.; et al. Structure and function of neonatal social communication in a genetic mouse model of autism. Mol. Psychiatry 2016, 21, 1208–1214. [Google Scholar] [CrossRef]
- Hiramoto, T.; Sumiyoshi, A.; Yamauchi, T.; Tanigaki, K.; Shi, Q.; Kang, G.; Ryoke, R.; Nonaka, H.; Enomoto, S.; Izumi, T.; et al. Tbx1, a gene encoded in 22q11.2 copy number variant, is a link between alterations in fimbria myelination and cognitive speed in mice. Mol. Psychiatry 2022, 27, 929–938. [Google Scholar] [CrossRef]
- Chen, L.; Mupo, A.; Huynh, T.; Cioffi, S.; Woods, M.; Jin, C.; McKeehan, W.; Thompson-Snipes, L.; Baldini, A.; Illingworth, E. Tbx1 regulates Vegfr3 and is required for lymphatic vessel development. J. Cell Biol. 2010, 189, 417–424. [Google Scholar] [CrossRef]
- Hoeffding, L.K.; Trabjerg, B.B.; Olsen, L.; Mazin, W.; Sparsø, T.; Vangkilde, A.; Mortensen, P.B.; Pedersen, C.B.; Werge, T. Risk of Psychiatric Disorders Among Individuals With the 22q11.2 Deletion or Duplication: A Danish Nationwide, Register-Based Study. JAMA Psychiatry 2017, 74, 282–290. [Google Scholar] [CrossRef]
- Greene, C.; Kealy, J.; Humphries, M.M.; Gong, Y.; Hou, J.; Hudson, N.; Cassidy, L.M.; Martiniano, R.; Shashi, V.; Hooper, S.R.; et al. Dose-dependent expression of claudin-5 is a modifying factor in schizophrenia. Mol. Psychiatry 2018, 23, 2156–2166. [Google Scholar] [CrossRef] [PubMed]
- Nitta, T.; Hata, M.; Gotoh, S.; Seo, Y.; Sasaki, H.; Hashimoto, N.; Furuse, M.; Tsukita, S. Size-selective loosening of the blood-brain barrier in claudin-5-deficient mice. J. Cell Biol. 2003, 161, 653–660. [Google Scholar] [CrossRef] [PubMed]
- Korenberg, J.R.; Chen, X.N.; Schipper, R.; Sun, Z.; Gonsky, R.; Gerwehr, S.; Carpenter, N.; Daumer, C.; Dignan, P.; Disteche, C. Down syndrome phenotypes: The consequences of chromosomal imbalance. Proc. Natl. Acad. Sci. USA 1994, 91, 4997–5001. [Google Scholar] [CrossRef] [PubMed]
- Ross, M.H.; Galaburda, A.M.; Kemper, T.L. Down’s syndrome: Is there a decreased population of neurons? Neurology 1984, 34, 909–916. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Sidor, B.; Wisniewski, K.E.; Shepard, T.H.; Sersen, E.A. Brain growth in Down syndrome subjects 15 to 22 weeks of gestational age and birth to 60 months. Clin. Neuropathol. 1990, 4, 181–190. [Google Scholar]
- Guidi, S.; Bonasoni, P.; Ceccarelli, C.; Santini, D.; Gualtieri, F.; Ciani, E.; Bartesaghi, R. Neurogenesis impairment and increased cell death reduce total neuron number in the hippocampal region of fetuses with Down syndrome. Brain Pathol. 2008, 18, 180–197. [Google Scholar] [CrossRef]
- Chakrabarti, L.; Galdzicki, Z.; Haydar, H.F. Defects in embryonic neurogenesis and initial synapse formation in the forebrain of the Ts65Dn mouse model of Down syndrome. J. Neurosci. 2007, 27, 11483–11495. [Google Scholar] [CrossRef]
- Ishihara, K.; Amano, K.; Takaki, E.; Shimohata, A.; Sago, H.; Epstein, C.J.; Yamakawa, K. Enlarged brain ventricles and impaired neurogenesis in the Ts1Cje and Ts2Cje mouse models of Down syndrome. Cereb. Cortex 2010, 20, 1131–1143. [Google Scholar] [CrossRef]
- Ishihara, K. Genes Associated with Disturbed Cerebral Neurogenesis in the Embryonic Brain of Mouse Models of Down Syndrome. Genes 2021, 12, 1598. [Google Scholar] [CrossRef]
- Baek, K.H.; Zaslavsky, A.; Lynch, R.C.; Britt, C.; Okada, Y.; Siarey, R.J.; Lensch, M.W.; Park, I.H.; Yoon, S.S.; Minami, T.; et al. Down’s syndrome suppression of tumour growth and the role of the calcineurin inhibitor DSCR1. Nature 2009, 459, 1126–1130. [Google Scholar] [CrossRef]
- Reynolds, L.E.; Watson, A.R.; Baker, M.; Jones, T.A.; D’Amico, G.; Robinson, S.D.; Joffre, C.; Garrido-Urbani, S.; Rodriguez-Manzaneque, J.C.; Martino-Echarri, E.; et al. Tumour angiogenesis is reduced in the Tc1 mouse model of Down’s syndrome. Nature 2010, 465, 813–817. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, K.; Shimizu, R.; Takata, K.; Kawashita, E.; Amano, K.; Shimohata, A.; Low, D.; Nabe, T.; Sago, H.; Alexander, W.S.; et al. Perturbation of the immune cells and prenatal neurogenesis by the triplication of the Erg gene in mouse models of Down syndrome. Brain Pathol. 2020, 30, 75–91. [Google Scholar] [CrossRef] [PubMed]
- Vlaeminck-Guillem, V.; Carrere, S.; Dewitte, F.; Stehelin, D.; Desbiens, X.; Duterque-Coquillaud, M. The Ets family member Erg gene is expressed in mesodermal tissues and neural crests at fundamental steps during mouse embryogenesis. Mech. Dev. 2000, 91, 331–335. [Google Scholar] [CrossRef]
- Shah, A.V.; Birdsey, G.M.; Randi, A.M. Regulation of endothelial homeostasis, vascular development and angiogenesis by the transcription factor ERG. Vascul. Pharmacol. 2016, 86, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, R.; Ishihara, K.; Kawashita, E.; Sago, H.; Yamakawa, K.; Mizutani, K.I.; Akiba, S. Decrease in the T-box1 gene expression in embryonic brain and adult hippocampus of down syndrome mouse models. Biochem. Biophys. Res. Commun. 2021, 535, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.E.; Lawrence, J.B. Chromosome silencing in vitro reveals trisomy 21 causes cell-autonomous deficits in angiogenesis and early dysregulation in Notch signaling. Cell Rep. 2022, 40, 111174. [Google Scholar] [CrossRef]
- Arion, D.; Horváth, S.; Lewis, D.A.; Mirnics, K. Infragranular gene expression disturbances in the prefrontal cortex in schizophrenia: Signature of altered neural development? Neurobiol. Dis. 2010, 37, 738–746. [Google Scholar] [CrossRef]
- Lewis, D.A.; Pierri, J.N.; Volk, D.W.; Melchitzky, D.S.; Woo, T.U. Altered GABA neurotransmission and prefrontal cortical dysfunction in schizophrenia. Biol. Psychiatry 1999, 46, 616–626. [Google Scholar] [CrossRef]
- Wang, M.; Zhang, L.; Gage, F.H. Microglia, complement and schizophrenia. Nat. Neurosci. 2019, 22, 333–334. [Google Scholar] [CrossRef]
- Hakak, Y.; Walker, J.R.; Li, C.; Wong, W.H.; Davis, K.L.; Buxbaum, J.D.; Haroutunian, V.; Fienberg, A.A. Genome-wide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia. Proc. Natl. Acad. Sci. USA 2001, 98, 4746–4751. [Google Scholar] [CrossRef]
- Meyer-Lindenberg, A. From maps to mechanisms through neuroimaging of schizophrenia. Nature 2010, 468, 194–202. [Google Scholar] [CrossRef]
- Kaar, S.J.; Angelescu, I.; Marques, T.R.; Howes, O.D. Pre-frontal parvalbumin interneurons in schizophrenia: A meta-analysis of post-mortem studies. J. Neural. Transm. 2019, 126, 1637–1651. [Google Scholar] [CrossRef] [PubMed]
- Berdenis van Berlekom, A.; Muflihah, C.H.; Snijders, G.J.L.J.; MacGillavry, H.D.; Middeldorp, J.; Hol, E.M.; Kahn, R.S.; de Witte, L.D. Synapse Pathology in Schizophrenia: A Meta-analysis of Postsynaptic Elements in Postmortem Brain Studies. Schizophr. Bull. 2020, 46, 374–386. [Google Scholar] [CrossRef] [PubMed]
- Wagstyl, K.; Ronan, L.; Whitaker, K.J.; Goodyer, I.M.; Roberts, N.; Crow, T.J.; Fletcher, P.C. Multiple markers of cortical morphology reveal evidence of supragranular thinning in schizophrenia. Transl. Psychiatry 2016, 6, e780. [Google Scholar] [CrossRef] [PubMed]
- Kempton, M.J.; Stahl, D.; Williams, S.C.; DeLisi, L.E. Progressive lateral ventricular enlargement in schizophrenia: A meta-analysis of longitudinal MRI studies. Schizophr. Res. 2010, 120, 54–62. [Google Scholar] [CrossRef]
- Srikanth, P.; Han, K.; Callahan, D.G.; Makovkina, E.; Muratore, C.R.; Lalli, M.A.; Zhou, H.; Boyd, J.D.; Kosik, K.S.; Selkoe, D.J.; et al. Genomic DISC1 Disruption in hiPSCs Alters Wnt Signaling and Neural Cell Fate. Cell Rep. 2015, 12, 1414–1429. [Google Scholar] [CrossRef]
- Brennand, K.; Simone, A.; Jou, J.; Gelboin-Burkhart, C.; Tran, N.; Sangar, S.; Li, Y.; Mu, Y.; Chen, G.; Yu, D.; et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature 2011, 473, 221–225. [Google Scholar] [CrossRef]
- Srikanth, P.; Lagomarsino, V.N.; Muratore, C.R.; Ryu, S.C.; He, A.; Taylor, W.M.; Zhou, C.; Arellano, M.; Young-Pearse, T.L. Shared effects of DISC1 disruption and elevated WNT signaling in human cerebral organoids. Transl. Psychiatry 2018, 8, 77. [Google Scholar] [CrossRef]
- Topol, A.; Zhu, S.; Tran, N.; Simone, A.; Fang, G.; Brennand, K.J. Altered WNT Signaling in Human Induced Pluripotent Stem Cell Neural Progenitor Cells Derived from Four Schizophrenia Patients. Biol. Psychiatry 2015, 78, e29–e34. [Google Scholar] [CrossRef]
- Hirabayashi, Y.; Itoh, Y.; Tabata, H.; Nakajima, K.; Akiyama, T.; Masuyama, N.; Gotoh, Y. The Wnt/beta-catenin pathway directs neuronal differentiation of cortical neural precursor cells. Development 2004, 131, 2791–2801. [Google Scholar] [CrossRef]
- Mutch, C.A.; Funatsu, N.; Monuki, E.S.; Chenn, A. Beta-catenin signaling levels in progenitors influence the laminar cell fates of projection neurons. J. Neurosci. 2009, 29, 13710–13719. [Google Scholar] [CrossRef] [PubMed]
- Sawada, T.; Chater, T.E.; Sasagawa, Y.; Yoshimura, M.; Fujimori-Tonou, N.; Tanaka, K.; Benjamin, K.J.M.; Paquola, A.C.M.; Erwin, J.A.; Goda, Y.; et al. Developmental excitation-inhibition imbalance underlying psychoses revealed by single-cell analyses of discordant twins-derived cerebral organoids. Mol. Psychiatry 2020, 25, 2695–2711. [Google Scholar] [CrossRef] [PubMed]
- Harris, L.W.; Wayland, M.; Lan, M.; Ryan, M.; Giger, T.; Lockstone, H.; Wuethrich, I.; Mimmack, M.; Wang, L.; Kotter, M.; et al. The cerebral microvasculature in schizophrenia: A laser capture microdissection study. PLoS ONE 2008, 3, e3964. [Google Scholar] [CrossRef]
- Shalev, H.; Serlin, Y.; Friedman, A. Breaching the blood–brain barrier as a gate to psychiatric disorder. Cardiovasc. Psychiatry Neurol. 2009, 2009, 278531. [Google Scholar]
- Bechter, K.; Reiber, H.; Herzog, S.; Fuchs, D.; Tumani, H.; Maxeiner, H.G. Cerebrospinal fluid analysis in affective and schizophrenic spectrum disorders: Identification of subgroups with immune responses and blood-CSF barrier dysfunction. J. Psychiatr. Res. 2010, 44, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Wang, T.; Zhang, T.; Yi, S.; Zhao, S.; Li, N.; Yang, Y.; Zhang, F.; Xu, L.; Shan, B.; et al. Increased blood-brain barrier permeability of the thalamus and the correlation with symptom severity and brain volume alterations in schizophrenia patients. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2022, 7, 1025–1034. [Google Scholar]
- Puvogel, S.; Alsema, A.; Kracht, L.; Webster, M.J.; Weickert, C.S.; Sommer, I.E.C.; Eggen, B.J.L. Single-nucleus RNA sequencing of midbrain blood-brain barrier cells in schizophrenia reveals subtle transcriptional changes with overall preservation of cellular proportions and phenotypes. Mol. Psychiatry 2022, 27, 4731–4740. [Google Scholar] [CrossRef] [PubMed]
- Casas, B.S.; Vitória, G.; Prieto, C.P.; Casas, M.; Chacón, C.; Uhrig, M.; Ezquer, F.; Ezquer, M.; Rehen, S.K.; Palma, V. Schizophrenia-derived hiPSC brain microvascular endothelial-like cells show impairments in angiogenesis and blood-brain barrier function. Mol. Psychiatry 2022, 27, 4731–4740. [Google Scholar] [CrossRef]
- Lichtermann, D.; Ekelund, J.; Pukkala, E.; Tanskanen, A.; Lönnqvist, J. Incidence of cancer among persons with schizophrenia and their relatives. Arch. Gen. Psychiatry 2001, 58, 573–578. [Google Scholar] [CrossRef]
- Barak, Y.; Achiron, A.; Mandel, M.; Mirecki, I.; Aizenberg, D. Reduced cancer incidence among patients with schizophrenia. Cancer 2005, 104, 2817–2821. [Google Scholar] [CrossRef]
- Hippisley-Cox, J.; Vinogradova, Y.; Coupland, C.; Parker, C. Risk of malignancy in patients with schizophrenia or bipolar disorder: Nested case-control study. Arch. Gen. Psychiatry 2007, 64, 1368–1376. [Google Scholar] [CrossRef] [PubMed]
- Chou, F.H.; Tsai, K.Y.; Su, C.Y.; Lee, C.C. The incidence and relative risk factors for developing cancer among patients with schizophrenia: A nine-year follow-up study. Schizophr. Res. 2011, 129, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Sundquist, K.; Ning, Y.; Kendler, K.S.; Sundquist, J.; Chen, X. Incidence of cancer in patients with schizophrenia and their first-degree relatives: A population-based study in Sweden. Schizophr. Bull. 2013, 39, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.M.; Chen, Y.J.; Kuo, D.J.; Jaiteh, L.E.; Wu, Y.C.; Lo, T.S.; Li, Y.H. Cancer incidence in patients with schizophrenia or bipolar disorder: A nationwide population-based study in Taiwan, 1997-2009. Schizophr. Bull. 2013, 39, 407–416. [Google Scholar] [CrossRef]
- Corrada, M.M.; Brookmeyer, R.; Paganini-Hill, A.; Berlau, D.; Kawas, C.H. Dementia incidence continues to increase with age in the oldest old: The 90+ study. Ann. Neurol. 2010, 67, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Kugler, E.C.; Greenwood, J.; MacDonald, R.B. The “Neuro-Glial-Vascular” Unit: The Role of Glia in Neurovascular Unit Formation and Dysfunction. Front. Cell Dev. Biol. 2021, 27, 732820. [Google Scholar] [CrossRef]
- Buée, L.; Hof, P.R.; Bouras, C.; Delacourte, A.; Perl, D.P.; Morrison, J.H.; Fillit, H.M. Pathological alterations of the cerebral microvasculature in Alzheimer’s disease and related dementing disorders. Acta Neuropathol. 1994, 87, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Biron, K.E.; Dickstein, D.L.; Gopaul, R.; Jefferies, W.A. Amyloid triggers extensive cerebral angiogenesis causing blood brain barrier permeability and hypervascularity in Alzheimer’s disease. PLoS ONE 2011, 6, e23789. [Google Scholar] [CrossRef]
- Bennett, R.E.; Robbins, A.B.; Hu, M.; Cao, X.; Betensky, R.A.; Clark, T.; Das, S.; Hyman, B.T. Tau induces blood vessel abnormalities and angiogenesis-related gene expression in P301L transgenic mice and human Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2018, 115, E1289–E1298. [Google Scholar] [CrossRef]
- Brown, W.R. A review of string vessels or collapsed, empty basement membrane tubes. J. Alzheimers Dis. 2010, 21, 725–739. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.P.; Bae, D.G.; Kang, H.J.; Gwag, B.J.; Gho, Y.S.; Chae, C.B. Co-accumulation of vascular endothelial growth factor with beta-amyloid in the brain of patients with Alzheimer’s disease. Neurobiol. Aging 2004, 25, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Religa, P.; Cao, R.; Religa, D.; Xue, Y.; Bogdanovic, N.; Westaway, D.; Marti, H.H.; Winblad, B.; Cao, Y. VEGF significantly restores impaired memory behavior in Alzheimer’s mice by improvement of vascular survival. Sci. Rep. 2013, 3, 2053. [Google Scholar] [CrossRef]
- Alitalo, K.; Tammela, T.; Petrova, T.V. Lymphangiogenesis in development and human disease. Nature 2005, 438, 946–953. [Google Scholar] [CrossRef]
- Aspelund, A.; Antila, S.; Proulx, S.T.; Karlsen, T.V.; Karaman, S.; Detmar, M.; Wiig, H.; Alitalo, K. A dural lymphatic vascular system that drains brain interstitial fluid and macromolecules. J. Exp. Med. 2015, 212, 991–999. [Google Scholar] [CrossRef]
- Louveau, A.; Smirnov, I.; Keyes, T.J.; Eccles, J.D.; Rouhani, S.J.; Peske, J.D.; Derecki, N.C.; Castle, D.; Mandell, J.W.; Lee, K.S.; et al. Structural and functional features of central nervous system lymphatic vessels. Nature 2015, 523, 337–341. [Google Scholar] [CrossRef]
- Sengillo, J.D.; Winkler, E.A.; Walker, C.T.; Sullivan, J.S.; Johnson, M.; Zlokovic, B.V. Deficiency in mural vascular cells coincides with blood-brain barrier disruption in Alzheimer’s disease. Brain Pathol. 2013, 23, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Miners, J.S.; Schulz, I.; Love, S. Differing associations between Aβ accumulation, hypoperfusion, blood-brain barrier dysfunction and loss of PDGFRB pericyte marker in the precuneus and parietal white matter in Alzheimer’s disease. J. Cereb. Blood Flow Metab. 2018, 38, 103–115. [Google Scholar] [CrossRef]
- Van de Haar, H.J.; Burgmans, S.; Jansen, J.F.; van Osch, M.J.; van Buchem, M.A.; Muller, M.; Hofman, P.A.; Verhey, F.R.; Backes, W.H. Blood-Brain Barrier Leakage in Patients with Early Alzheimer Disease. Radiology 2016, 281, 527–535. [Google Scholar] [CrossRef]
- Choi, S.H.; Lee, D.Y.; Kim, S.U.; Jin, B.K. Thrombin-induced oxidative stress contributes to the death of hippocampal neurons in vivo: Role of microglial NADPH oxidase. J. Neurosci. 2005, 25, 4082–4090. [Google Scholar] [CrossRef]
- Choi, M.S.; Kim, Y.E.; Lee, W.J.; Choi, J.W.; Park, G.H.; Kim, S.D.; Jeon, S.J.; Go, H.S.; Shin, S.M.; Kim, W.K.; et al. Activation of protease-activated receptor1 mediates induction of matrix metalloproteinase-9 by thrombin in rat primary astrocytes. Brain Res. Bull. 2008, 76, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Grammas, P.; Samany, P.G.; Thirumangalakudi, L. Thrombin and inflammatory proteins are elevated in Alzheimer’s disease microvessels: Implications for disease pathogenesis. J. Alzheimers Dis. 2006, 9, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Wright, J.; Wall, T.; Grammas, P. Brain endothelial cells synthesize neurotoxic thrombin in Alzheimer’s disease. Am. J. Pathol. 2010, 176, 1600–1606. [Google Scholar] [CrossRef]
- Machida, T.; Takata, F.; Matsumoto, J.; Takenoshita, H.; Kimura, I.; Yamauchi, A.; Dohgu, S.; Kataoka, Y. Brain pericytes are the most thrombin-sensitive matrix metalloproteinase-9-releasing cell type constituting the blood-brain barrier in vitro. Neurosci. Lett. 2015, 599, 109–114. [Google Scholar] [CrossRef]
- Vukic, V.; Callaghan, D.; Walker, D.; Lue, L.F.; Liu, Q.Y.; Couraud, P.O.; Romero, I.A.; Weksler, B.; Stanimirovic, D.B.; Zhang, W. Expression of inflammatory genes induced by beta-amyloid peptides in human brain endothelial cells and in Alzheimer’s brain is mediated by the JNK-AP1 signaling pathway. Neurobiol. Dis. 2009, 34, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.; Maloney, A.J. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet 1976, 2, 1403. [Google Scholar] [CrossRef]
- Bowen, D.M.; Smith, C.B.; White, P.; Davison, A.N. Neurotransmitter-related enzymes and indices of hypoxia in senile dementia and other abiotrophies. Brain 1976, 99, 459–496. [Google Scholar] [CrossRef]
- Shimohama, S.; Taniguchi, T.; Fujiwara, M.; Kameyama, M. Changes in nicotinic and muscarinic cholinergic receptors in Alzheimer-type dementia. J. Neurochem. 1986, 46, 288–293. [Google Scholar] [CrossRef]
- Whitehouse, P.J.; Price, D.L.; Struble, R.G.; Clark, A.W.; Coyle, J.T.; Delon, M.R. Alzheimer’s disease and senile dementia: Loss of neurons in the basal forebrain. Science 1982, 215, 1237–1239. [Google Scholar] [CrossRef]
- Giacobini, E.; Cuello, A.C.; Fisher, A. Reimagining cholinergic therapy for Alzheimer’s disease. Brain 2022, 145, 2250–2275. [Google Scholar] [CrossRef]
- Bartus, R.T.; Dean, R.L.; Beer, B.; Lippa, A.S. The cholinergic hypothesis of geriatric memory dysfunction. Science 1982, 217, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Hampel, H.; Mesulam, M.M.; Cuello, A.C.; Khachaturian, A.S.; Vergallo, A.; Farlow, M.R.; Snyder, P.J.; Giacobini, E.; Khachaturian, Z.S. Revisiting the Cholinergic Hypothesis in Alzheimer’s Disease: Emerging Evidence from Translational and Clinical Research. J. Prev. Alzheimers Dis. 2019, 6, 2–15. [Google Scholar] [PubMed]
- Pozzi, F.E.; Conti, E.; Appollonio, I.; Ferrarese, C.; Tremolizzo, L. Predictors of response to acetylcholinesterase inhibitors in dementia: A systematic review. Front. Neurosci. 2022, 20, 998224. [Google Scholar] [CrossRef] [PubMed]
- Grothe, M.J.; Ewers, M.; Krause, B.; Heinsen, H.; Teipel, S.J. Alzheimer’s Disease Neuroimaging Initiative. Basal forebrain atrophy and cortical amyloid deposition in nondemented elderly subjects. Alzheimers. Dement. 2014, 10, S344–S353. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Rodriguez, J.J.; Pacheco-Herrero, M.; Thyssen, D.; Murillo-Carretero, M.I.; Berrocoso, E.; Spires-Jones, T.L.; Bacskai, B.J.; Garcia-Alloza, M. Rapid β-amyloid deposition and cognitive impairment after cholinergic denervation in APP/PS1 mice. J. Neuropathol. Exp. Neurol. 2013, 72, 272–285. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.R.; Hunter, R.; Wyper, D.J.; Patterson, J.; Kelly, R.C.; Montaldi, D.; McCullouch, J. Longitudinal changes in cognitive function and regional cerebral function in Alzheimer’s disease: A SPECT blood flow study. J. Psychiatr. Res. 1996, 30, 109–126. [Google Scholar] [CrossRef] [PubMed]
- Austin, S.A.; Katusic, Z.S. Partial loss of endothelial nitric oxide leads to increased cerebrovascular beta amyloid. J. Cereb. Blood Flow Metab. 2020, 40, 392–403. [Google Scholar] [CrossRef]
- Zecchin, H.G.; Priviero, F.B.; Souza, C.T.; Zecchin, K.G.; Prada, P.O.; Carvalheira, J.B.; Velloso, L.A.; Antunes, E.; Saad, M.J. Defective insulin and acetylcholine induction of endothelial cell-nitric oxide synthase through insulin receptor substrate/Akt signaling pathway in aorta of obese rats. Diabetes 2007, 56, 1014–1024. [Google Scholar] [CrossRef]
- Rikitake, Y.; Kim, H.H.; Huang, Z.; Seto, M.; Yano, K.; Asano, T.; Moskowitz, M.A.; Liao, J.K. Inhibition of Rho kinase (ROCK) leads to increased cerebral blood flow and stroke protection. Stroke 2005, 36, 2251–2257. [Google Scholar] [CrossRef]
- Nizari, S.; Wells, J.A.; Carare, R.O.; Romero, I.A.; Hawkes, C.A. Loss of cholinergic innervation differentially affects eNOS-mediated blood flow, drainage of Aβ and cerebral amyloid angiopathy in the cortex and hippocampus of adult mice. Acta Neuropathol. Commun. 2021, 9, 12. [Google Scholar] [CrossRef]
- Arber, C.; Toombs, J.; Lovejoy, C.; Ryan, N.S.; Paterson, R.W.; Willumsen, N.; Gkanatsiou, E.; Portelius, E.; Blennow, K.; Heslegrave, A.; et al. Familial Alzheimer’s disease patient-derived neurons reveal distinct mutation-specific effects on amyloid beta. Mol. Psychiatry. 2020, 25, 2919–2931. [Google Scholar] [CrossRef] [PubMed]
- Novikova, G.; Kapoor, M.; Tcw, J.; Abud, E.M.; Efthymiou, A.G.; Chen, S.X.; Cheng, H.; Fullard, J.F.; Bendl, J.; Liu, Y.; et al. Integration of Alzheimer’s disease genetics and myeloid genomics identifies disease risk regulatory elements and genes. Nat. Commun. 2021, 12, 1610. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.F.; Smith, A.V.; Aspelund, T.; Betensky, R.A.; Smoller, J.W.; Gudnason, V.; Launer, L.J.; Blacker, D. Genetic overlap between vascular pathologies and Alzheimer’s dementia and potential causal mechanisms. Alzheimers Dement. 2019, 15, 65–75. [Google Scholar] [CrossRef]
- Yang, A.C.; Vest, R.T.; Kern, F.; Lee, D.P.; Agam, M.; Maat, C.A.; Losada, P.M.; Chen, M.B.; Schaum, N.; Khoury, N.; et al. A human brain vascular atlas reveals diverse mediators of Alzheimer’s risk. Nature 2022, 603, 885–892. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.J.; Raghavan, N.S.; Bhattarai, P.; Siddiqui, T.; Sariya, S.; Reyes-Dumeyer, D.; Flowers, X.E.; Cardoso, S.A.L.; De Jager, P.L.; Bennett, D.A.; et al. FMNL2 regulates gliovascular interactions and is associated with vascular risk factors and cerebrovascular pathology in Alzheimer’s disease. Acta Neuropathol. 2022, 144, 59–79. [Google Scholar] [CrossRef]
- Ngandu, T.; Lehtisalo, J.; Solomon, A.; Levälahti, E.; Ahtiluoto, S.; Antikainen, R.; Bäckman, L.; Hänninen, T.; Jula, A.; Laatikainen, T.; et al. A 2 year multidomain intervention of diet, exercise, cognitive training, and vascular risk monitoring versus control to prevent cognitive decline in at-risk elderly people (FINGER): A randomised controlled trial. Lancet 2015, 385, 2255–2263. [Google Scholar] [CrossRef] [PubMed]
- Garbuzova-Davis, S.; Sanberg, P.R. Blood-CNS Barrier Impairment in ALS patients versus an animal model. Front. Cell Neurosci. 2014, 8, 21. [Google Scholar] [CrossRef]
- Winkler, E.A.; Sengillo, J.D.; Sagare, A.P.; Zhao, Z.; Ma, Q.; Zuniga, E.; Wang, Y.; Zhong, Z.; Sullivan, J.S.; Griffin, J.H.; et al. Blood-spinal cord barrier disruption contributes to early motor-neuron degeneration in ALS-model mice. Proc. Natl. Acad. Sci. USA 2014, 111, E1035–E1042. [Google Scholar] [CrossRef]
- Padel, T.; Roth, M.; Gaceb, A.; Li, J.Y.; Bjorkqvist, M.; Paul, G. Brain pericyte activation occurs early in Huntington’s disease. Exp. Neurol. 2018, 305, 139–150. [Google Scholar] [CrossRef]
- Faucheux, B.A.; Agid, Y.; Hirsch, E.C.; Bonnet, A.-M. Blood vessels change in the mesencephalon of patients with Parkinson’s disease. Lancet 1999, 353, 981–982. [Google Scholar] [CrossRef]
- Zhong, Z.; Deane, R.; Ali, Z.; Parisi, M.; Shapovalov, Y.; O’Banion, M.K.; Stojanovic, K.; Sagare, A.; Boillee, S.; Cleveland, D.W.; et al. ALS-causing SOD1 mutants generate vascular changes prior to motor neuron degeneration. Nat. Neurosci. 2008, 11, 420–422. [Google Scholar] [CrossRef] [PubMed]
- Drouin-Ouellet, J.; Sawiak, S.J.; Cisbani, G.; Lagace, M.; Kuan, W.L.; Saint-Pierre, M.; Dury, R.J.; Alata, W.; St-Amour, I.; Mason, S.L.; et al. Cerebrovascular and blood-brain barrier impairments in Huntington’s disease: Potential implications for its pathophysiology. Ann. Neurol. 2015, 78, 160–177. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.Y.; Hsu, Y.H.; Lin, M.H.; Yang, T.H.; Chen, H.M.; Chen, Y.C.; Hsiao, H.Y.; Chen, C.C.; Chern, Y.; Chang, C. Neurovascular abnormalities in humans and mice with Huntington’s disease. Exp. Neurol. 2013, 250, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, H.Y.; Chen, Y.C.; Huang, C.H.; Chen, C.C.; Hsu, Y.H.; Chen, H.M.; Chiu, F.L.; Kuo, H.C.; Chang, C.; Chern, Y. Aberrant astrocytes impair vascular reactivity in Huntington disease. Ann. Neurol. 2015, 78, 178–192. [Google Scholar] [CrossRef]
- Di Pardo, A.; Amico, E.; Scalabri, F.; Pepe, G.; Castaldo, S.; Elifani, F.; Capocci, L.; De Sanctis, C.; Comerci, L.; Pompeo, F.; et al. Impairment of blood-brain barrier is an early event in R6/2 mouse model of Huntington Disease. Sci. Rep. 2017, 7, 41316. [Google Scholar] [CrossRef]
- Lim, R.G.; Quan, C.; Reyes-Ortiz, A.M.; Lutz, S.E.; Kedaigle, A.J.; Gipson, T.A.; Wu, J.; Vatine, G.D.; Stocksdale, J.; Casale, M.S.; et al. Huntington’s disease iPSC-derived brain microvascular endothelial cells reveal WNT-mediated angiogenic and blood-brain barrier deficits. Cell Rep. 2017, 19, 1365–1377. [Google Scholar] [CrossRef] [PubMed]
- Desai Bradaric, B.; Patel, A.; Schneider, J.A.; Carvey, P.M.; Hendey, B. Evidence for angiogenesis in Parkinson’s disease, incidental Lewy body disease, and progressive supranuclear palsy. J. Neural Transm. 2012, 119, 59–71. [Google Scholar] [CrossRef]
- Carvey, P.M.; Zhao, C.H.; Hendey, B.; Lum, H.; Trachtenberg, J.; Desai, B.S.; Snyder, J.; Zhu, Y.G.; Ling, Z.D. 6-Hydroxydopamine-induced alterations in blood-brain barrier permeability. Eur. J. Neurosci. 2005, 22, 1158–1168. [Google Scholar] [CrossRef]
- Elabi, O.F.; Cunha, J.; Gaceb, A.; Fex, M.; Paul, G. High-fat diet-induced diabetes leads to vascular alterations, pericyte reduction, and perivascular depletion of microglia in a 6-OHDA toxin model of Parkinson disease. J. Neuroinflam. 2021, 18, 175. [Google Scholar] [CrossRef]
- Westin, J.E.; Lindgren, H.S.; Gardi, J.; Nyengaard, J.R.; Brundin, P.; Mohapel, P.; Cenci, M.A. Endothelial proliferation and increased blood-brain barrier permeability in the basal ganglia in a rat model of 3,4-dihydroxyphenyl-L-alanine-induced dyskinesia. J. Neurosci. 2006, 26, 9448–9461. [Google Scholar] [CrossRef]
- Zhao, C.H.; Ling, Z.D.; Newman, M.B.; Bhatia, A.; Carvey, P.M. TNF-alpha knockout and minocycline treatment attenuates blood-brain barrier leakage in MPTP-treated mice. Neurobiol. Dis. 2007, 26, 36–46. [Google Scholar] [CrossRef]
- Chen, X.; Lan, X.; Roche, I.; Liu, R.; Geiger, J.D. Caffeine protects against MPTP-induced blood-brain barrier dysfunction in mouse striatum. J. Neurochem. 2008, 107, 1147–1157. [Google Scholar] [CrossRef] [PubMed]
- Kortekaas, R.; Leenders, K.L.; van Oostrom, J.C.; Vaalburg, W.; Bart, J.; Willemsen, A.T.; Hendrikse, N.H. Blood-brain barrier dysfunction in parkinsonian midbrain in vivo. Ann. Neurol. 2005, 57, 176–179. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.T.; Woulfe, J.M. Striatal blood-brain barrier permeability in Parkinson’s disease. J. Cereb. Blood Flow Metab. 2015, 35, 747–750. [Google Scholar] [CrossRef] [PubMed]
- Al-Bachari, S.; Naish, J.H.; Parker, G.J.M.; Emsley, H.C.A.; Parkes, L.M. Blood-Brain Barrier Leakage Is Increased in Parkinson’s Disease. Front. Physiol. 2020, 11, 593026. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Peng, S.; Ma, Y.; Tang, C.C.; Hellman, M.; Feigin, A.; Eidelberg, D.; Dhawan, V. Blood-brain barrier permeability in Parkinson’s disease patients with and without dyskinesia. J. Neurol. 2021, 268, 2246–2255. [Google Scholar] [CrossRef]
- Ping, L.Y.; Chuang, Y.A.; Hsu, S.H.; Tsai, H.Y.; Cheng, M.C. Screening for Mutations in the TBX1 Gene on Chromosome 22q11.2 in Schizophrenia. Genes 2016, 7, 102. [Google Scholar] [CrossRef]
- Hartley, D.; Blumenthal, T.; Carrillo, M.; DiPaolo, G.; Esralew, L.; Gardiner, K.; Granholm, A.C.; Iqbal, K.; Krams, M.; Lemere, C.; et al. Down syndrome and Alzheimer’s disease: Common pathways, common goals. Alzheimers. Dement. 2015, 11, 700–709. [Google Scholar] [CrossRef]
- Lai, F.; Williams, R.S. A prospective study of Alzheimer disease in Down syndrome. Arch. Neurol. 1989, 46, 849–853. [Google Scholar] [CrossRef]
- Abi-Ghanem, C.; Robison, L.S.; Zuloaga, K.L. Androgens’ effects on cerebrovascular function in health and disease. Biol. Sex Differ. 2020, 11, 35. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).