
Multimodal Ophthalmic Imaging in Spinocerebellar Ataxia Type 7

 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Case Series
2.2. Neurological Examination
2.3. Ophthalmological Examination
2.4. Literature Review
3. Case Series
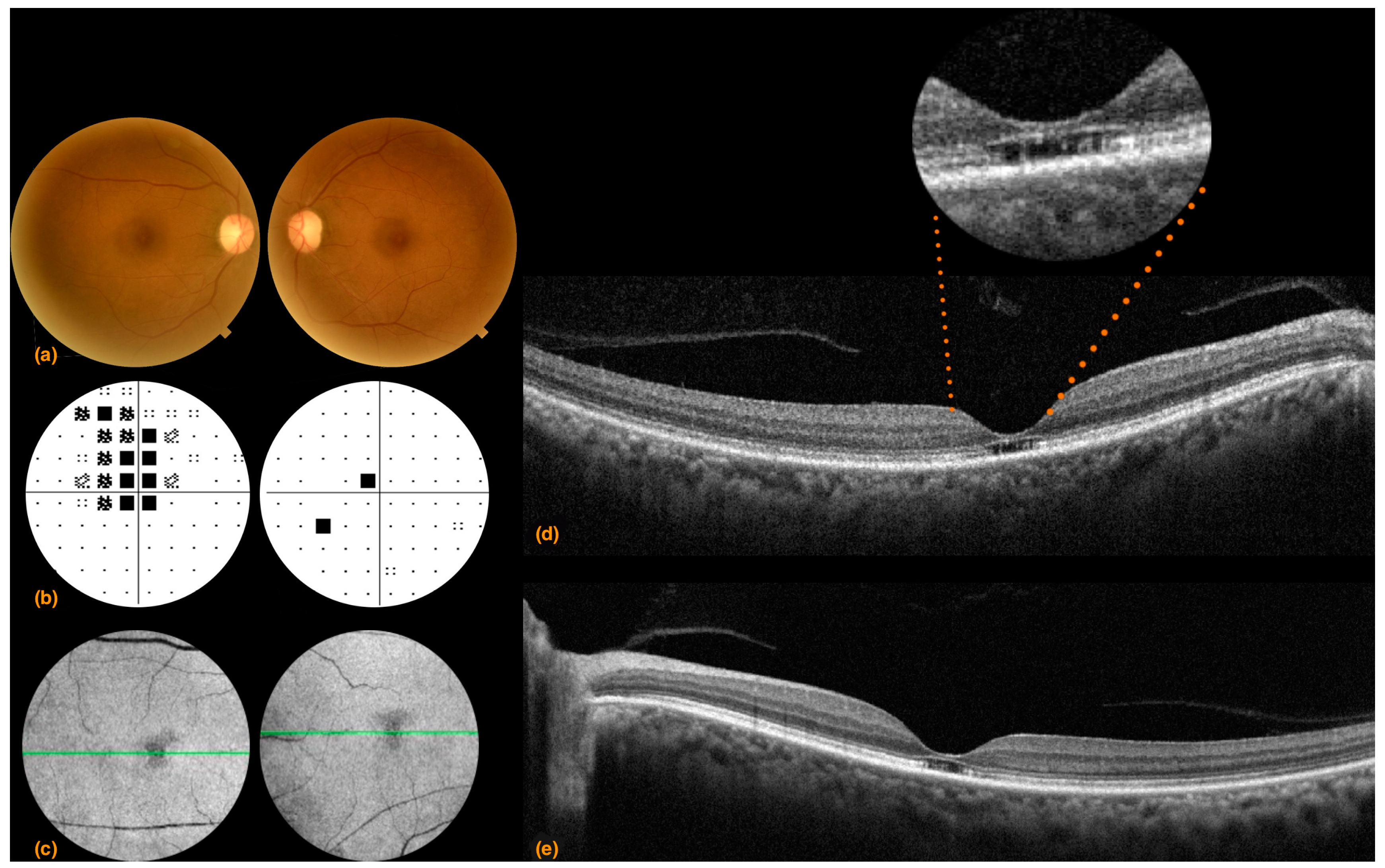
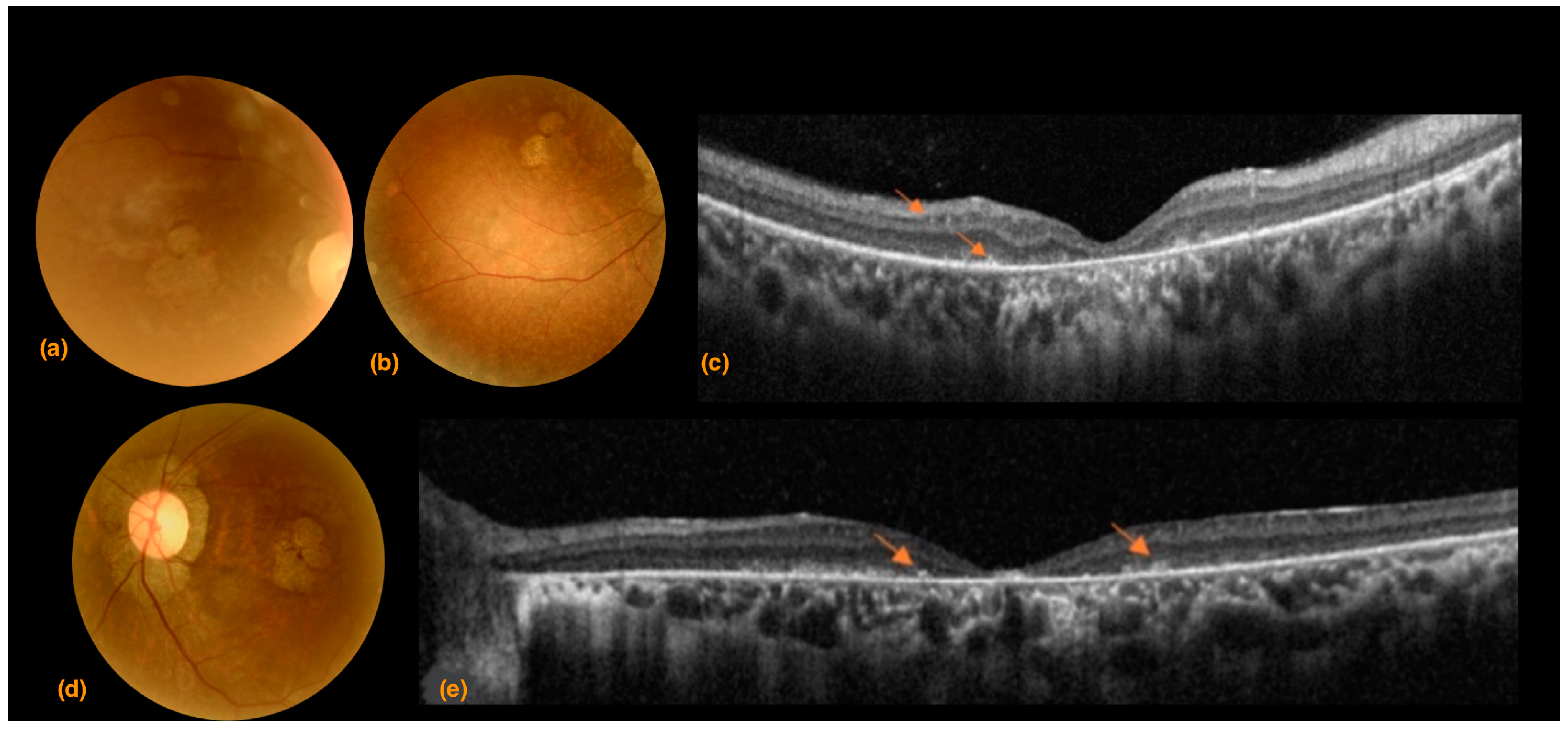
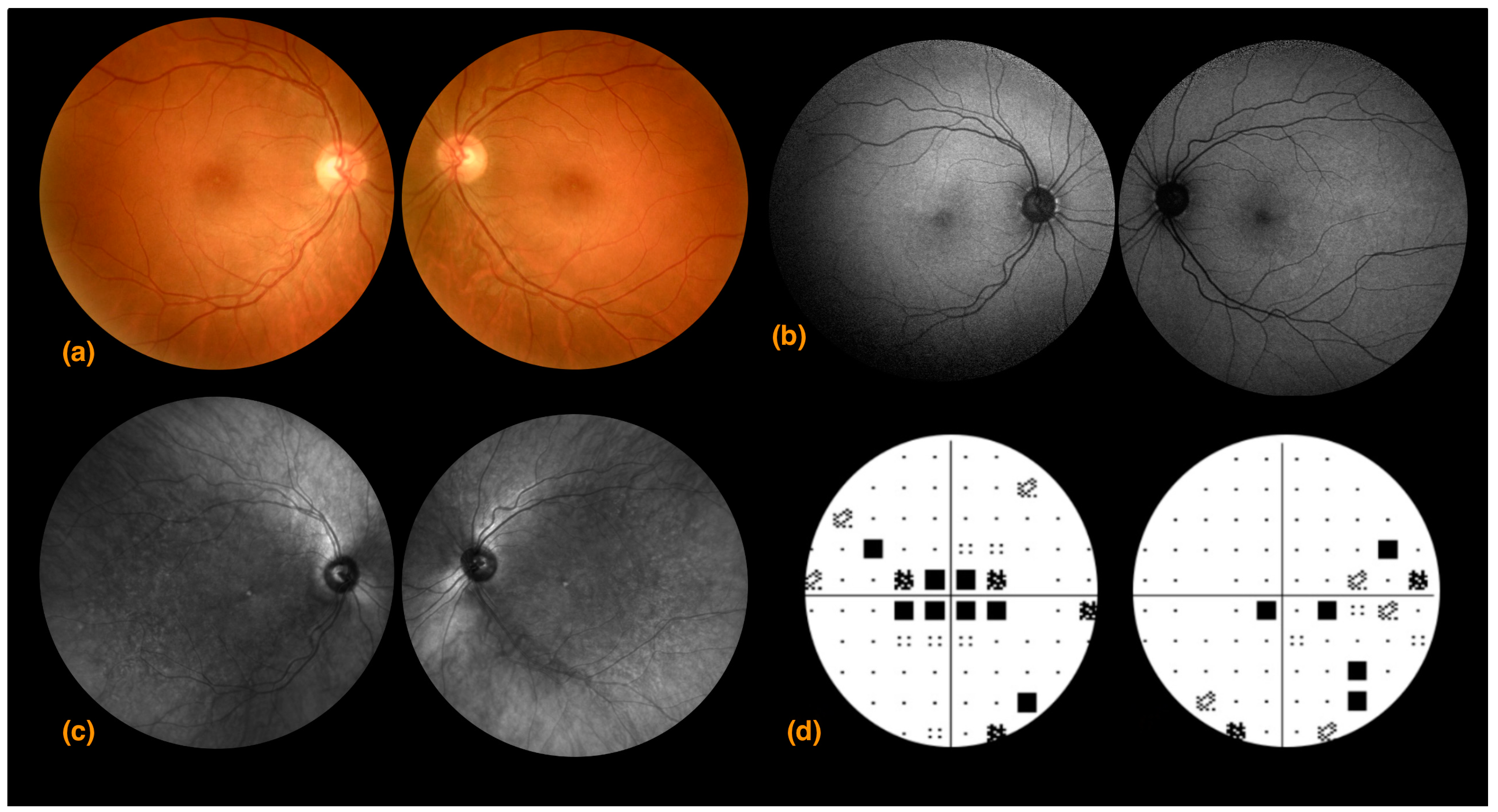
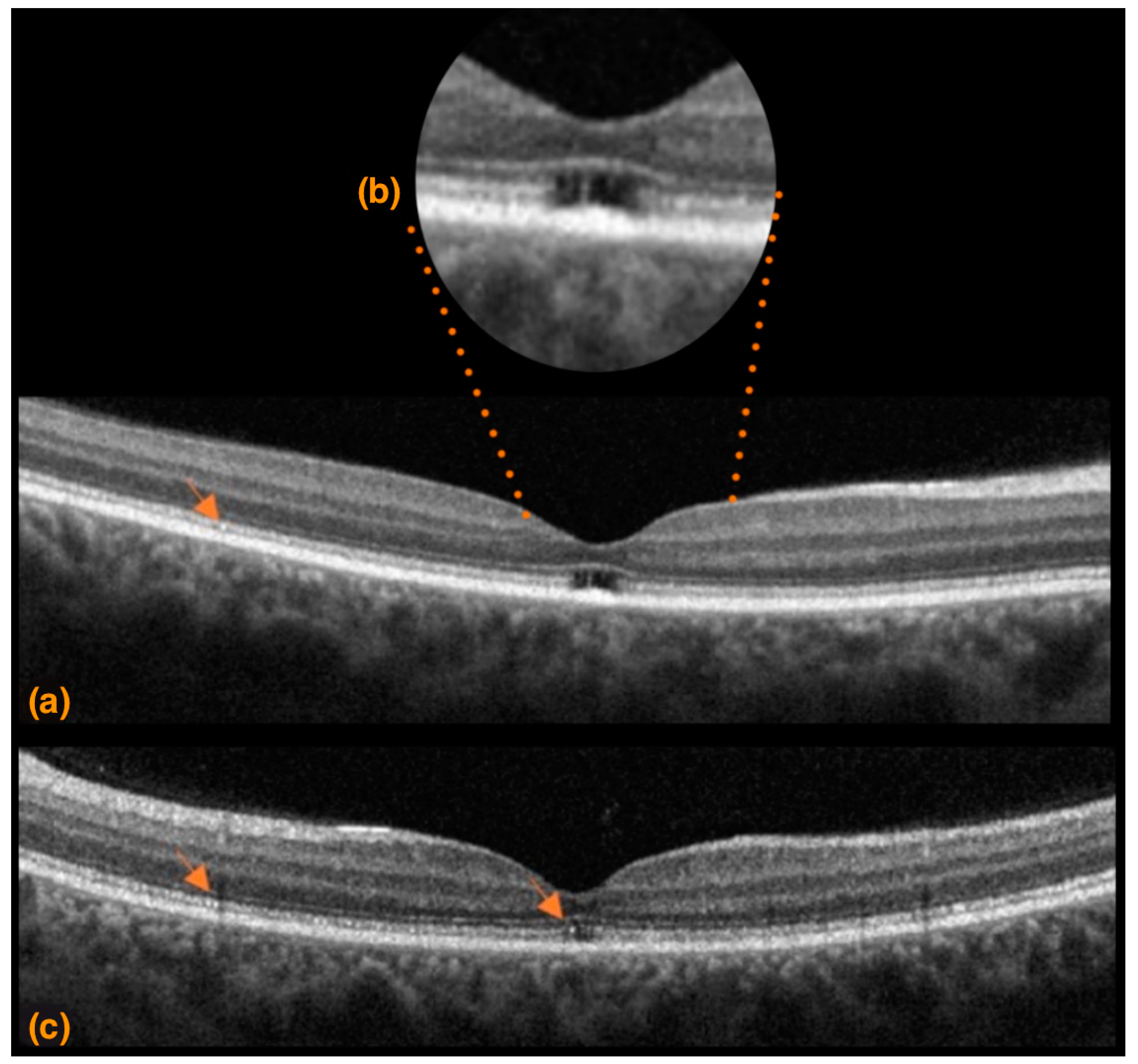
3.1. Case 1
3.2. Case 2
3.3. Case 3
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Paulson, H.L. The spinocerebellar ataxias. J. Neuro-Ophthalmol. Off. J. N. Am. Neuro-Ophthalmol. Soc. 2009, 29, 227–237. [Google Scholar] [CrossRef]
- Niewiadomska-Cimicka, A.; Doussau, F.; Perot, J.B.; Roux, M.J.; Keime, C.; Hache, A.; Piguet, F.; Novati, A.; Weber, C.; Yalcin, B.; et al. SCA7 Mouse Cerebellar Pathology Reveals Preferential Downregulation of Key Purkinje Cell-Identity Genes and Shared Disease Signature with SCA1 and SCA2. J. Neurosci. 2021, 41, 4910–4936. [Google Scholar] [CrossRef]
- Michalik, A.; Martin, J.J.; Van Broeckhoven, C. Spinocerebellar Ataxia Type 7 Associated with Pigmentary Retinal Dystrophy. Eur. J. Hum. Genet. 2004, 12, 2–15. [Google Scholar] [CrossRef]
- Goswami, R.; Bello, A.I.; Bean, J.; Costanzo, K.M.; Omer, B.; Cornelio-Parra, D.; Odah, R.; Ahluwalia, A.; Allan, S.K.; Nguyen, N.; et al. The Molecular Basis of Spinocerebellar Ataxia Type 7. Front. Neurosci. 2022, 16, 818757. [Google Scholar] [CrossRef]
- Van de Warrenburg, B.P.; Frenken, C.W.; Ausems, M.G.; Kleefstra, T.; Sinke, R.J.; Knoers, N.V.; Kremer, H.P. Striking Anticipation in Spinocerebellar Ataxia Type 7: The Infantile Phenotype. J. Neurol. 2001, 248, 911–914. [Google Scholar] [CrossRef]
- Karam, A.; Trottier, Y. Molecular Mechanisms and Therapeutic Strategies in Spinocerebellar Ataxia Type 7. Adv. Exp. Med. Biol. 2018, 1049, 197–218. [Google Scholar]
- Johansson, J. Expanded CAG Repeats in Swedish Spinocerebellar Ataxia Type 7 (SCA7) Patients: Effect of CAG Repeat Length on the Clinical Manifestation. Hum. Mol. Genet. 1998, 7, 171–176. [Google Scholar] [CrossRef]
- Helmlinger, D.; Abou-Sleymane, G.; Yvert, G.; Rousseau, S.; Weber, C.; Trottier, Y.; Mandel, J.L.; Devys, D. Disease progression despite early loss of polyglutamine protein expression in SCA7 mouse model. J. Neurosci. 2004, 24, 1881–1887. [Google Scholar] [CrossRef]
- La Spada, A.R.; Fu, Y.H.; Sopher, B.L.; Libby, R.L.; Wang, X.; Li, L.Y.; Einum, D.D.; Huang, J.; Possin, D.E.; Smith, A.C.; et al. Polyglutamine-Expanded Ataxin-7 Antagonizes CRX Function and Induces Cone-Rod Dystrophy in a Mouse Model of SCA7. Neuron 2001, 31, 913–927. [Google Scholar] [CrossRef]
- Mohan, R.D.; Dialynas, G.; Weake, V.M.; Liu, J.; Martin-Brown, S.; Florens, L.; Washburn, M.P.; Workman, J.L.; Abmayr, S.M. Loss of Drosophila Ataxin-7, a SAGA subunit, reduces H2B ubiquitination and leads to neural and retinal degeneration. Genes Dev. 2014, 28, 259–272. [Google Scholar] [CrossRef]
- Yefimova, M.G.; Messaddeq, N.; Karam, A.; Jacquard, C.; Weber, C.; Jonet, L.; Wolfrum, U.; Jeanny, J.C.; Trottier, Y. Polyglutamine toxicity induces rod photoreceptor division, morphological transformation or death in Spinocerebellar ataxia 7 mouse retina. Neurobiol. Dis. 2010, 40, 311–324. [Google Scholar] [CrossRef]
- Charles, P. Factors Influencing Disease Progression in Autosomal Dominant Cerebellar Ataxia and Spastic Paraplegia. Arch. Neurol. 2012, 69, 500–508. [Google Scholar] [CrossRef]
- Campos-Romo, A.; Graue-Hernandez, E.O.; Pedro-Aguilar, L.; Hernandez-Camarena, J.C.; Rivera-De la Parra, D.; Galvez, V.; Diaz, R.; Jimenez-Corona, A.; Fernandez-Ruiz, J. Ophthalmic Features of Spinocerebellar Ataxia Type 7. Eye 2018, 32, 120–127. [Google Scholar] [CrossRef]
- Abe, T.; Tsuda, T.; Yoshida, M.; Wada, Y.; Kano, T.; Itoyama, Y.; Tamai, M. Macular Degeneration Associated with Aberrant Expansion of Trinucleotide Repeat of the SCA7 Gene in 2 Japanese Families. Arch. Ophthalmol. 2000, 118, 1415. [Google Scholar] [CrossRef]
- Ahn, J.K.; Seo, J.K.; Chung, H.; Yu, H.G. Anatomical and Functional Characteristics in Atrophic Maculopathy Associated with Spinocerebellar Ataxia Type 7. Am. J. Ophthalmol. 2005, 139, 923–925. [Google Scholar] [CrossRef]
- AlHilali, S.; AlMadhi, N.H.; AlBalawi, E.D. Ophthalmic Features of Spinocerebellar Ataxia Type 7: A Case Report. Am. J. Case Rep. 2021, 22, e932279-1–e932279-7. [Google Scholar] [CrossRef]
- Gu, W.; Wang, Y.; Liu, X.; Zhou, B.; Zhou, Y.; Wang, G. Molecular and Clinical Study of Spinocerebellar Ataxia Type 7 in Chinese Kindreds. Arch. Neurol. 2000, 57, 10. [Google Scholar] [CrossRef]
- Hugosson, T.; Gränse, L.; Ponjavic, V.; Andréasson, S. Macular dysfunction and morphology in spinocerebellar ataxia type 7 (SCA 7). Ophthalmic Genet. 2009, 30, 1–6. [Google Scholar] [CrossRef]
- Katagiri, S.; Hayashi, T.; Takeuchi, T.; Yamada, H.; Gekka, T.; Kawabe, K.; Kurita, A.; and Tsuneok, H. Somatic Instability of Expanded CAG Repeats of ATXN7 in Japanese Patients with Spinocerebellar Ataxia Type 7. Doc. Ophthalmol. 2015, 130, 189–195. [Google Scholar] [CrossRef]
- Miller, R.C.; Tewari, A.; Miller, J.A.; Garbern, J.; Van Stavern, G.P. Neuro-Ophthalmologic Features of Spinocerebellar Ataxia Type 7. J. Neuro-Ophthalmol. 2009, 29, 180–186. [Google Scholar] [CrossRef]
- Velázquez-Pérez, L.; Cerecedo-Zapata, C.M.; Hernández-Hernández, O.; Martínez-Cruz, E.; Tapia-Guerrero, Y.S.; González-Piña, P.; Salas-Vargas, J. A Comprehensive Clinical and Genetic Study of a Large Mexican Population with Spinocerebellar Ataxia Type 7. Neurogenetics 2015, 1, 11–21. [Google Scholar] [CrossRef]
- Marianelli, B.F.; Filho, F.M.R.; Salles, M.V.; Clares de Andrade, J.B.; Pedroso, J.L.; Sallum, J.M.; Barsottini, O.G. A Proposal for Classification of Retinal Degeneration in Spinocerebellar Ataxia Type 7. Cerebellum 2021, 20, 384–391. [Google Scholar] [CrossRef]
- Aleman, T.S.; Cideciyan, A.V.; Volpe, N.J.; Stevanin, G.; Brice, A.; Jacobson, S.G. Spinocerebellar Ataxia Type 7 (SCA7) Shows a Cone–Rod Dystrophy Phenotype. Exp. Eye Res. 2002, 74, 737–745. [Google Scholar] [CrossRef]
- Atadzhanov, M.; Smith, D.C.; Mwaba, M.H.; Siddiqi, O.K.; Bryer, A.; Greenberg, L.J. Clinical and Genetic Analysis of Spinocerebellar Ataxia Type 7 (SCA7) in Zambian Families. Cerebellum Ataxias 2017, 4, 17. [Google Scholar] [CrossRef]
- Kim, B.C.; Kim, M.K.; Cho, K.H.; Jeon, B.S. Spinocerebellar ataxia type 7 without retinal degeneration: A case report. J. Korean Med. Sci. 2002, 17, 577–579. [Google Scholar] [CrossRef]
- Italiano, D.; Tarantino, P.; De Marco, E.V.; Calabrò, R.S.; Bramanti, P.; Quattrone, A.; Annesi, G. Spinocerebellar Ataxia Type 7: Report of a New Italian Family. Intern. Med. 2012, 51, 2953–2955. [Google Scholar] [CrossRef]
- Manrique, R.K.; Noval, S.; Aguilar-Amat, M.J.; Arpa, J.; Rosa, I.; Contreras, I. Ophthalmic Features of Spinocerebellar Ataxia Type 7. J. Neuro-Ophthalmol. 2009, 29, 3. [Google Scholar] [CrossRef]
- Thurtell, M.J.; Fraser, A.J.; Bala, E.; Tomsak, R.l.; Biousse, V.; Leigh, J.R.; Newman, N.J. Two Patients with Spinocerebellar Ataxia Type 7 Presenting With Profound Binocular Visual Loss Yet Minimal Ophthalmoscopic Findings. J. Neuro-Ophthalmol. 2009, 29, 187–191. [Google Scholar] [CrossRef]
- Wali, G.M. Spinocerebellar Ataxia Type 7: Report of an Indian Family. Ann. Indian Acad. Neurol. 2013, 4, 708. [Google Scholar] [CrossRef]
- Park, J.Y.; Wy, S.Y.; Joo, K.; Woo, J.S. Spinocerebellar Ataxia Type 7 with RP1L1 -Negative Occult Macular Dystrophy as Retinal Manifestation. Ophthalmic Genet. 2019, 40, 282–285. [Google Scholar] [CrossRef]
- Zou, X.; Yao, F.; Li, F.; Wu, S.; Li, H.; Sun, Z.; Zhu, T.; Wei, X.; Li, D.; Sui, R. Clinical Characterization and the Improved Molecular Diagnosis of Autosomal Dominant Cone-Rod Dystrophy in Patients with SCA7. Mol. Vis. 2021, 12, 221–232. [Google Scholar]
- Pawar, N.; Manayath, G.J.; Verghese, S.; Chandrakanth, P.; Shah, V.; Raut, A.; Gaikwad, S.; Patil, P.A.; Daswani, M.; Meenakshi, R.; et al. Potpourri of retinopathies in rare eye disease—A case series. Indian J. Ophthalmol. 2022, 70, 2605–2609. [Google Scholar]
- Costello, F.; Hodge, W.; Pan, Y.; Eggenberger, E.; Coupland, S.; Kardon, R. Tracking Retinal Nerve Fiber Layer Loss after Optic Neuritis: A Prospective Study Using Optical Coherence Tomography. Mult. Scler. J. 2014, 14, 893–905. [Google Scholar] [CrossRef]
- Contreras, I.; Noval, S.; Rebolleda, G.; Muñoz-Negrete, F.J. Follow-up of Nonarteritic Anterior Ischemic Optic Neuropathy with Optical Coherence Tomography. Ophthalmology 2007, 114, 2338–2344. [Google Scholar] [CrossRef]
- Abdolrahimzadeh, S.; Ciancimino, C.; Grassi, F.; Sordi, E.; Fragiotta, S.; Scuderi, G. Near-Infrared Reflectance Imaging in Retinal Diseases Affecting Young Patients. J. Ophthalmol. 2021, 2021, 5581851. [Google Scholar] [CrossRef]
- Horton, L.C.; Frosch, M.P.; Vangel, M.G.; Weigel-DiFranco, C.; Eliot, L.; Berson, E.L.; Schmahmann, J.D. Spinocerebellar Ataxia Type 7: Clinical Course, Phenotype–Genotype Correlations, and Neuropathology. Cerebellum 2013, 12, 176–193. [Google Scholar] [CrossRef]
- Buijisen, R.A.M.; Toonen, L.J.A.; Gardiner, S.L.; van Roon-Mom, W.M.C. Genetics, Mechanisms, and Therapeutic Progress in Polyglutamine Spinocerebellar Ataxias. Neurotherapeutics 2019, 16, 263–286. [Google Scholar] [CrossRef]
- Niu, C.; Prakash, T.P.; Kim, A.; Quach, J.L.; Huryn, L.A.; Yang, Y.; Lopez, E. Antisense Oligonucleotides Targeting Mutant Ataxin-7 Restore Visual Function in a Mouse Model of Spinocerebellar Ataxia Type 7. Sci. Transl. Med. 2018, 10, 465. [Google Scholar] [CrossRef]
- Levinson, J.D.; Yan, J.; Lambert, S.R.; Shankar, S.P. Multimodal imaging of a family with spinocerebellar ataxia type 7 demonstrating phenotypic variation and progression of retinal degeneration. Retin. Cases Brief Rep. 2016, 10, 267–272. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ciancimino, C.; Di Pippo, M.; Manco, G.A.; Romano, S.; Ristori, G.; Scuderi, G.; Abdolrahimzadeh, S. Multimodal Ophthalmic Imaging in Spinocerebellar Ataxia Type 7. Life 2023, 13, 2169. https://doi.org/10.3390/life13112169
Ciancimino C, Di Pippo M, Manco GA, Romano S, Ristori G, Scuderi G, Abdolrahimzadeh S. Multimodal Ophthalmic Imaging in Spinocerebellar Ataxia Type 7. Life. 2023; 13(11):2169. https://doi.org/10.3390/life13112169
Chicago/Turabian StyleCiancimino, Chiara, Mariachiara Di Pippo, Gregorio Antonio Manco, Silvia Romano, Giovanni Ristori, Gianluca Scuderi, and Solmaz Abdolrahimzadeh. 2023. "Multimodal Ophthalmic Imaging in Spinocerebellar Ataxia Type 7" Life 13, no. 11: 2169. https://doi.org/10.3390/life13112169