

Metastasis in Neuroblastoma and Its Link to Autophagy

Abstract
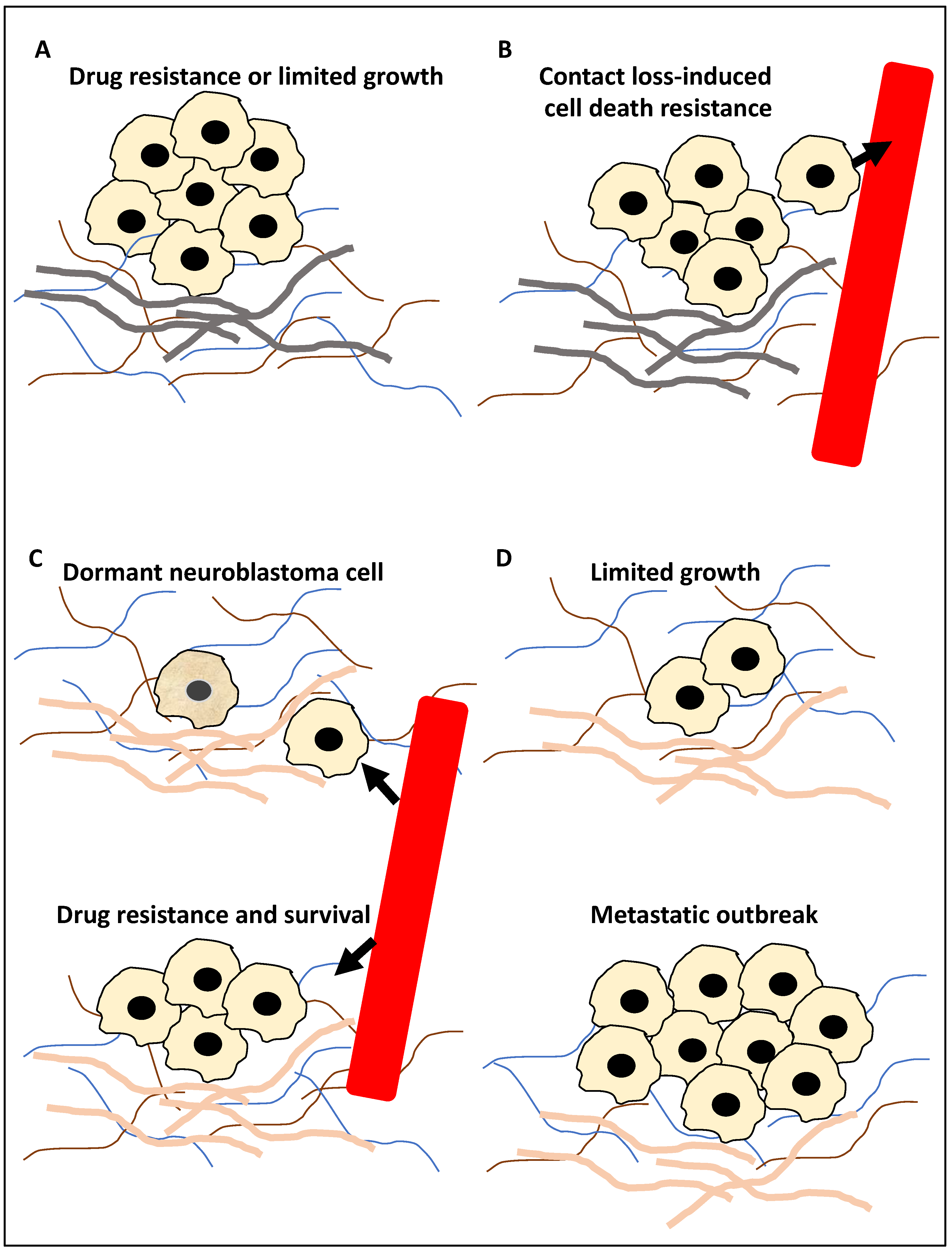
:1. Introduction
2. Autophagy in NB Metastasis
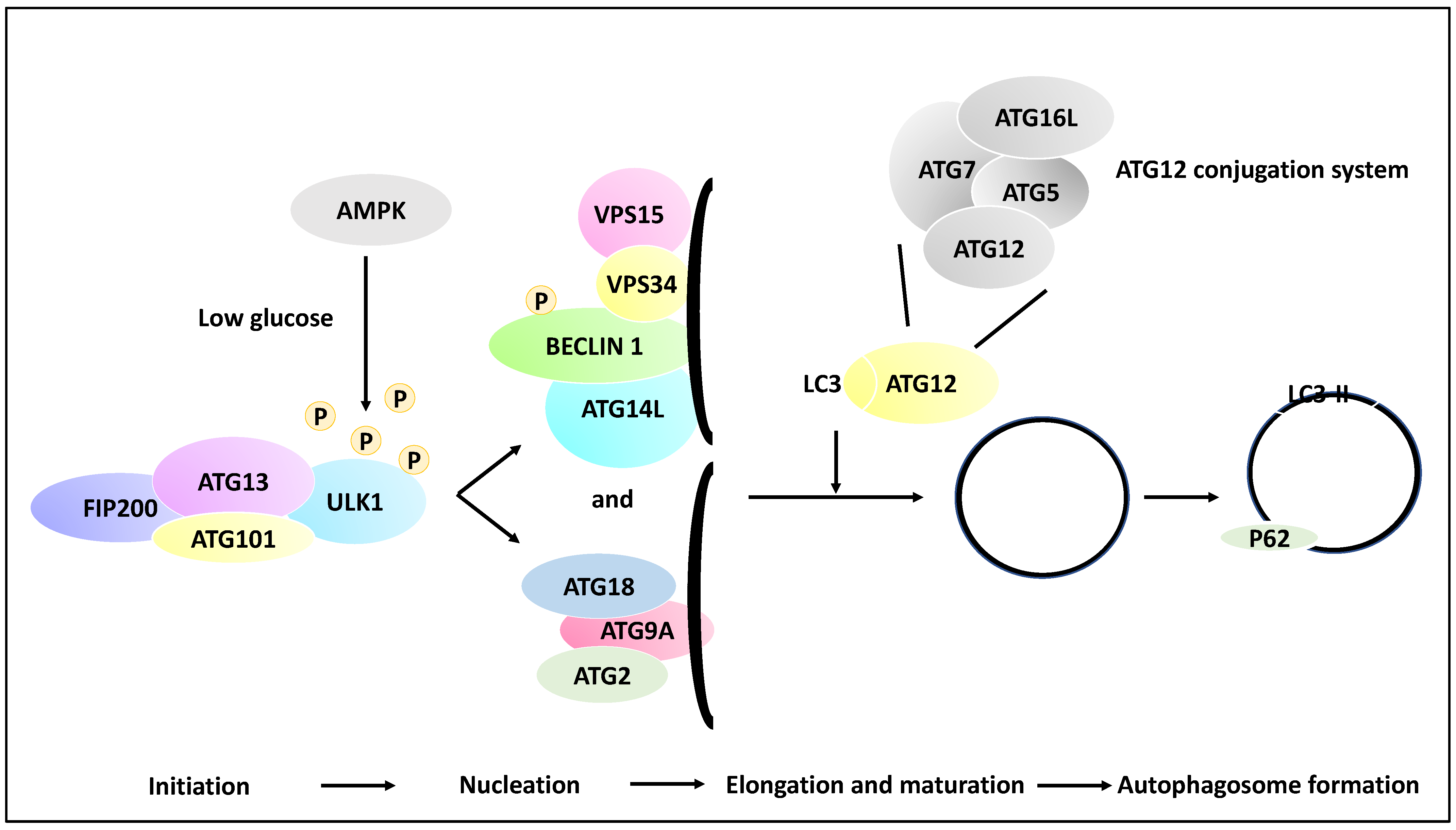
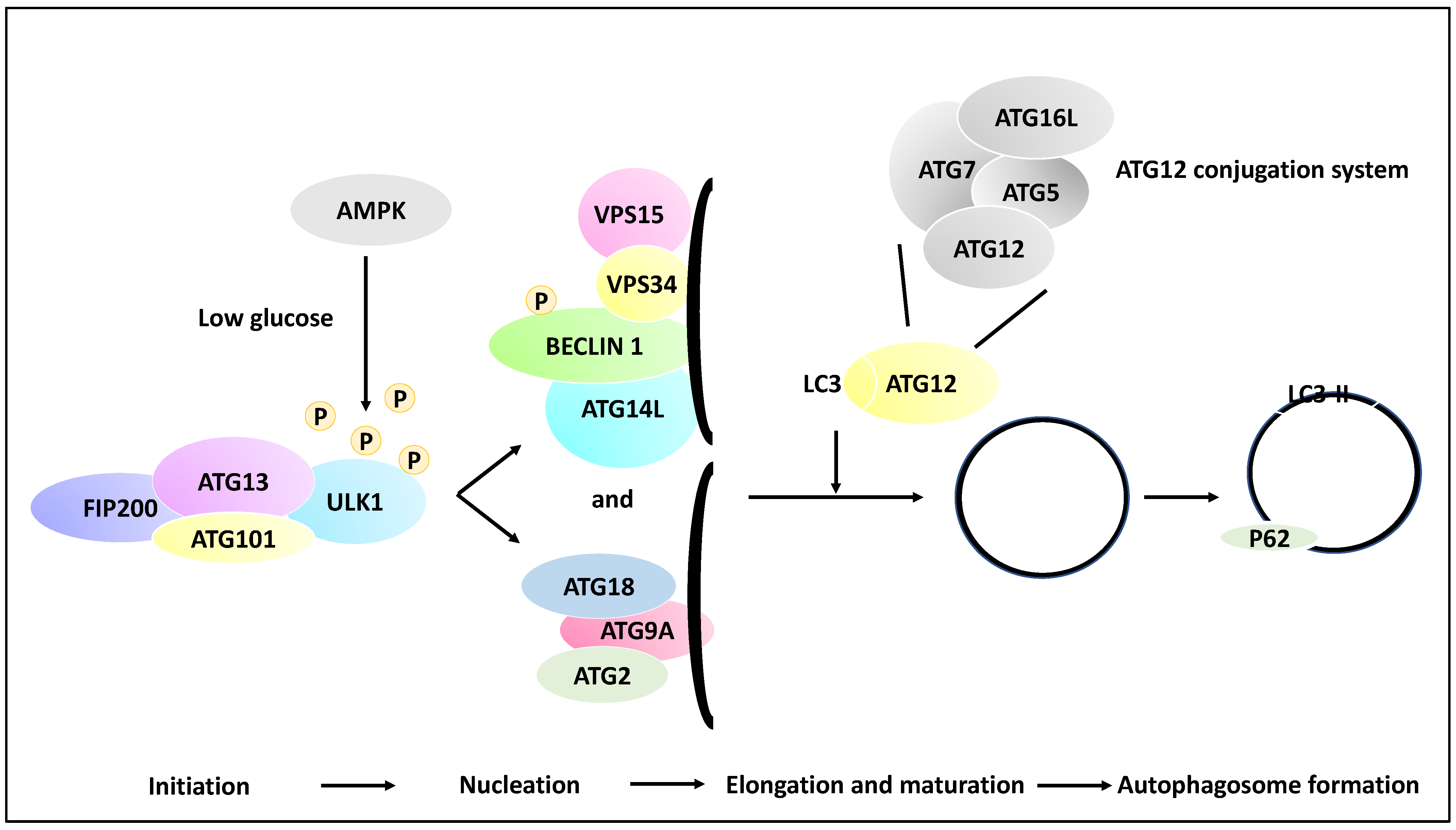
2.1. The Molecular Basis of Autophagy
2.2. Molecular Players and Compounds Linking Autophagy to Metastasis
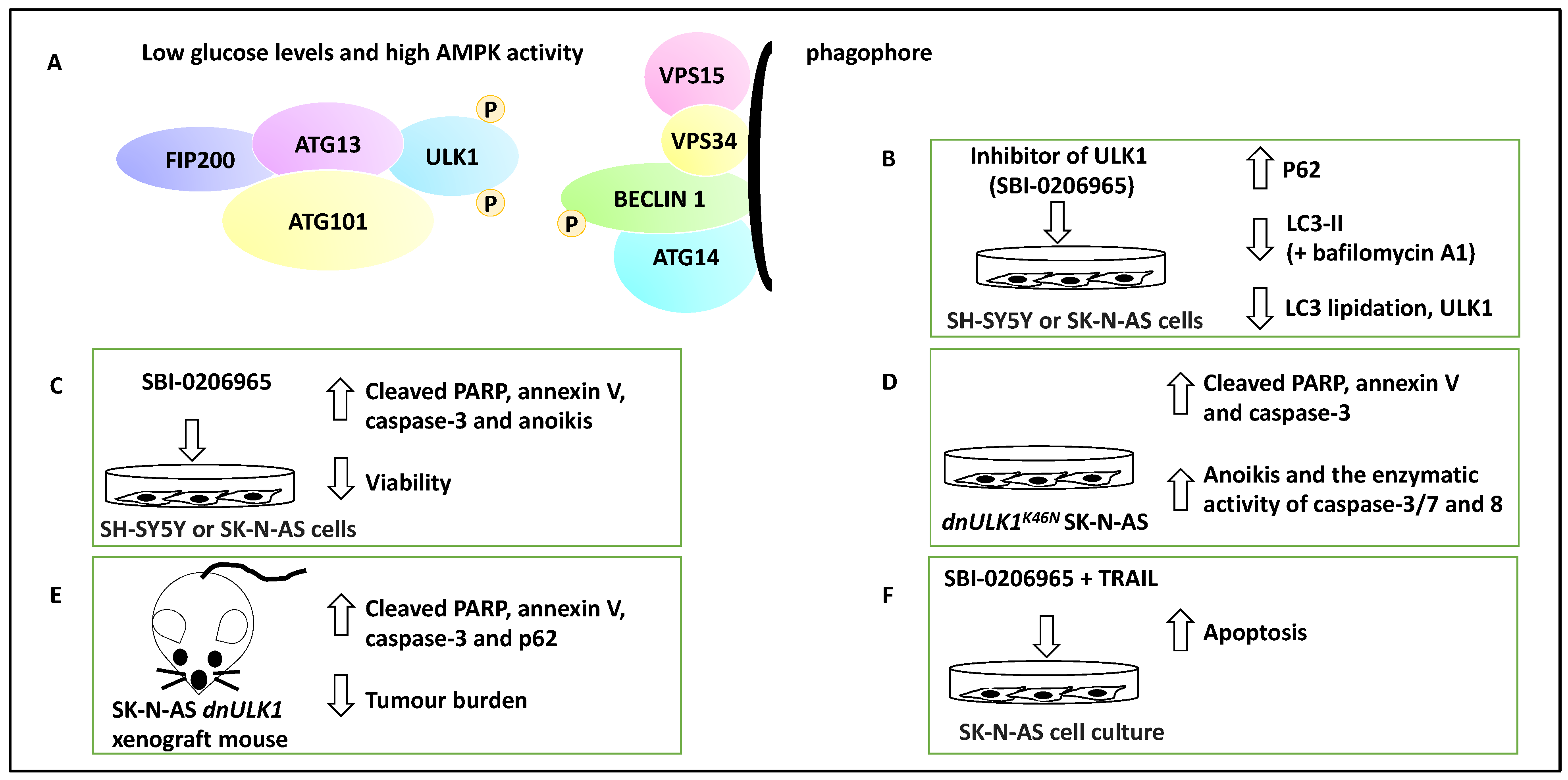
2.2.1. Mediators of Autophagy and Their Link to NB Metastasis
2.2.2. Non-Coding RNA with Oncogenic Roles Linked to Autophagy in NB Metastasis
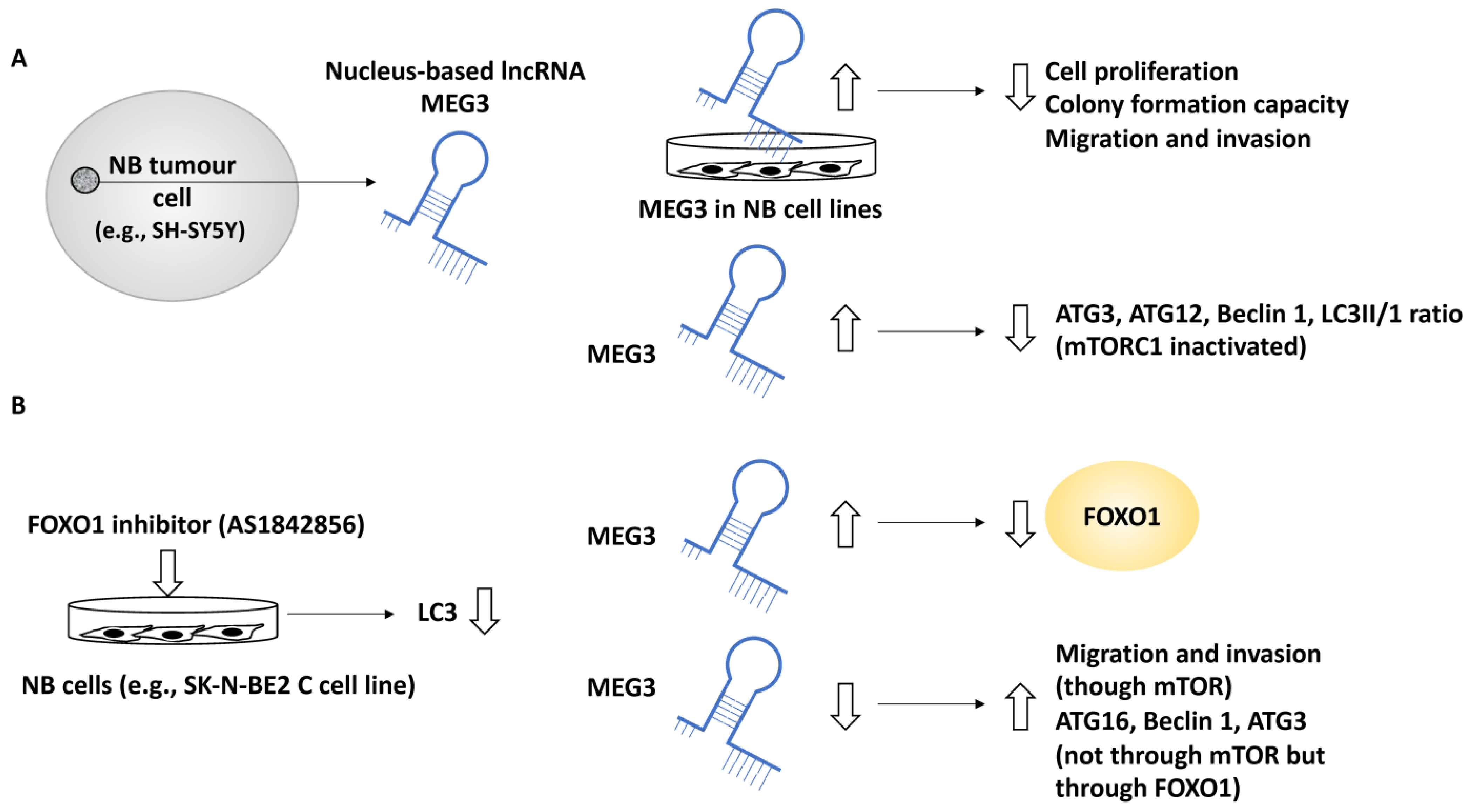
2.2.3. Non-Coding RNA with Tumour Suppressor Roles That Affect Autophagy in NB Metastasis
2.2.4. Compounds and Small Molecule Inhibitors That Link Autophagy in NB Metastasis
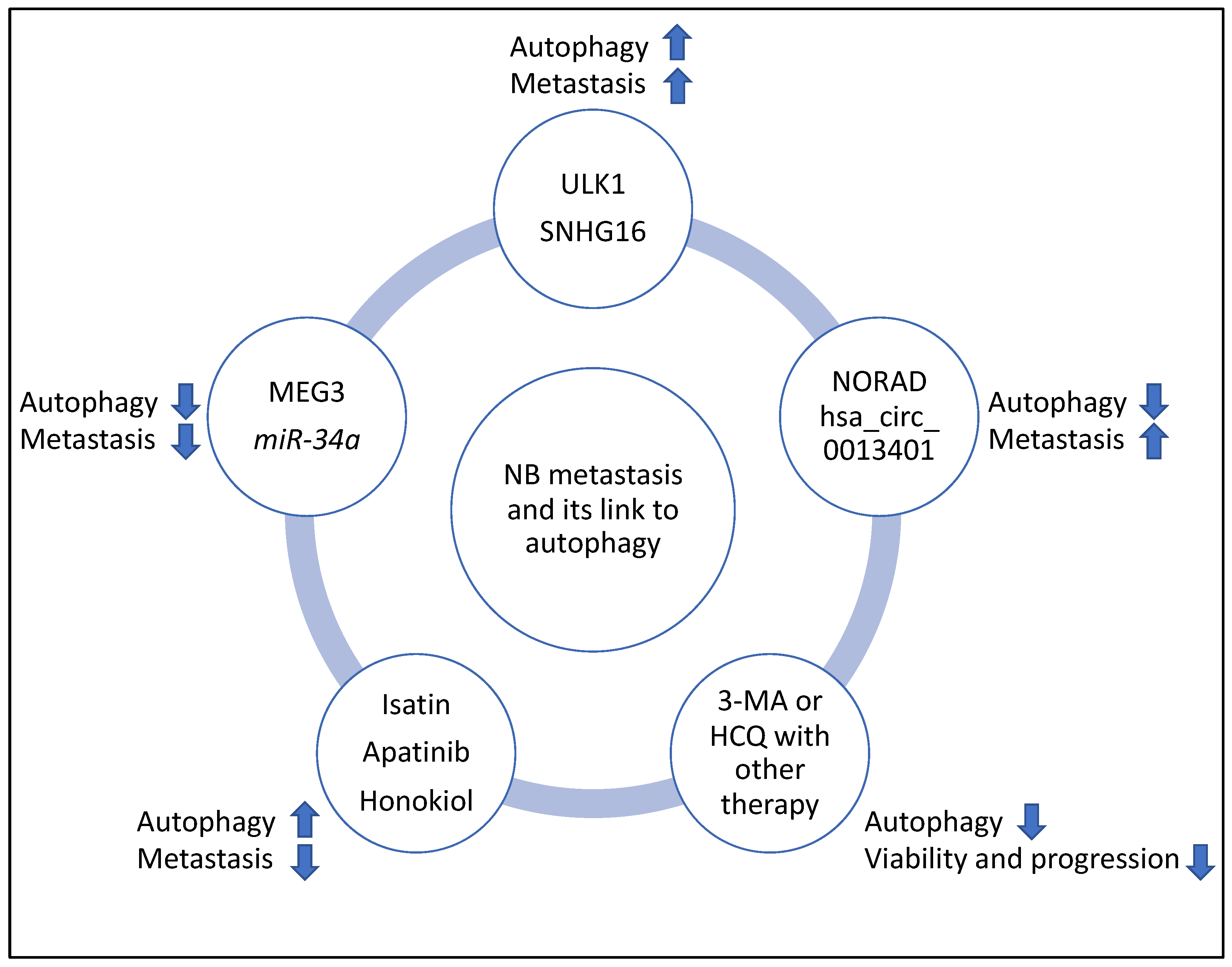
3. Discussion; Molecular Players and Compounds That May Link NB Metastasis to Autophagy
3.1. Mapping Various Molecular Players and Compounds to Metastasis Steps
3.2. The Link between the Multiple Molecular Players and Compounds with Autophagy in NB and Other Cancers, the Potential for New Biomarkers and Evidence from Clinical Trials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ALT | Alternating length of telomeres |
| AMPK | AMP-activated kinase |
| ATGs | Autophagy-related genes |
| DOX | Doxorubicin |
| EFS | Event-free survival |
| EMT | Mesenchymal transition |
| HCQ | Hydroxychloroquine (HCQ) |
| HDAC8 | Histone deacetylase 8 |
| IC50 | Half-maximal inhibitory concentration |
| IDRF | Image-defined risk factors |
| INRGSS | International neuroblastoma risk group system |
| INSS | International neuroblastoma staging system |
| LC3 | microtubule-associated protein light chain 3 |
| LncRNAs | Long non-coding RNAs |
| LRPPRC | Mitochondrion-associated autophagy inhibitor |
| 3-MA | 3-Methyladenine |
| NB | Neuroblastoma |
| NSG | NOD/SCID-gamma |
| OS | Overall survival |
| PARP | Poly-ADP ribose polymerase |
| TERT | Telomere reverse transcriptase |
| TRAIL | TNF-related apoptosis-inducing ligand |
| VEGFR-2 | Vascular endothelial growth factor receptor 2 |
| ULK1 | UNC-51-like kinase1 |
References
- Bosse, K.R.; Maris, J.M. Advances in the translational genomics of neuroblastoma: From improving risk stratification and revealing novel biology to identifying actionable genomic alterations. Cancer 2016, 122, 20–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maris, J.M.; Hogarty, M.D.; Bagatell, R.; Cohn, S.L. Neuroblastoma. Lancet 2007, 369, 2106–2120. [Google Scholar] [CrossRef] [PubMed]
- Cohn, S.L.; Pearson, A.D.J.; London, W.B.; Monclair, T.; Ambros, P.F.; Brodeur, G.M.; Faldum, A.; Hero, B.; Iehara, T.; Machin, D.; et al. The International Neuroblastoma Risk Group (INRG) classification system: An INRG Task Force report. J. Clin. Oncol. 2009, 27, 289–297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brodeur, G.M. Neuroblastoma: Biological insights into a clinical enigma. Nat. Rev. Cancer 2003, 3, 203–216. [Google Scholar] [CrossRef]
- Brodeur, G.M.; Pritchard, J.; Berthold, F.; Carlsen, N.L.; Castel, V.; Castelberry, R.P.; De Bernardi, B.; Evans, A.E.; Favrot, M.; Hedborg, F. Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J. Clin. Oncol. 1993, 11, 1466–1477. [Google Scholar] [CrossRef]
- Morgenstern, D.A.; London, W.B.; Stephens, D.; Volchenboum, S.L.; Simon, T.; Nakagawara, A.; Shimada, H.; Schleiermacher, G.; Matthay, K.K.; Cohn, S.L.; et al. Prognostic significance of pattern and burden of metastatic disease in patients with stage 4 neuroblastoma: A study from the International Neuroblastoma Risk Group database. Eur. J. Cancer 2016, 65, 1–10. [Google Scholar] [CrossRef] [PubMed]
- DuBois, S.G.; Kalika, Y.; Lukens, J.N.; Brodeur, G.M.; Seeger, R.C.; Atkinson, J.B.; Haase, G.M.; Black, C.T.; Perez, C.; Shimada, H.; et al. Metastatic sites in stage IV and IVS neuroblastoma correlate with age, tumor biology, and survival. J. Pediatr. Hematol. Oncol. 1999, 21, 181–189. [Google Scholar] [CrossRef]
- Ara, T.; DeClerck, Y.A. Mechanisms of invasion and metastasis in human neuroblastoma. Cancer Metastasis Rev. 2006, 25, 645–657. [Google Scholar] [CrossRef]
- Dagg, R.A.; Pickett, H.A.; Neumann, A.A.; Napier, C.E.; Henson, J.D.; Teber, E.T.; Arthur, J.W.; Reynolds, C.P.; Murray, J.; Haber, M.; et al. Extensive Proliferation of Human Cancer Cells with Ever-Shorter Telomeres. Cell Rep. 2017, 19, 2544–2556. [Google Scholar] [CrossRef] [Green Version]
- Brisse, H.J.; McCarville, M.B.; Granata, C.; Krug, K.B.; Wootton-Gorges, S.L.; Kanegawa, K.; Giammarile, F.; Schmidt, M.; Shulkin, B.; Matthay, K.K.; et al. Guidelines for imaging and staging of neuroblastic tumors: Consensus report from the International Neuroblastoma Risk Group Project. Radiology 2011, 261, 243–257. [Google Scholar] [CrossRef] [Green Version]
- Nakagawara, A.; Li, Y.; Izumi, H.; Muramori, K.; Inada, H.; Nishi, M. Neuroblastoma. Jpn. J. Clin. Oncol. 2018, 48, 214–241. [Google Scholar] [CrossRef] [Green Version]
- Castleberry, R.P.; Pritchard, J.; Ambros, P.; Berthold, F.; Brodeur, G.M.; Castel, V.; Cohn, S.; De Bernardi, B.; Dicks-Mireaux, C.; Frappaz, D.; et al. The International Neuroblastoma Risk Groups (INRG): A preliminary report. Eur. J. Cancer 1997, 33, 2113–2116. [Google Scholar] [CrossRef] [PubMed]
- Irwin, M.S.; Naranjo, A.; Zhang, F.F.; Cohn, S.L.; London, W.B.; Gastier-Foster, J.M.; Ramirez, N.C.; Pfau, R.; Reshmi, S.; Wagner, E.; et al. Revised Neuroblastoma Risk Classification System: A Report from the Children’s Oncology Group. J. Clin. Oncol. 2021, 39, 3229–3241. [Google Scholar] [CrossRef] [PubMed]
- Maris, J.M. Recent advances in neuroblastoma. N. Engl. J. Med. 2010, 362, 2202–2211. [Google Scholar] [CrossRef] [Green Version]
- Peifer, M.; Hertwig, F.; Roels, F.; Dreidax, D.; Gartlgruber, M.; Menon, R.; Krämer, A.; Roncaioli, J.L.; Sand, F.; Heuckmann, J.M.; et al. Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature 2015, 526, 700–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valentijn, L.; Koster, J.; Zwijnenburg, D.; Hasselt, N.; van Sluis, P.; Volckmann, R.; van Noesel, M.; George, R.; Tytgat, G.; Molenaar, J.; et al. TERT rearrangements are frequent in neuroblastoma and identify aggressive tumors. Nat. Genet. 2015, 47, 1411–1414. [Google Scholar] [CrossRef] [PubMed]
- Tolbert, V.P.; Matthay, K.K. Neuroblastoma: Clinical and biological approach to risk stratification and treatment. Cell Tissue Res. 2018, 372, 195–209. [Google Scholar] [CrossRef]
- Steeg, P.S.; Theodorescu, D. Metastasis: A therapeutic target for cancer. Nat. Clin. Pract. Oncol. 2008, 5, 206–219. [Google Scholar] [CrossRef]
- Morandi, F.; Corrias, M.V.; Pistoia, V. Evaluation of bone marrow as a metastatic site of human neuroblastoma. Ann. N. Y. Acad. Sci. 2015, 1335, 23–31. [Google Scholar] [CrossRef]
- Popov, A.M.; Shorikov, E.V.; Verzhditskaia, T.I.; Tsaur, G.A.; Druĭ, A.E.; Solodovnikov, A.G.; Savel’ev, L.I.; Fechina, L.G. Prognostic value of bone marrow lesions in children with neuroblastoma detected by flow cytometry. Vopr. Onkol. 2014, 60, 469–475. [Google Scholar]
- Kenific, C.M.; Thorburn, A.; Debnath, J. Autophagy and metastasis: Another double-edged sword. Curr. Opin. Cell Biol. 2010, 22, 241–245. [Google Scholar] [CrossRef] [Green Version]
- Russell, R.C.; Yuan, H.X.; Guan, K.L. Autophagy regulation by nutrient signaling. Cell Res. 2014, 24, 42–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef]
- Chavez-Dominguez, R.; Perez-Medina, M.; Lopez-Gonzalez, J.S.; Galicia-Velasco, M.; Aguilar-Cazares, D. The Double-Edge Sword of Autophagy in Cancer: From Tumor Suppression to Pro-tumor Activity. Front. Oncol. 2020, 10, 578418. [Google Scholar] [CrossRef]
- Lock, R.; Debnath, J. Extracellular matrix regulation of autophagy. Curr. Opin. Cell Biol. 2008, 20, 583–588. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Yang, Z.; Xie, L.; Xu, L.; Xu, D.L.X. Statins, autophagy and cancer metastasis. Int. J. Biochem. Cell Biol. 2013, 45, 745–752. [Google Scholar] [CrossRef]
- Kaur, J.; Debnath, J. Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell Biol. 2015, 16, 461–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, Q.-H.; Liu, F.; Yang, Z.-L.; Fu, X.-H.; Yang, Z.-H.; Liu, Q.; Fan, X.J. Prognostic value of autophagy related proteins ULK1, Beclin 1, ATG3, ATG5, ATG7, ATG9, ATG10, ATG12, LC3B and p62/SQSTM1 in gastric cancer. Am. J. Transl. Res. 2016, 8, 3831–3847. [Google Scholar] [PubMed]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; He, S.; Ma, B. Autophagy and autophagy-related proteins in cancer. Mol. Cancer 2020, 19, 12. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T.; Ohsumi, Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol. 2011, 27, 107–132. [Google Scholar] [CrossRef] [PubMed]
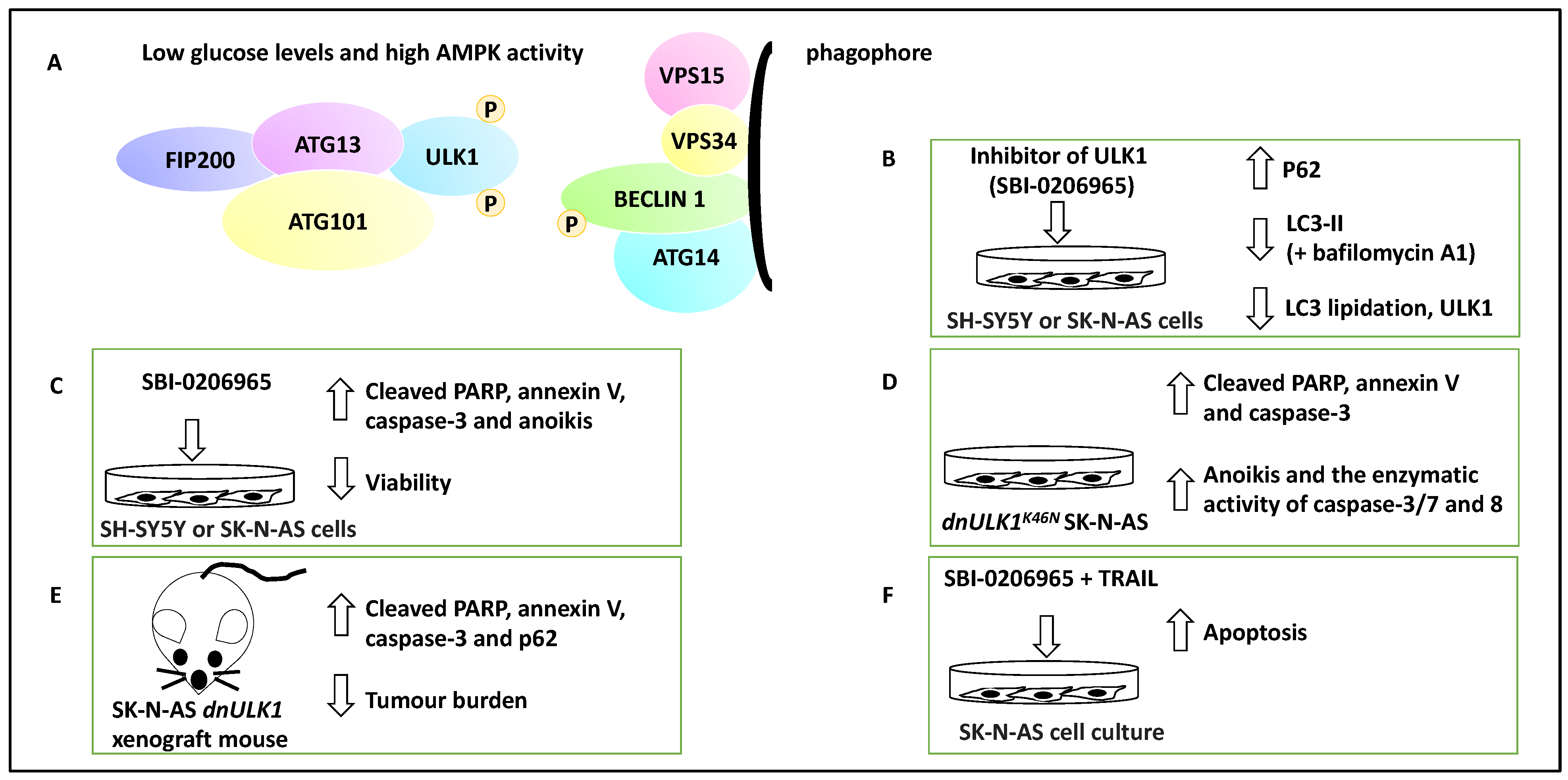
- Dower, C.M.; Bhat, N.; Gebru, M.T.; Chen, L.; Wills, C.A.; Miller, B.A.; Wang, H.-G. Targeted Inhibition of ULK1 Promotes Apoptosis and Suppresses Tumor Growth and Metastasis in Neuroblastoma. Mol. Cancer Ther. 2018, 17, 2365–2376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
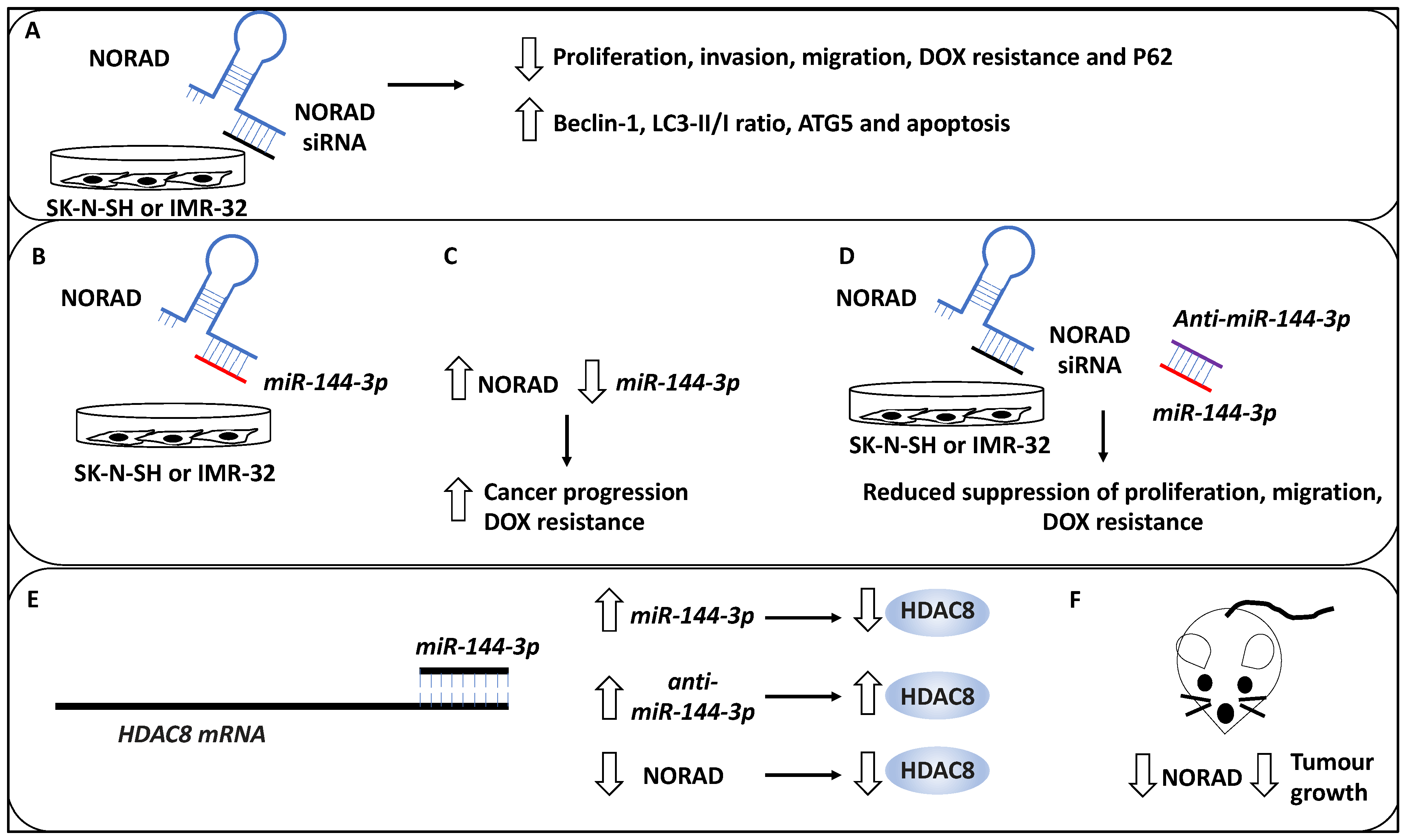
- Wang, B.; Xu, L.; Zhang, J.; Cheng, X.; Xu, Q.; Wang, J.; Mao, F. LncRNA NORAD accelerates the progression and doxorubicin resistance of neuroblastoma through up-regulating HDAC8 via sponging miR-144-3p. Biomed. Pharmacother. 2020, 129, 110268. [Google Scholar] [CrossRef]
- Zhu, S.; Tang, X.; Gao, X.; Zhang, J.; Cui, Y.; Li, D.; Jia, W. hsa_circ_0013401 Accelerates the Growth and Metastasis and Prevents Apoptosis and Autophagy of Neuroblastoma Cells by Sponging miR-195 to Release PAK2. Oxid. Med. Cell Longev. 2021, 2021, 9936154. [Google Scholar] [CrossRef]
- Wen, Y.; Gong, X.; Dong, Y.; Tang, C. Long Non Coding RNA SNHG16 Facilitates Proliferation, Migration, Invasion and Autophagy of Neuroblastoma Cells via Sponging miR-542-3p and Upregulating ATG5 Expression. OncoTargets Ther. 2020, 13, 263–275. [Google Scholar] [CrossRef] [Green Version]
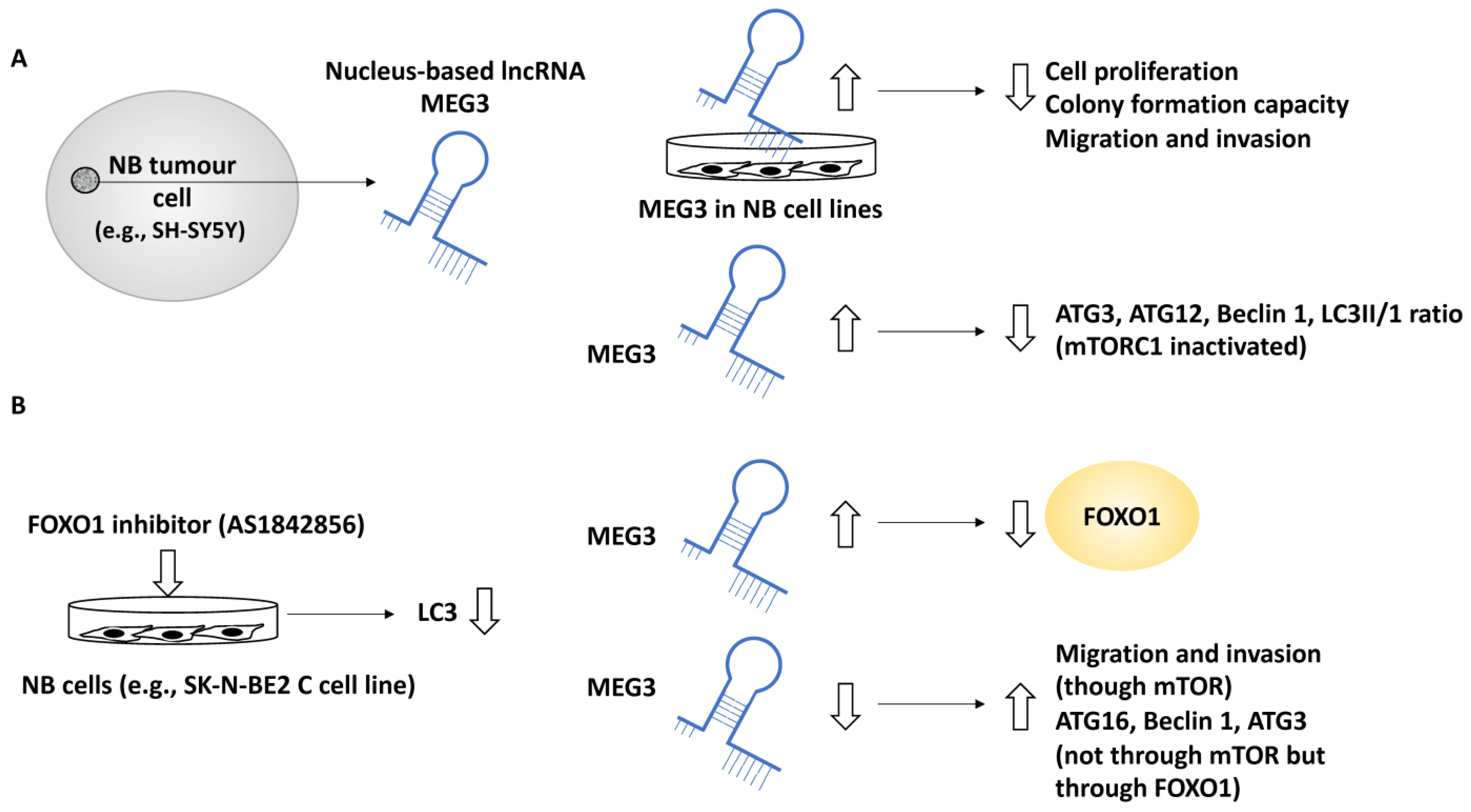
- Ye, M.; Lu, H.; Tang, W.; Jing, T.; Chen, S.; Wei, M.; Zhang, J.; Wang, J.; Ma, J.; Ma, D.; et al. Downregulation of MEG3 promotes neuroblastoma development through FOXO1-mediated autophagy and mTOR-mediated epithelial-mesenchymal transition. Int. J. Biol. Sci. 2020, 16, 3050–3061. [Google Scholar] [CrossRef]
- Cheng, X.; Xu, Q.; Zhang, Y.; Shen, M.; Zhang, S.; Mao, F.; Li, B.; Yan, X.; Shi, Z.; Wang, L.; et al. miR-34a inhibits progression of neuroblastoma by targeting autophagy-related gene 5. Eur. J. Pharmacol. 2019, 850, 53–63. [Google Scholar] [CrossRef]
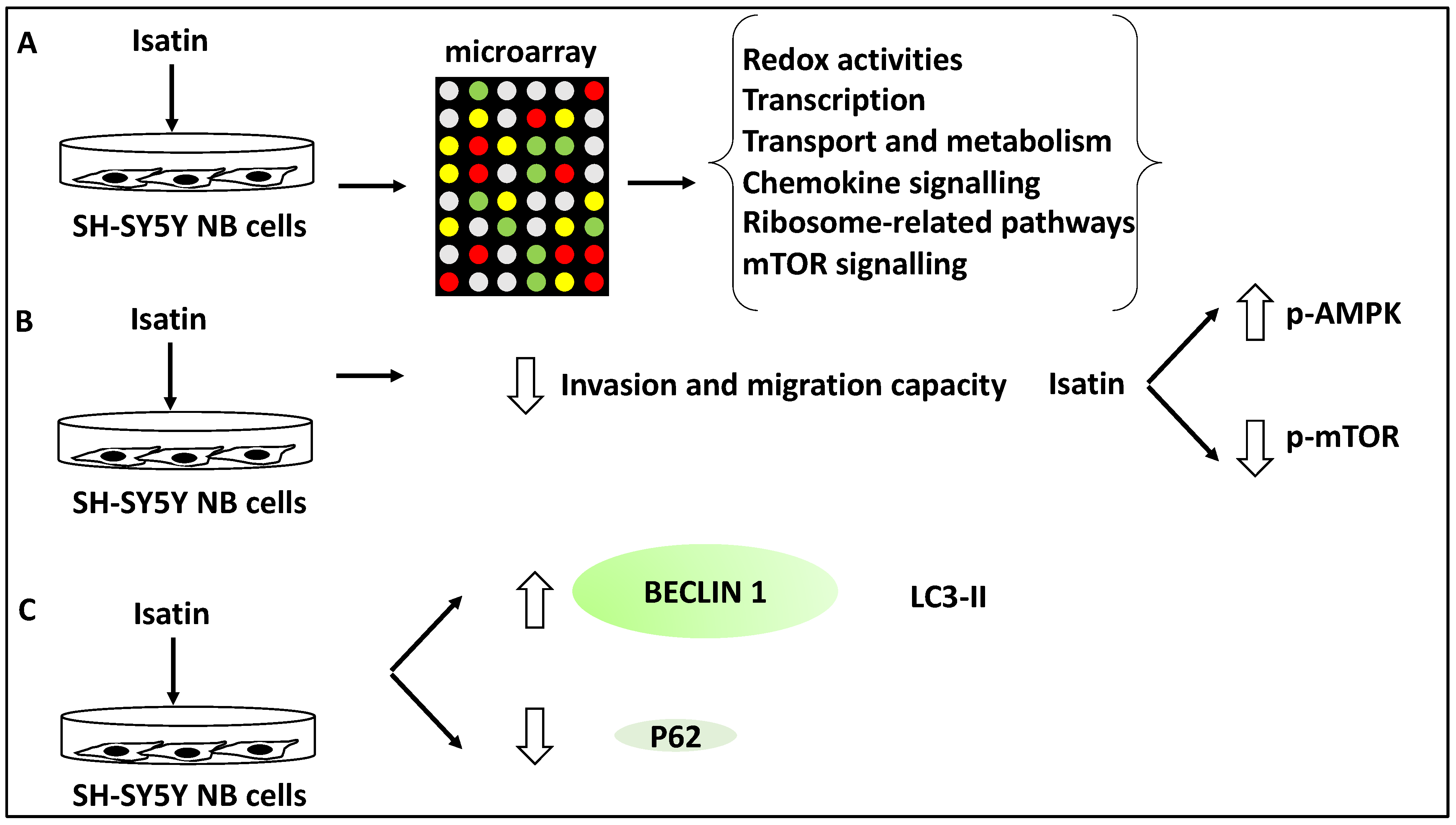
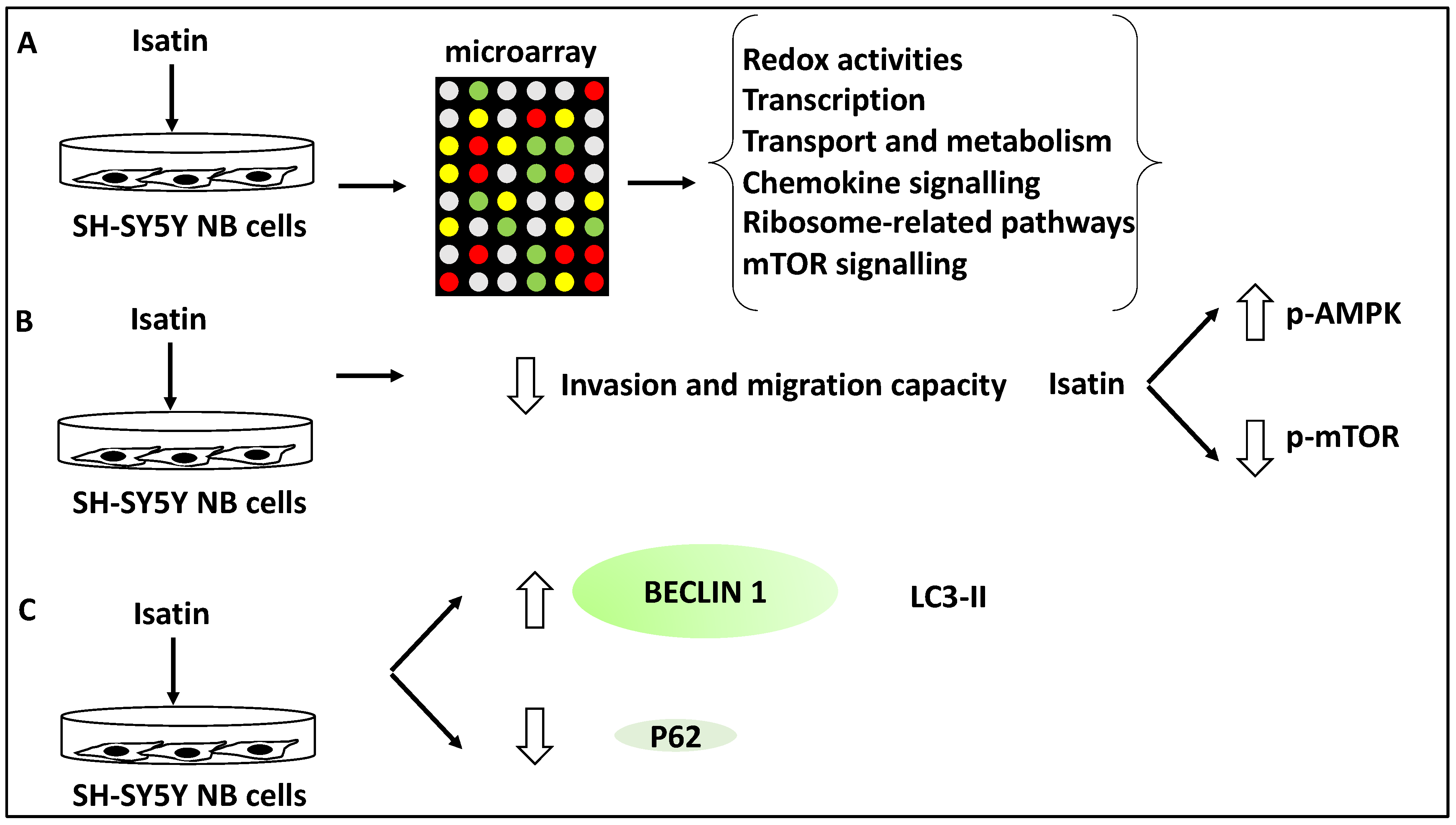
- Zhang, L.; Sun, W.; Cao, Y.; Hou, L.; Ju, C.; Wang, X. Isatin inhibits the invasion of human neuroblastoma SH-SY5Y cells, based on microarray analysis. Mol. Med. Rep. 2019, 20, 1700–1706. [Google Scholar] [CrossRef] [Green Version]
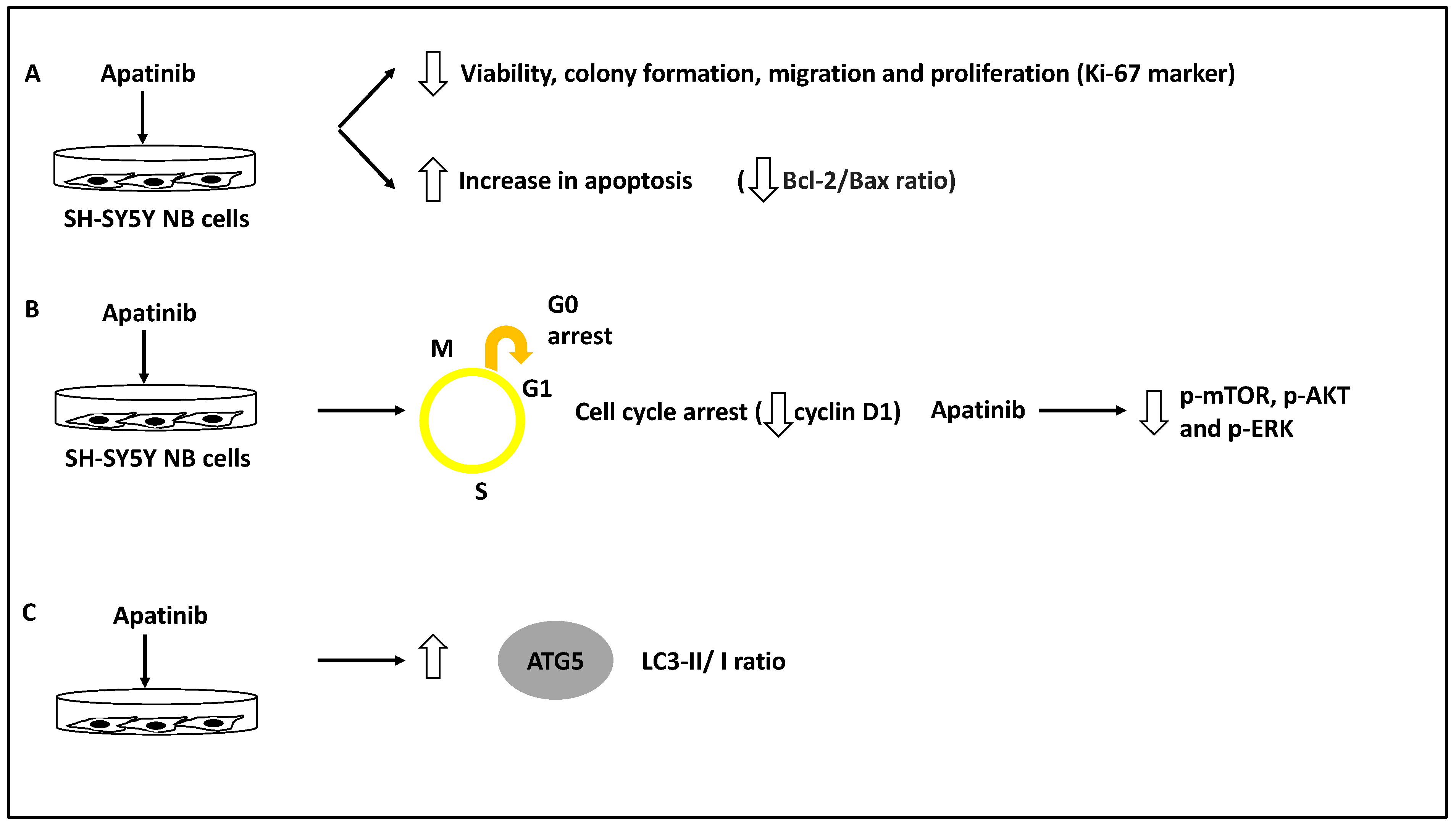
- Yu, X.; Fan, H.; Jiang, X.; Zheng, W.; Yang, Y.; Jin, M.; Ma, X.; Jiang, W. Apatinib induces apoptosis and autophagy via the PI3K/AKT/mTOR and MAPK/ERK signaling pathways in neuroblastoma. Oncol. Lett. 2020, 20, 52. [Google Scholar] [CrossRef] [PubMed]
- Yeh, P.-S.; Wang, W.; Chang, Y.-A.; Lin, C.-J.; Wang, J.-J.; Chen, R.-M. Honokiol induces autophagy of neuroblastoma cells through activating the PI3K/Akt/mTOR and endoplasmic reticular stress/ERK1/2 signaling pathways and suppressing cell migration. Cancer Lett. 2016, 370, 66–77. [Google Scholar] [CrossRef]
- Horwacik, I.; Gaik, M.; Durbas, M.; Boratyn, E.; Zając, G.; Szychowska, K.; Szczodrak, M.; Kołoczek, H.; Rokita, H. Inhibition of autophagy by 3-methyladenine potentiates sulforaphane-induced cell death of BE(2)-C human neuroblastoma cells. Mol. Med. Rep. 2015, 12, 535–542. [Google Scholar] [CrossRef] [Green Version]
- Belounis, A.; Nyalendo, C.; Le Gall, R.; Imbriglio, T.V.; Mahma, M.; Teira, P.; Beaunoyer, M.; Cournoyer, S.; Haddad, E.; Vassal, G.; et al. Autophagy is associated with chemoresistance in neuroblastoma. BMC Cancer 2016, 16, 891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egan, D.; Kim, J.; Shaw, R.J.; Guan, K.-L. The autophagy initiating kinase ULK1 is regulated via opposing phosphorylation by AMPK and mTOR. Autophagy 2011, 7, 643–644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjørkøy, G.; Lamark, T.; Pankiv, S.; Øvervatn, A.; Brech, A.; Johansen, T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009, 452, 181–197. [Google Scholar] [PubMed]
- Swarbrick, A.; Woods, S.L.; Shaw, A.; Balakrishnan, A.; Phua, Y.; Nguyen, A.; Chanthery, Y.; Lim, L.; Ashton, L.J.; Judson, R.L.; et al. miR-380-5p represses p53 to control cellular survival and is associated with poor outcome in MYCN-amplified neuroblastoma. Nat. Med. 2010, 16, 1134–1140. [Google Scholar] [CrossRef]
- Wang, Y.; Guan, E.; Li, D.; Sun, L. miRNA-34a-5p regulates progression of neuroblastoma via modulating the Wnt/β-catenin signaling pathway by targeting SOX4. Medicine 2021, 100, e25827. [Google Scholar] [CrossRef]
- Chen, W.; Hao, X.; Yang, B.; Zhang, Y.; Sun, L.; Hua, Y.; Yang, L.; Yu, J.; Zhao, J.; Hou, L.; et al. MYCN-amplified neuroblastoma cell-derived exosomal miR-17-5p promotes proliferation and migration of non-MYCN amplified cells. Mol. Med. Rep. 2021, 23, 245. [Google Scholar] [CrossRef]
- Mizushima, N.; Yoshimori, T. How to interpret LC3 immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef]
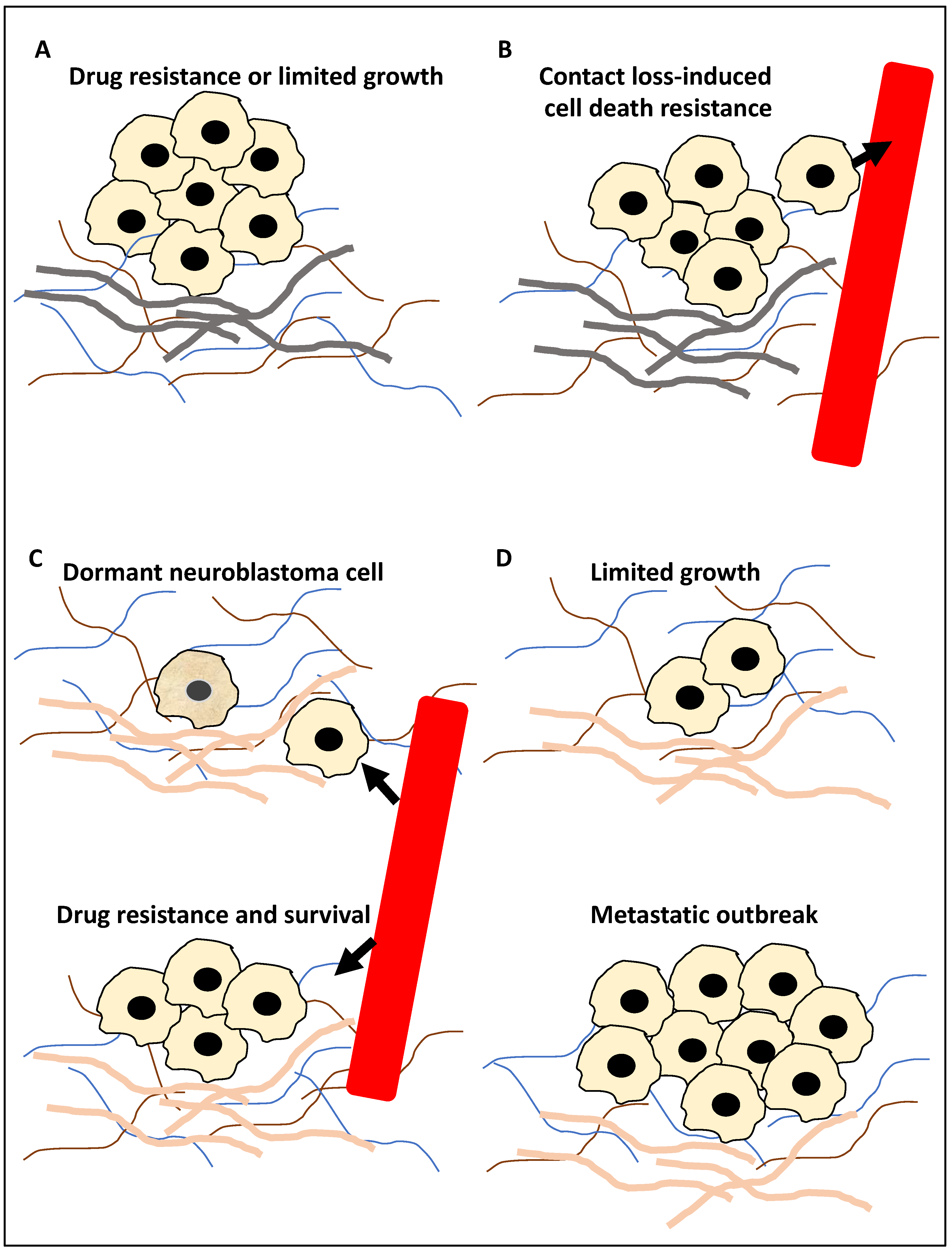
- Jahangiri, L.; Ishola, T. Dormancy in Breast Cancer, the Role of Autophagy, lncRNAs, miRNAs and Exosomes. Int. J. Mol. Sci. 2022, 23, 5271. [Google Scholar] [CrossRef]
- Bildik, G.; Liang, X.; Sutton, M.N.; Bast, R.C.J.; Lu, Z. DIRAS3: An Imprinted Tumor Suppressor Gene that Regulates RAS and PI3K-driven Cancer Growth, Motility, Autophagy, and Tumor Dormancy. Mol. Cancer Ther. 2022, 21, 25–37. [Google Scholar] [CrossRef]
- Vera-Ramirez, L.; Vodnala, S.K.; Nini, R.; Hunter, K.W.; Green, J.E. Autophagy promotes the survival of dormant breast cancer cells and metastatic tumour recurrence. Nat. Commun. 2018, 9, 1944. [Google Scholar] [CrossRef] [Green Version]
- Amaravadi, R.K. Autophagy-induced tumor dormancy in ovarian cancer. J. Clin. Investig. 2008, 118, 3837–3840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.-Y.; Ma, Y.-D.; Zhang, Y.; Cui, J.; Wang, Z.-M. Elevated levels of autophagy-related marker ULK1 and mitochondrion-associated autophagy inhibitor LRPPRC are associated with biochemical progression and overall survival after androgen deprivation therapy in patients with metastatic prostate cancer. J. Clin. Pathol. 2017, 70, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sun, Y.; Zhang, X.; Cai, H.; Zhang, C.; Qu, H.; Liu, L.; Zhang, M.; Fu, J.; Zhang, J.; et al. Oxidative stress activates NORAD expression by H3K27ac and promotes oxaliplatin resistance in gastric cancer by enhancing autophagy flux via targeting the miR-433-3p. Cell Death Dis. 2021, 12, 90. [Google Scholar] [CrossRef] [PubMed]
- Jing, Z.; Ye, X.; Ma, X.; Hu, X.; Yang, W.; Shi, J.; Chen, G.; Gong, L. SNGH16 regulates cell autophagy to promote Sorafenib Resistance through suppressing miR-23b-3p via sponging EGR1 in hepatocellular carcinoma. Cancer Med. 2020, 9, 4324–4338. [Google Scholar] [CrossRef] [Green Version]
- Xia, H.; Qu, X.-L.; Liu, L.-Y.; Qian, D.-H.; Jing, H.-Y. LncRNA MEG3 promotes the sensitivity of vincristine by inhibiting autophagy in lung cancer chemotherapy. Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 1020–1027. [Google Scholar]
- Liu, F.; Ai, F.-Y.; Zhang, D.-C.; Tian, L.; Yang, Z.-Y.; Liu, S.-J. LncRNA NEAT1 knockdown attenuates autophagy to elevate 5-FU sensitivity in colorectal cancer via targeting miR-34a. Cancer Med. 2020, 9, 1079–1091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, W.; Zhang, L.; Hou, L.; Ju, C.; Zhao, S.; Wei, Y. Isatin inhibits SH-SY5Y neuroblastoma cell invasion and metastasis through MAO/HIF-1α/CXCR4 signaling. Anticancer Drugs 2017, 28, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Wang, H.; Zhao, J.; Hu, L.; Zhi, J.; Wei, S.; Ruan, X.; Hou, X.; Li, D.; Zhang, J.; et al. Apatinib Inhibits Cell Proliferation and Induces Autophagy in Human Papillary Thyroid Carcinoma via the PI3K/Akt/mTOR Signaling Pathway. Front. Oncol. 2020, 10, 217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Dong, H.; Li, M.; Wu, Y.; Liu, Y.; Zhao, Y.; Chen, X.; Ma, M. Honokiol induces autophagy and apoptosis of osteosarcoma through PI3K/Akt/mTOR signaling pathway. Mol. Med. Rep. 2018, 17, 2719–2723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mauthe, M.; Orhon, I.; Rocchi, C.; Zhou, X.; Luhr, M.; Hijlkema, K.-J.; Coppes, R.P.; Engedal, N.; Mari, M.; Reggiori, F. Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 2018, 14, 1435–1455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chude, C.I.; Amaravadi, R.K. Targeting Autophagy in Cancer: Update on Clinical Trials and Novel Inhibitors. Int. J. Mol. Sci. 2017, 18, 1279. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
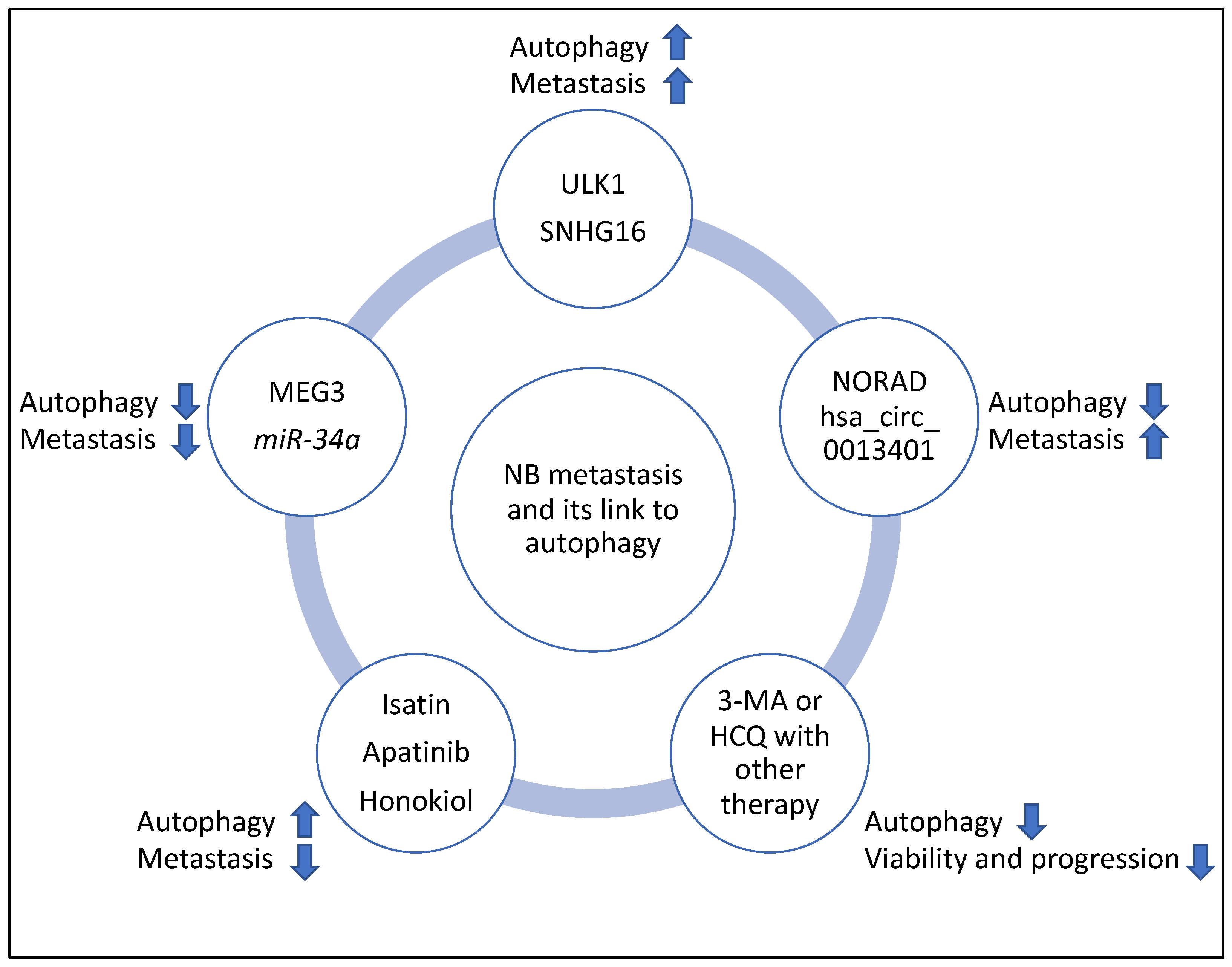
| Compounds or Molecule | Cancer Type | Effect on Autophagy | Effect on Metastasis | Reference |
|---|---|---|---|---|
| ULK1 | NB | Mediated autophagy since SBI-0206965 (an inhibitor of ULK1) reduced autophagic flux and LC3 lipidation | Promoted metastasis | [32] |
| NORAD | NB | Suppressed autophagy since in NORAD knockdown, Beclin 1, ATG5, and LC3B-II/I ratios were increased | Promoted metastasis | [33] |
| hsa_circ_0013401 | NB | Suppressed autophagy since hsa_circ_0013401 knockdown led to increased LC3B-II/I ratio and Beclin 1 levels | Promoted metastasis | [34] |
| SNHG16 | NB | Induced autophagy through upregulating ATG5 | Promoted metastasis | [35] |
| MEG3 | NB | Suppressed autophagy, since silencing of MEG3 increased autophagy markers including ATG16, Beclin 1, and ATG3 | Suppressed metastasis | [36] |
| miR-34a | NB | Suppressed autophagy since miR-34a negatively targeted ATG5 | Suppressed metastasis | [37] |
| Isatin | NB | Induced autophagy since Isatin increased Beclin 1, and LC3-II levels | Suppressed metastasis | [38] |
| Apatinib | NB | Induced autophagy since Apatinib increased ATG5 and LC3B-II/I ratio | Suppressed metastasis | [39] |
| Honokiol | NB | Induced autophagy since Honokiol increased LC3-II levels | Suppressed metastasis | [40] |
| 3-Methyladenine (3-MA) with sulforaphane | NB | Suppressed autophagy | Reduced tumour viability | [41] |
| Hydroxychloroquine (HCQ) with vincristine | NB | Suppressed autophagy | Reduced tumour progression | [42] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jahangiri, L. Metastasis in Neuroblastoma and Its Link to Autophagy. Life 2023, 13, 818. https://doi.org/10.3390/life13030818
Jahangiri L. Metastasis in Neuroblastoma and Its Link to Autophagy. Life. 2023; 13(3):818. https://doi.org/10.3390/life13030818
Chicago/Turabian StyleJahangiri, Leila. 2023. "Metastasis in Neuroblastoma and Its Link to Autophagy" Life 13, no. 3: 818. https://doi.org/10.3390/life13030818
APA StyleJahangiri, L. (2023). Metastasis in Neuroblastoma and Its Link to Autophagy. Life, 13(3), 818. https://doi.org/10.3390/life13030818