


Allopolyploidy: An Underestimated Driver in Juniperus Evolution

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. DNA Extraction
2.3. Amplified Fragment Length Polymorphism (AFLP) Analysis
2.4. AFLP Scoring
2.5. AFLP Data Analysis
3. Results
3.1. Polymorphism of AFLP Markers between Juniperus Species
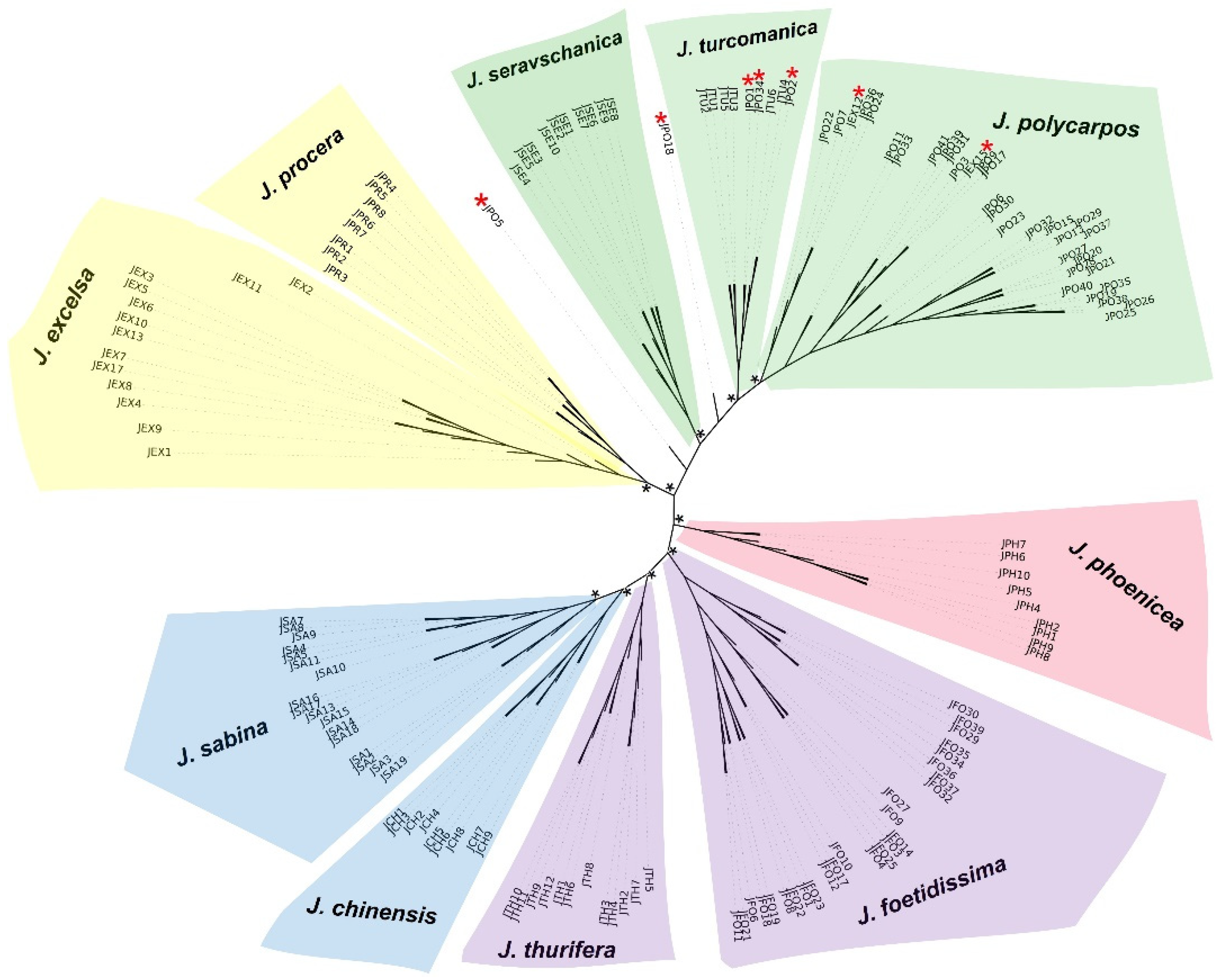
3.2. Phylogenetic Relationships among Juniperus Taxa
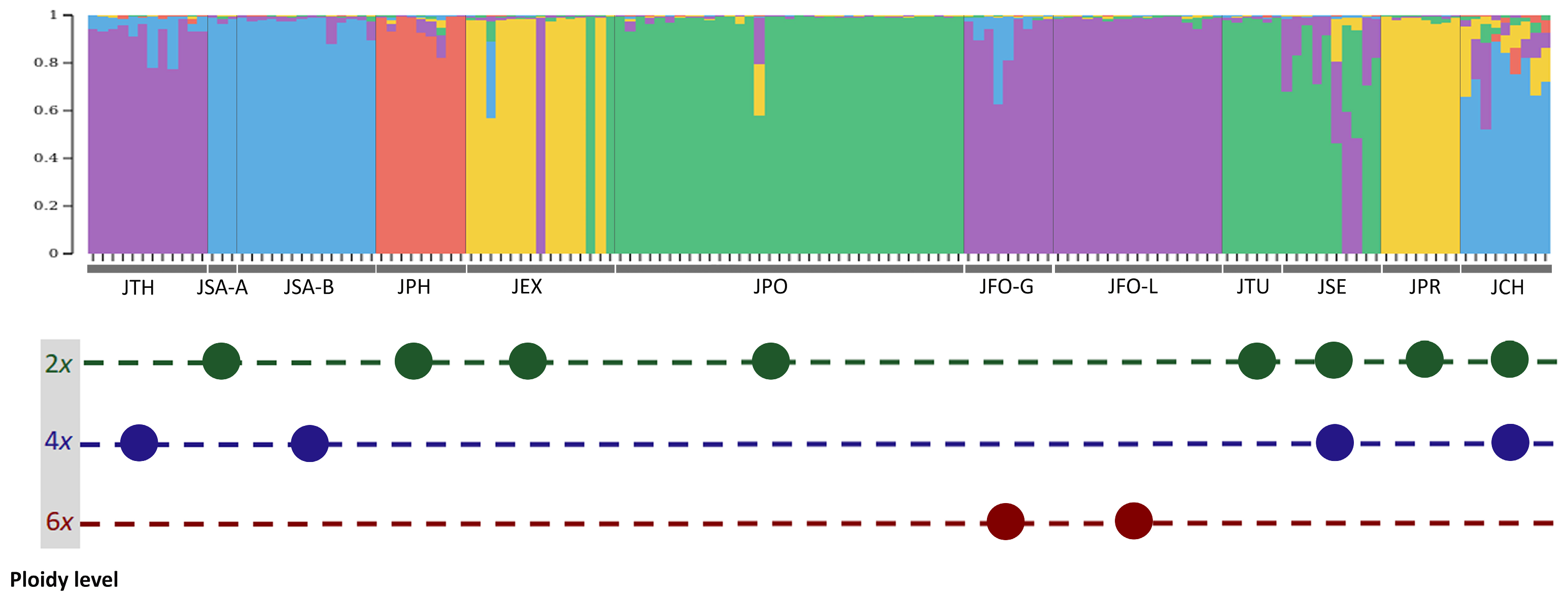
3.3. Admixture Patterns between Juniperus Taxa
4. Discussion
4.1. Genetic Delimitation of Diploid Juniperus Taxa
4.2. Potential Origin of the Tetraploid Junipers
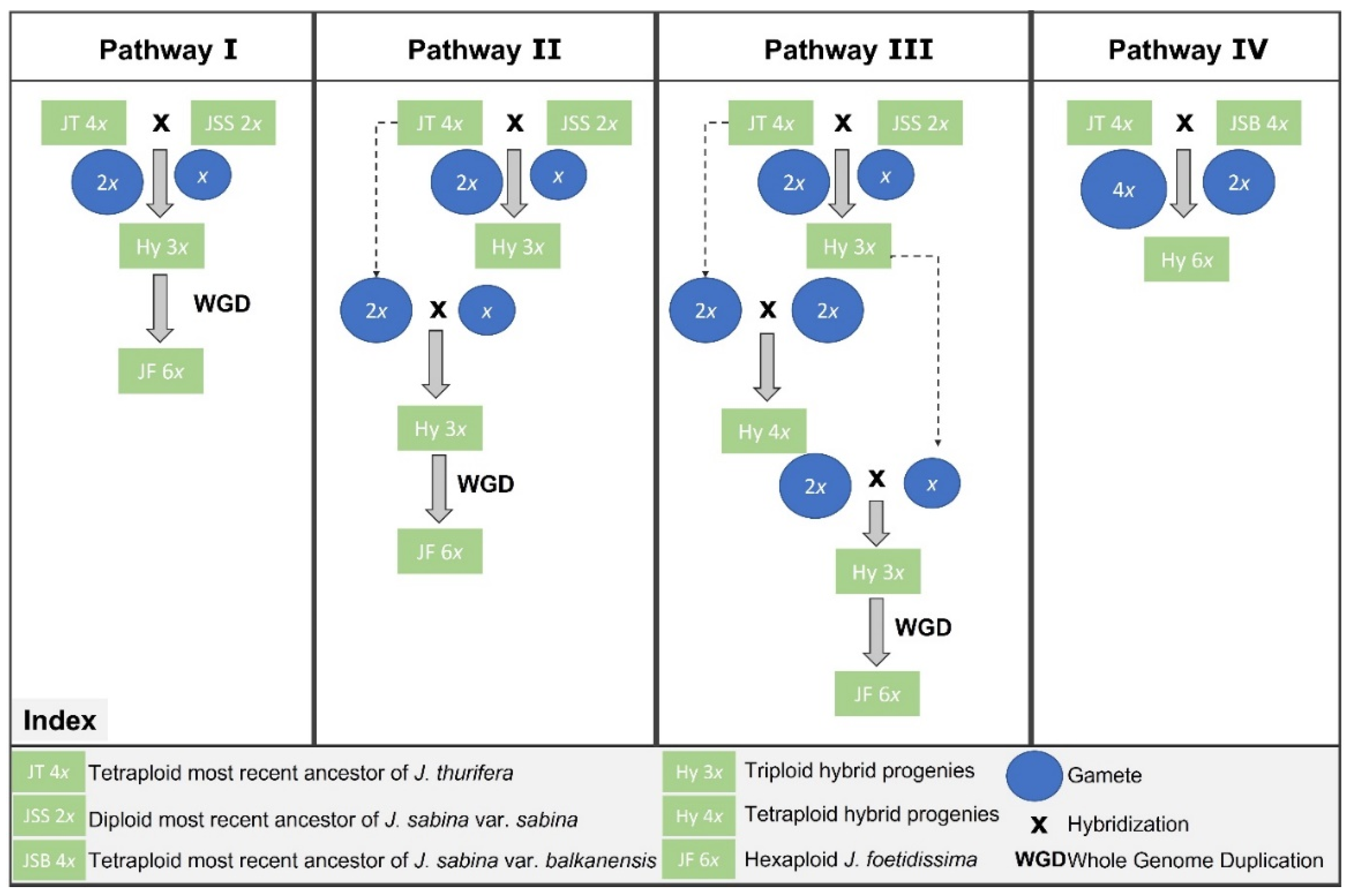
4.3. Insights into the Origin and Hypothetical Hexaploidy Pathway in J. foetidisima
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alix, K.; Gérard, P.R.; Schwarzacher, T.; Heslop-Harrison, J.S. Polyploidy and interspecific hybridization: Partners for adaptation, speciation and evolution in plants. Ann. Bot. 2017, 120, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Mallet, J. Hybrid Speciation. Nature 2007, 446, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Paun, O.; Forest, F.; Fay, M.F.; Chase, M.W. Hybrid speciation in angiosperms: Parental divergence drives ploidy. New Phytol. 2009, 182, 507–518. [Google Scholar] [CrossRef]
- Soltis, P.S.; Soltis, D.E. The role of hybridization in plant speciation. Annu. Rev. Plant Biol. 2009, 60, 561–588. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Ma, Z.; Wang, M.M.; Qin, A.L.; Ran, J.H.; Wang, X.Q. A high frequency of allopolyploid speciation in the gymnospermous genus Ephedra and its possible association with some biological and ecological features. Mol. Ecol. 2016, 25, 1192–1210. [Google Scholar] [CrossRef]
- Wu, H.; Yu, Q.; Ran, J.H.; Wang, X.Q. Unbiased subgenome evolution in allotetraploid species of Ephedra and its implications for the evolution of large genomes in gymnosperms. Genome Biol. Evol. 2021, 13, evaa236. [Google Scholar] [CrossRef]
- Farhat, P.; Hidalgo, O.; Robert, T.; Siljak-Yakovlev, S.; Leitch, I.J.; Adams, R.P.; Bou Dagher-Kharrat, M. Polyploidy in the conifer genus Juniperus: An unexpectedly high rate. Front. Plant Sci. 2019, 10, 676. [Google Scholar] [CrossRef]
- Adams, R.P. Junipers of the World: The Genus Juniperus, 4th ed.; Trafford Publishing Co: Bloomington, IN, USA, 2014; ISBN 978-1-4907-2325-9. [Google Scholar]
- Nagano, K.; Matoba, H.; Yonemura, K.; Matsuda, Y.; Murata, T.; Hoshi, Y. Karyotype analysis of three Juniperus species using Fluorescence in Situ Hybridization (FISH) with two ribosomal RNA genes. Cytologia 2007, 72, 37–42. [Google Scholar] [CrossRef]
- Romo, A.; Hidalgo, O.; Boratyński, A.; Sobierajska, K.; Jasińska, A.K.; Vallès, J.; Garnatje, T. Genome size and ploidy levels in highly fragmented habitats: The case of western mediterranean Juniperus (Cupressaceae) with special emphasis on J. thurifera L. Tree Genet. Genomes 2013, 9, 587–599. [Google Scholar] [CrossRef]
- Vallès, J.; Garnatje, T.; Robin, O.; Siljak-Yakovlev, S. Molecular cytogenetic studies in western Mediterranean Juniperus (Cupressaceae): A constant model of GC-rich chromosomal regions and rDNA loci with evidences for paleopolyploidy. Tree Genet. Genomes 2015, 11, 43. [Google Scholar] [CrossRef]
- Farhat, P.; Siljak-Yakovlev, S.; Adams, R.P.; Bou Dagher Kharrat, M.; Robert, T. Genome size variation and polyploidy in the geographical range of Juniperus sabina L. (Cupressaceae). Bot. Lett. 2019, 166, 134–143. [Google Scholar] [CrossRef]
- Bou Dagher-Kharrat, M.; Abdel-Samad, N.; Douaihy, B.; Bourge, M.; Fridlender, A.; Siljak-Yakovlev, S.; Brown, S.C. Nuclear DNA C-Values for Biodiversity Screening: Case of the Lebanese Flora. Plant Biosyst. Int. J. Deal. Asp. Plant Biol. 2013, 147, 1228–1237. [Google Scholar] [CrossRef]
- Hirayoshi, I.; Nakamura, Y. Chromosome number of Sequoia sempervirens. Bot. Zool. 1943, 11, 73–75. [Google Scholar]
- Mouterde, P. Nouvelle Flore du Liban et de La Syrie; Editions de l’Impr. Catholique: Beyrouth, Lebanon, 1966; ISBN 978-2-7214-5807-0. [Google Scholar]
- Meudt, H.M.; Clarke, A.C. Almost forgotten or latest practice? AFLP applications, analyses and advances. Trends Plant Sci. 2007, 12, 106–117. [Google Scholar] [CrossRef] [PubMed]
- Kokotovic, B.; Friis, N.F.; Jensen, J.S.; Ahrens, P. Amplified-Fragment Length Polymorphism Fingerprinting of Mycoplasma species. J. Clin. Microbiol. 1999, 37, 3300–3307. [Google Scholar] [CrossRef]
- Bou Dagher-Kharrat, M.; Mariette, S.; Lefèvre, F.; Fady, B.; Grenier-de March, G.; Plomion, C.; Savouré, A. Geographical diversity and genetic relationships among Cedrus species estimated by AFLP. Tree Genet. Genomes 2007, 3, 275–285. [Google Scholar] [CrossRef]
- Jacobs, M.M.; van den Berg, R.G.; Vleeshouwers, V.G.; Visser, M.; Mank, R.; Sengers, M.; Hoekstra, R.; Vosman, B. AFLP analysis reveals a lack of phylogenetic structure within Solanum section Petota. BMC Evol. Biol. 2008, 8, 145. [Google Scholar] [CrossRef] [PubMed]
- Dasmahapatra, K.K.; Hoffman, J.I.; Amos, W. Pinniped phylogenetic relationships inferred using AFLP markers. Heredity 2009, 103, 168–177. [Google Scholar] [CrossRef]
- Prebble, J.; Meudt, H.; Garnock-Jones, P. Phylogenetic relationships and species delimitation of New Zealand Bluebells (Wahlenbergia, Campanulaceae) based on analyses of AFLP data. N. Z. J. Bot. 2012, 50, 365–378. [Google Scholar] [CrossRef]
- Adams, R.P.; Farhat, P.; Shuka, L.; Siljak-Yakovlev, S. Discovery of Juniperus sabina var. balkanensis R. P. Adams and A. N. Tashev in Albania and relictual polymorphisms found in nrDNA. Phytologia 2018, 100, 187–194. [Google Scholar]
- Farhat, P.; Takvorian, N.; Avramidou, M.; Garraud, L.; Adams, R.P.; Siljak-Yakovlev, S.; Bou Dagher-Kharrat, M.; Robert, T. First evidence for allotriploid hybrids between Juniperus thurifera and J. sabina in a sympatric area in the French Alps. Ann. For. Sci. 2020, 77, 93. [Google Scholar] [CrossRef]
- Farhat, P.; Siljak-Yakovlev, S.; Valentin, N.; Fabregat, C.; Lopez-Udias, S.; Salazar-Mendias, C.; Altarejos, J.; Adams, R.P. Gene flow between diploid and tetraploid junipers—Two contrasting evolutionary pathways in two Juniperus populations. BMC Evol. Biol. 2020, 20, 148. [Google Scholar] [CrossRef] [PubMed]
- Douaihy, B.C. Caractérisation Écogéographique et Génétique de Juniperius excelsa Dans l’est Du Bassin Méditerranée. Ph.D. Thesis, Préparée en co-tutelle entre la Faculté des sciences de l’Université Saint-Joseph de Beyrouth et Museum National D’Histoire Naturelle, Paris, France, 2011. [Google Scholar]
- Doyle, J.J.; Doyle, J.L. Isolation of plant DNA from fresh tissue. Focus 1990, 12, 39–40. [Google Scholar]
- Vos, P.; Hogers, R.; Bleeker, M.; Reijans, M.; van de Lee, T.; Hornes, M.; Friters, A.; Pot, J.; Paleman, J.; Kuiper, M.; et al. AFLP: A New Technique for DNA Fingerprinting. Nucleic Acids Res. 1995, 23, 4407–4414. [Google Scholar] [CrossRef] [PubMed]
- Bonin, A.; Bellemain, E.; Bronken Eidesen, P.; Pompanon, F.; Brochmann, C.; Taberlet, P. How to track and assess genotyping errors in population genetics studies: Tracking and assessing genotyping errors. Mol. Ecol. 2004, 13, 3261–3273. [Google Scholar] [CrossRef] [PubMed]
- Vekemans, X.; Beauwens, T.; Lemaire, M.; Roldan-Ruiz, I. Data from Amplified Fragment Length Polymorphism (AFLP) markers show indication of size homoplasy and of a relationship between degree of homoplasy and fragment size. Mol. Ecol. 2002, 11, 139–151. [Google Scholar] [CrossRef]
- Nei, M.; Li, W.H. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. USA 1979, 76, 5269–5273. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (ITOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, 293–296. [Google Scholar] [CrossRef]
- Adams, R.P.; Schwarzbach, A.E.; Tashev, A.N. Chloroplast capture by a new variety, Juniperus sabina var. balkanensis R. P. Adams and A. N. Tashev, from the Balkan Peninsula: A putative stabilized relictual hybrid between J. sabina and ancestral J. thurifera. Phytologia 2016, 98, 100–111. [Google Scholar]
- Adams, R.P.; Boratynski, A.; Marcysiak, K.; Roma-Marzio, F.; Peruzzi, L.; Bartolucci, F.; Conti, F.; Mataraci, T.; Schwarzbach, A.E.; Tashev, A.N.; et al. Discovery of Juniperus sabina var. balkanensis R. P. Adams and A. N. Tashev in Macedonia, Bosnia-Herzegovina, Croatia and Central and Southern Italy and relictual polymorphisms found in nrDNA. Phytologia 2018, 100, 117–127. [Google Scholar]
- Chen, C.; Durand, E.; Forbes, F.; François, O. Bayesian clustering algorithms ascertaining spatial population structure: A new computer program and a comparison study. Mol. Ecol. Notes 2007, 7, 747–756. [Google Scholar] [CrossRef]
- Jakobsson, M.; Rosenberg, N.A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 2007, 23, 1801–1806. [Google Scholar] [CrossRef]
- Schenk, M.F.; Thienpont, C.-N.; Koopman, W.J.M.; Gilissen, L.J.W.J.; Smulders, M.J.M. Phylogenetic relationships in Betula (Betulaceae) based on AFLP markers. Tree Genet. Genomes 2008, 4, 911. [Google Scholar] [CrossRef]
- Shasany, A.K.; Darokar, M.P.; Dhawan, S.; Gupta, A.K.; Gupta, S.; Shukla, A.K.; Patra, N.K.; Khanuja, S.P.S. Use of RAPD and AFLP markers to identify inter- and intraspecific hybrids of Mentha. J. Hered. 2005, 96, 542–549. [Google Scholar] [CrossRef] [PubMed]
- Divakaran, M.; Babu, K.N.; Ravindran, P.N.; Peter, K.V. Interspecific hybridization in Vanilla and molecular characterization of hybrids and selfed progenies using RAPD and AFLP markers. Sci. Hortic. 2006, 108, 414–422. [Google Scholar] [CrossRef]
- Lazarević, M.; Siljak-Yakovlev, S.; Sanino, A.; Niketić, M.; Lamy, F.; Hinsinger, D.D.; Tomović, G.; Stevanović, B.; Stevanović, V.; Robert, T. Genetic variability in Balkan paleoendemic resurrection plants Ramonda serbica and R. nathaliae across their range and in the zone of sympatry. Front. Plant Sci. 2022, 13, 873471. [Google Scholar] [CrossRef] [PubMed]
- Douaihy, B.; Vendramin, G.G.; Boratyński, A.; Machon, N.; Bou Dagher-Kharrat, M. High genetic diversity with moderate differentiation in Juniperus excelsa from Lebanon and the Eastern Mediterranean Region. AoB Plants 2011, 2011, plr003. [Google Scholar] [CrossRef]
- Adams, R.P.; Hojjati, F.; Schwarzbach, A.E. Taxonomy of Juniperus in Iran: DNA sequences of nrDNA plus three cpDNAs reveal Juniperus polycarpos var. turcomanica and J. seravschanica in southern Iran. Phytologia 2014, 96, 19–25. [Google Scholar]
- Hojjati, F.; Kazempour-Osaloo, S.; Adams, R.P.; Assadi, M. Molecular phylogeny of Juniperus in Iran with special reference to the J. excelsa complex, focusing on J. seravschanica. Phytotaxa 2018, 375, 135. [Google Scholar] [CrossRef]
- Elshire, R.J.; Glaubitz, J.C.; Sun, Q.; Poland, J.A.; Kawamoto, K.; Buckler, E.S.; Mitchell, S.E. A robust, simple Genotyping-by-Sequencing (GBS) approach for high diversity species. PLoS ONE 2011, 6, e19379. [Google Scholar] [CrossRef]
- Weitemier, K.; Straub, S.C.K.; Cronn, R.C.; Fishbein, M.; Schmickl, R.; McDonnell, A.; Liston, A. Hyb-Seq: Combining target enrichment and genome skimming for plant phylogenomics. Appl. Plant Sci. 2014, 2, 1400042. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.P. Two new cases of chloroplast capture in incongruent topologies in the Juniperus excelsa complex: J. excelsa var. turcomanica Comb. Nov. and J. excelsa var. seravschanica Comb. Nov. Phytologia 2016, 98, 219–231. [Google Scholar]
- Xiong, Z.; Gaeta, R.T.; Edger, P.P.; Cao, Y.; Zhao, K.; Zhang, S.; Pires, J.C. Chromosome inheritance and meiotic stability in allopolyploid Brassica napus. G3-Genes Genom Genet. 2021, 11, jkaa011. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Taxon | Population | Somatic Ploidy Level | Country | Fresh/Dried Leaves | Number of Individuals | GPS Coordinates |
|---|---|---|---|---|---|---|
| Juniperus excelsa | Afqa * | 2x | Lebanon | Fresh | 4 | N 34°4′25″, E 35°54′20″ |
| Barqa * | Lebanon | Fresh | 1 | N 34°11′48″, E 36°8′15″ | ||
| Devas Mountain, Agios Georgious Forest | Greece | Fresh | 1 | N 39°45′0.4716″, E 19°40′59.8944″ | ||
| Qammoua forest * | Lebanon | Fresh | 5 | N 34°29′34″, E 36°15′14″ | ||
| Mrebbine | Lebanon | Fresh | 2 | N 34°34′4.008″, E 36°7′33.996″ | ||
| Nebha, Alkedam, Wadi ghned, Arsal | Lebanon | Fresh | 2 | N 34°11′19.7952″, E 36°12′57.6684″ | ||
| J. chinensis | Japan | 4xi | Japan | Dried | 3 | N 35°01.44′, E 138°47.30′ |
| Lanzhou | 2x, 4x | China | Dried | 4 | N 36°6′0″, E 103°43′59.88″ | |
| Xian | China | Dried | 2 | N 34°08′50.2”, E 109°34′41.3” | ||
| J. foetidissima | Devas Mountain, Agios Georgious Forest | 6xi | Greece | Fresh | 9 | N 39°45′0.4716″, E 19°40′59.8944″ |
| Hermel | Lebanon | Fresh | 14 | N 34°19′8.7528″, E 36°15′29.7792″ | ||
| Qammoua forest | Lebanon | Fresh | 3 | N 34° 30′ 44.388″, E 36°16′30.36″ | ||
| J. phoenicea | Baćina | 2x | Croatia | Fresh | 9 | N 43°05′12.6”, E 17°22′36.2” |
| J. polycarpos | Arsal * | 2x | Lebanon | Fresh | 17 | N 34°4′57″, E 36°28′33.996″ |
| Azerbaijan | 2xi | Azerbaijan | Dried | 2 | N 40°44′41.064″, E 47°35′19.14″ | |
| Wadi El Njass * | 2x | Lebanon | Fresh | 8 | N 34°19′49″, E 36°3′16″ | |
| Wadi El Njass | Lebanon | Fresh | 8 | N 34°20′48″, E 36°5′17″ | ||
| J. procera | Ethiopia | 2xi | Ethiopia | Dried | 4 | N 9°1′59.9988″, E 38°23′60″ |
| near Abha | 2x | Saudi Arabia | Dried | 4 | N 18°16′59.988″, E 42°21′0″ | |
| J. sabina var. sabina | Austria, Alps | 2xii | Austria | Dried | 3 | N 46°56′6″, E 11°2′20.4″ |
| J. sabina var. balkanensis | Mts. Cvrsnica and Cabulja | 4xii | Bosnia-Herzegovina | Fresh | 7 | N 43°34′18.084″, E 17°30′39.888″ |
| Biokovo Mts. | Croatia | Fresh | 7 | N 43°19′31.1808″, E 17°2′57.0228″ | ||
| J. seravschanica | Kazakhstan | 4xi | Kazakhstan | Dried | 2 | N 42°24′31.788″, E 70°28′30″ |
| Kazakhstan | Kazakhstan | Dried | 3 | N 42°10′46.2″, E 70°20′0.816″ | ||
| Oman | 2x, 4x | Oman | Dried | 3 | N 23°7′41.016″, E 57°36′9.288″ | |
| Pakistan | Pakistan | Dried | 2 | N 30°13′0.012″, E 67°5′60″ | ||
| J. thurifera | Alps | 4xiii | France | Dried | 5 | N 44°43′7.986″, E 6°36′13.0392″ |
| Monegros region | 4xi | Spain | Fresh | 3 | N 41°36′19.3428″, W 0°15′7.092″ | |
| Southern Iberian central range | Spain | Fresh | 4 | N 40° 0′ 14.4936″, W 5°1′11.82″ | ||
| J. turcomanica | Turkmenistan | 2xi | Turkmenistan | Dried | 2 | N 38°25′7.212″, E 56° 58′ 48″ |
| Shahmirzad | 2x | Iran | Dried | 1 | N 35°50′54.996″, E 53°26′24.216″ | |
| Bajgirna | Iran | Dried | 1 | N 37°25′9.804″, E 58°32′0.204″ | ||
| Baladae | Iran | Dried | 1 | N 36°14′34.404″, E 51°50′20.4″ | ||
| Fasa | Iran | Dried | 1 | N 29°9′57.816″, E 53°40′7.788″ |
| Species | Replicate Number of Repetition Type 1 | Replicate Number of Repetition Type 2 |
|---|---|---|
| J. excelsa | 1 | 2 |
| J. polycarpos | 3 | 6 |
| J. foetidissima | 1 | 12 |
| J. seravschanica | 1 | 0 |
| J. turcomanica | 1 | 0 |
| J. chinensis | 1 | 0 |
| J. procera | 1 | 0 |
| J. sabina | 1 | 2 |
| J. thurifera | 1 | 0 |
| J. phoenicea | 1 | 0 |
| CODE | Primer (5′-> 3′) | Length | GC (%) | Tm | Label | |
|---|---|---|---|---|---|---|
| Adapter | EcoRI-adapter L | CTCGTAGACTGCGTACC | 17 | 58.8 | 55.2 | No label |
| EcoRI-adapter S | AATTGGTACGCAGTC | 15 | 46.7 | 45.1 | No label | |
| Tru1I-adapter L | GACGATGAGTCCTGAG | 16 | 56.3 | 51.7 | No label | |
| Tru1I-adapter S | TACTCAGGACTCAT | 14 | 42.9 | 40 | No label | |
| Preamplification | EcoRI-PA | ACTGCGTACCAATTCA | 16 | 43.8 | 46.6 | No label |
| Tru1I-PA | GATGAGTCCTGAGTAAC | 17 | 47.1 | 50.4 | No label | |
| Amplification | EcoRI-1 | ACTGCGTACCAATTCACG | 18 | 50 | 53.7 | FAM |
| EcoRI-2 | ACTGCGTACCAATTCACT | 18 | 44.4 | 51.4 | FAM | |
| EcoRI-4 | ACTGCGTACCAATTCACC | 18 | 50 | 53.7 | PET | |
| Tru1I-1 | GATGAGTCCTGAGTAACTA | 19 | 42.1 | 52.4 | No label | |
| Tru1I-2 | GATGAGTCCTGAGTAACTG | 19 | 47.4 | 54.5 | No label | |
| Tru1I-3 | GATGAGTCCTGAGTAACAG | 19 | 47.4 | 54.5 | No label | |
| Tru1I-4 | GATGAGTCCTGAGTAACTT | 19 | 42.1 | 52.4 | No label | |
| Amplification primers combinations | EcoRI-1/Tru1I-1 | |||||
| EcoRI-1/Tru1I-3 | ||||||
| EcoRI-2/Tru1I-4 | ||||||
| EcoRI-4/Tru1I-2 | ||||||
| Species | Primers EcoRI-1/Tru1I-1 | Primers EcoRI-1/Tru1I-3 | Primers EcoRI-2/Tru1I-4 | Primers EcoRI-4/Tru1I-2 |
|---|---|---|---|---|
| J. chinensis | 68.5 * | 58.5 * | 73.1 * | 59.9 * |
| J. excelsa | 46.8 | 48.1 | 60.6 | 44.2 |
| J. foetidissima | 50.9 | 42.2 | 48.5 | 41.6 |
| J. phoenicea | 39.1 | 42.5 | 49.8 | 37.2 |
| J. polycarpos | 38.4 * | 36.9 | 50.8 | 32 |
| J. procera | 40.4 | 38.7 | 50.8 | 32 |
| J. sabina | 49.1 | 41.1 | 53.2 | 37.2 |
| J. seravschanica | 56.8 | 46 | 56.6 | 43.9 |
| J. thurifera | 44.2 | 36.2 * | 43.8 * | 32.7 |
| J. turcomanica | 39.9 | 37.6 | 54.5 | 31.2 * |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Farhat, P.; Siljak-Yakovlev, S.; Takvorian, N.; Bou Dagher Kharrat, M.; Robert, T. Allopolyploidy: An Underestimated Driver in Juniperus Evolution. Life 2023, 13, 1479. https://doi.org/10.3390/life13071479
Farhat P, Siljak-Yakovlev S, Takvorian N, Bou Dagher Kharrat M, Robert T. Allopolyploidy: An Underestimated Driver in Juniperus Evolution. Life. 2023; 13(7):1479. https://doi.org/10.3390/life13071479
Chicago/Turabian StyleFarhat, Perla, Sonja Siljak-Yakovlev, Najat Takvorian, Magda Bou Dagher Kharrat, and Thierry Robert. 2023. "Allopolyploidy: An Underestimated Driver in Juniperus Evolution" Life 13, no. 7: 1479. https://doi.org/10.3390/life13071479
APA StyleFarhat, P., Siljak-Yakovlev, S., Takvorian, N., Bou Dagher Kharrat, M., & Robert, T. (2023). Allopolyploidy: An Underestimated Driver in Juniperus Evolution. Life, 13(7), 1479. https://doi.org/10.3390/life13071479