
Iron Chelation as a Potential Therapeutic Approach in Acute Lung Injury


Abstract
:1. Introduction
2. Iron in Health and Disease
2.1. Importance of Iron in Human Health
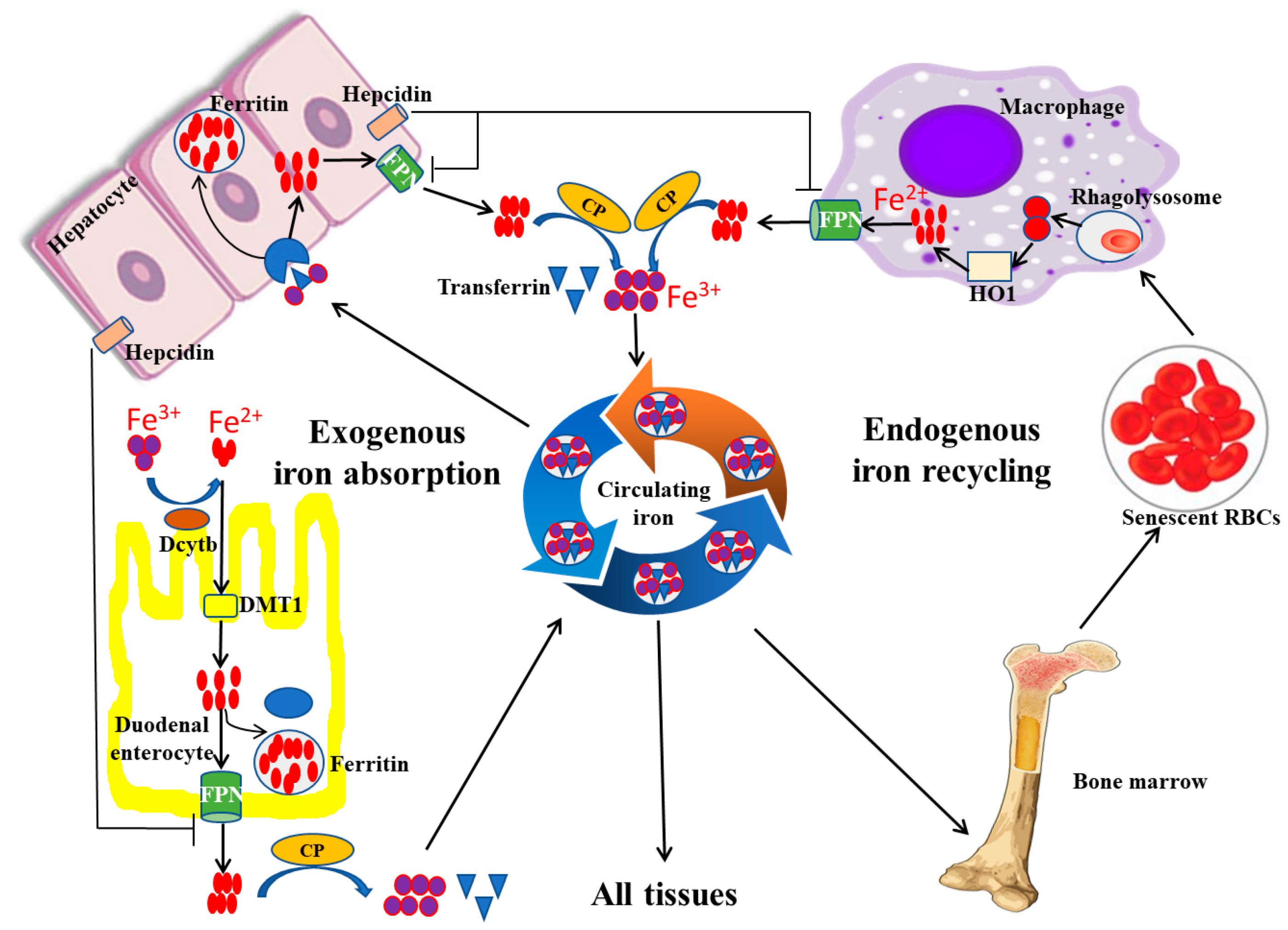
2.2. Iron Regulation
2.3. Iron in Pathology and Diseases
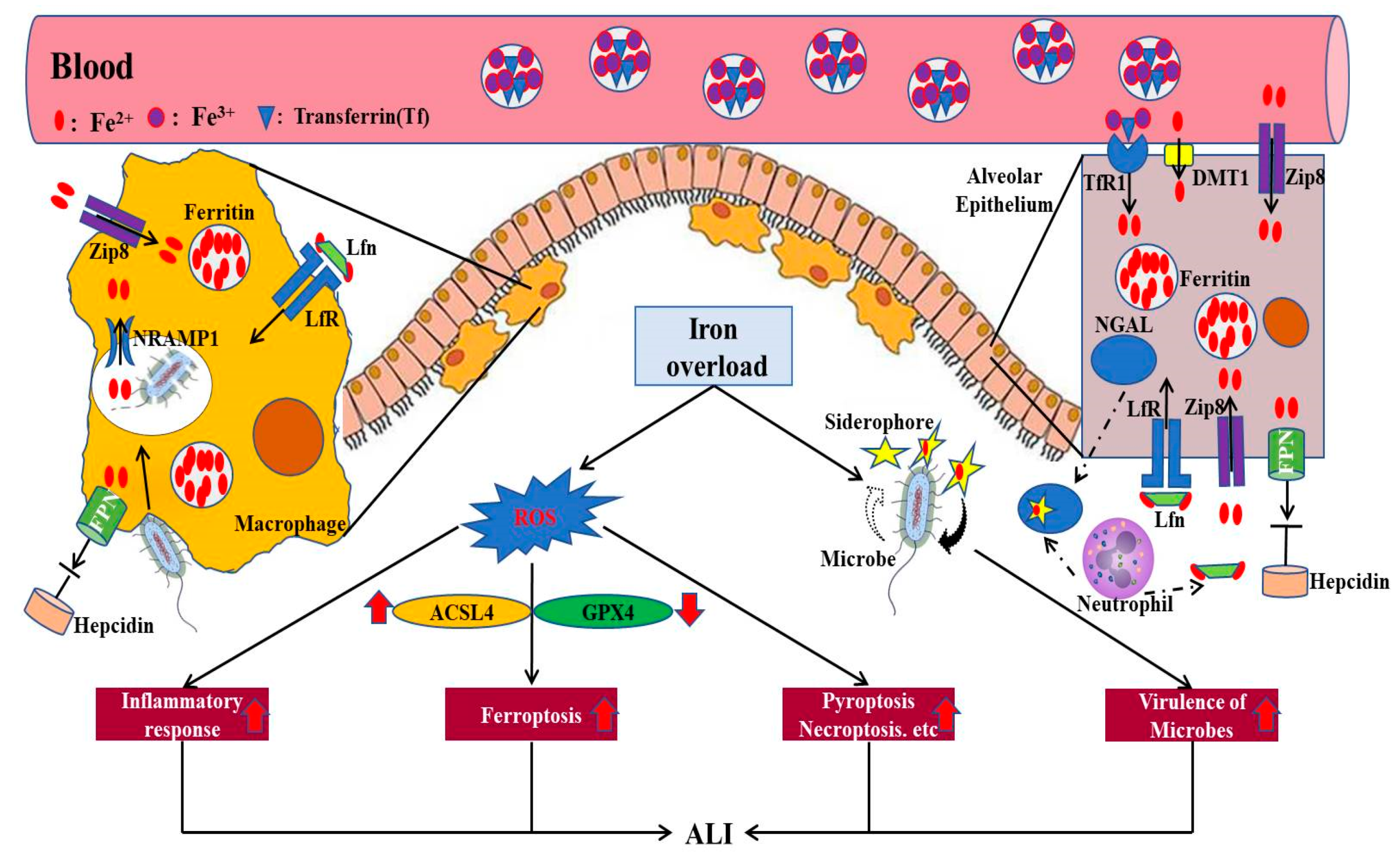
3. Iron and ALI
3.1. Direct ALI
3.2. Indirect ALI
4. Potential Therapeutics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ACSL4 | Acyl-CoA synthetase long-chain family 4 |
| AKI | Acute kidney injury |
| ALI | Acute lung injury |
| ARDS | Acute respiratory distress syndrome |
| CP | Ceruloplasmin |
| DcytB | Duodenal cytochrome B |
| DFO | Deferoxamine |
| DFP | Deferiprone |
| DFX | Deferasirox |
| DMT1 | Divalent metal transporter 1 |
| FPN | Ferroportin |
| GPX4 | Glutathione peroxidase 4 |
| HO-1 | Heme oxygenase 1 |
| IRE | Iron responsive elements |
| IRP | Iron regulatory protein |
| Lfn | Lactoferrin |
| LncRNAs | Long non-coding RNAs |
| LPS | Lipopolysaccharide |
| NET | Neutrophil extracellular trap |
| NGAL | Neutrophil gelatinase–associated lipocalin |
| NRAMP1 | Natural resistance–associated macrophage protein 1 |
| PLA2 | Phospholipase A2 |
| ROS | Reactive oxygen species |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| TBARs | Thiobarbituric acid reactive substances |
| Tf | Transferrin |
| TfR | Transferrin receptor |
| YAP1 | Yes-associated protein 1 |
| ZIP8 | Zinc transporter SLC39A8 |
References
- Yang, Y.; Ma, Y.; Li, Q.; Ling, Y.; Zhou, Y.; Chu, K.; Xue, L.; Tao, S. STAT6 inhibits ferroptosis and alleviates acute lung injury via regulating P53/SLC7A11 pathway. Cell Death Dis. 2022, 13, 530. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.R.; Matthay, M.A. Acute lung injury: Epidemiology, pathogenesis, and treatment. J. Aerosol Med. Pulm. Drug Deliv. 2010, 23, 243–252. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, J.; Xie, W. The role of ferroptosis in acute lung injury. Mol. Cell. Biochem. 2022, 477, 1453–1461. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Lu, Q.; Wang, K.; Lu, J.; Gu, X.; Zhu, D.; Liu, F.; Guo, Z. miR-34b-5p inhibition attenuates lung inflammation and apoptosis in an LPS-induced acute lung injury mouse model by targeting progranulin. J. Cell. Physiol. 2018, 233, 6615–6631. [Google Scholar] [CrossRef] [Green Version]
- Wu, G.; Xu, G.; Chen, D.W.; Gao, W.X.; Xiong, J.Q.; Shen, H.Y.; Gao, Y.Q. Hypoxia Exacerbates Inflammatory Acute Lung Injury via the Toll-Like Receptor 4 Signaling Pathway. Front. Immunol. 2018, 9, 1667. [Google Scholar] [CrossRef] [Green Version]
- Lei, J.; Wei, Y.; Song, P.; Li, Y.; Zhang, T.; Feng, Q.; Xu, G. Cordycepin inhibits LPS-induced acute lung injury by inhibiting inflammation and oxidative stress. Eur. J. Pharmacol. 2018, 818, 110–114. [Google Scholar] [CrossRef]
- Lagan, A.L.; Melley, D.D.; Evans, T.W.; Quinlan, G.J. Pathogenesis of the systemic inflammatory syndrome and acute lung injury: Role of iron mobilization and decompartmentalization. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L161–L174. [Google Scholar] [CrossRef]
- Neves, J.; Haider, T.; Gassmann, M.; Muckenthaler, M.U. Iron Homeostasis in the Lungs-A Balance between Health and Disease. Pharmaceuticals 2019, 12, 5. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, N.B.; Callaghan, K.D.; Ghio, A.J.; Haile, D.J.; Yang, F. Hepcidin expression and iron transport in alveolar macrophages. Am. J. Physiol Lung Cell. Mol. Physiol. 2006, 291, L417–L425. [Google Scholar] [CrossRef] [Green Version]
- Mao, K.; Tang, R.; Wu, Y.; Zhang, Z.; Gao, Y.; Huang, H. Prognostic markers of ferroptosis-related long non-coding RNA in lung adenocarcinomas. Front. Genet. 2023, 14, 1118273. [Google Scholar] [CrossRef]
- Yao, J.; Chen, X.; Liu, X.; Li, R.; Zhou, X.; Qu, Y. Characterization of a ferroptosis and iron-metabolism related lncRNA signature in lung adenocarcinoma. Cancer Cell Int. 2021, 21, 340. [Google Scholar] [CrossRef]
- Ward, J.L.; Torres-Gonzalez, M.; Ammons, M.C.B. The Influence of Viral Infections on Iron Homeostasis and the Potential for Lactoferrin as a Therapeutic in the Age of the SARS-CoV-2 Pandemic. Nutrients 2022, 14, 3090. [Google Scholar] [CrossRef]
- Cutone, A.; Rosa, L.; Lepanto, M.S.; Scotti, M.J.; Berlutti, F.; Bonaccorsi di Patti, M.C.; Musci, G.; Valenti, P. Lactoferrin Efficiently Counteracts the Inflammation-Induced Changes of the Iron Homeostasis System in Macrophages. Front. Immunol. 2017, 8, 705. [Google Scholar] [CrossRef] [Green Version]
- Cutone, A.; Lepanto, M.S.; Rosa, L.; Scotti, M.J.; Rossi, A.; Ranucci, S.; De Fino, I.; Bragonzi, A.; Valenti, P.; Musci, G.; et al. Aerosolized Bovine Lactoferrin Counteracts Infection, Inflammation and Iron Dysbalance in A Cystic Fibrosis Mouse Model of Pseudomonas aeruginosa Chronic Lung Infection. Int. J. Mol. Sci. 2019, 20, 2128. [Google Scholar] [CrossRef] [Green Version]
- Muñoz, M.; Villar, I.; García-Erce, J.A. An update on iron physiology. World J. Gastroenterol. 2009, 15, 4617–4626. [Google Scholar] [CrossRef]
- Mackenzie, B.; Garrick, M.D. Iron Imports. II. Iron uptake at the apical membrane in the intestine. Am. J. Physiol Gastrointest. Liver Physiol. 2005, 289, G981–G986. [Google Scholar] [CrossRef] [Green Version]
- Donovan, A.; Lima, C.A.; Pinkus, J.L.; Pinkus, G.S.; Zon, L.I.; Robine, S.; Andrews, N.C. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005, 1, 191–200. [Google Scholar] [CrossRef] [Green Version]
- Abbasi, U.; Abbina, S.; Gill, A.; Takuechi, L.E.; Kizhakkedathu, J.N. Role of Iron in the Molecular Pathogenesis of Diseases and Therapeutic Opportunities. ACS Chem. Biol. 2021, 16, 945–972. [Google Scholar] [CrossRef]
- Knutson, M.D. Iron transport proteins: Gateways of cellular and systemic iron homeostasis. J. Biol. Chem. 2017, 292, 12735–12743. [Google Scholar] [CrossRef] [Green Version]
- Kawabata, H. Transferrin and transferrin receptors update. Free Radic. Biol. Med. 2019, 133, 46–54. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, Y.; Guo, L.; Gao, W.; Tang, T.L.; Yan, M. Interaction between macrophages and ferroptosis. Cell Death Dis. 2022, 13, 355. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.; Hoque, N.; Nasrin, N.; Hossain, M.; Rizwan, F.; Biswas, K.; Asaduzzaman, M.; Rahman, S.; Hoskin, D.W.; Sultana, S.; et al. Iron Overload and Breast Cancer: Iron Chelation as a Potential Therapeutic Approach. Life 2022, 12, 963. [Google Scholar] [CrossRef] [PubMed]
- Parrow, N.L.; Fleming, R.E.; Minnick, M.F. Sequestration and scavenging of iron in infection. Infect. Immun. 2013, 81, 3503–3514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michels, K.R.; Zhang, Z.; Bettina, A.M.; Cagnina, R.E.; Stefanova, D.; Burdick, M.D.; Vaulont, S.; Nemeth, E.; Ganz, T.; Mehrad, B. Hepcidin-mediated iron sequestration protects against bacterial dissemination during pneumonia. JCI Insight 2017, 2, e92002. [Google Scholar] [CrossRef] [PubMed]
- Mayneris-Perxachs, J.; Moreno-Navarrete, J.M.; Fernández-Real, J.M. The role of iron in host-microbiota crosstalk and its effects on systemic glucose metabolism. Nat. Rev Endocrinol. 2022, 18, 683–698. [Google Scholar] [CrossRef] [PubMed]
- Bergamaschi, G.; Di Sabatino, A.; Pasini, A.; Ubezio, C.; Costanzo, F.; Grataroli, D.; Masotti, M.; Alvisi, C.; Corazza, G.R. Intestinal expression of genes implicated in iron absorption and their regulation by hepcidin. Clin. Nutr. (Edinb. Scotl.) 2017, 36, 1427–1433. [Google Scholar] [CrossRef]
- Anderson, C.P.; Shen, M.; Eisenstein, R.S.; Leibold, E.A. Mammalian iron metabolism and its control by iron regulatory proteins. Biochim. Et Biophys. Acta 2012, 1823, 1468–1483. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, N.; Pantopoulos, K. The IRP/IRE system in vivo: Insights from mouse models. Front. Pharmacol. 2014, 5, 176. [Google Scholar] [CrossRef] [Green Version]
- Li, L.X.; Guo, F.F.; Liu, H.; Zeng, T. Iron overload in alcoholic liver disease: Underlying mechanisms, detrimental effects, and potential therapeutic targets. Cell. Mol. Life Sci. CMLS 2022, 79, 201. [Google Scholar] [CrossRef]
- Wang, C.Y.; Jenkitkasemwong, S.; Duarte, S.; Sparkman, B.K.; Shawki, A.; Mackenzie, B.; Knutson, M.D. ZIP8 is an iron and zinc transporter whose cell-surface expression is up-regulated by cellular iron loading. J. Biol. Chem. 2012, 287, 34032–34043. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.J.; Bao, S.; Gálvez-Peralta, M.; Pyle, C.J.; Rudawsky, A.C.; Pavlovicz, R.E.; Killilea, D.W.; Li, C.; Nebert, D.W.; Wewers, M.D.; et al. ZIP8 regulates host defense through zinc-mediated inhibition of NF-κB. Cell Rep. 2013, 3, 386–400. [Google Scholar] [CrossRef] [Green Version]
- Pyle, C.J.; Akhter, S.; Bao, S.; Dodd, C.E.; Schlesinger, L.S.; Knoell, D.L. Zinc Modulates Endotoxin-Induced Human Macrophage Inflammation through ZIP8 Induction and C/EBPβ Inhibition. PLoS ONE 2017, 12, e0169531. [Google Scholar] [CrossRef] [Green Version]
- Devireddy, L.R.; Gazin, C.; Zhu, X.; Green, M.R. A cell-surface receptor for lipocalin 24p3 selectively mediates apoptosis and iron uptake. Cell 2005, 123, 1293–1305. [Google Scholar] [CrossRef] [Green Version]
- Ratledge, C.; Dover, L.G. Iron metabolism in pathogenic bacteria. Annu. Rev. Microbiol. 2000, 54, 881–941. [Google Scholar] [CrossRef]
- Zhang, V.; Nemeth, E.; Kim, A. Iron in lung pathology. Pharmaceuticals 2019, 12, 30. [Google Scholar] [CrossRef] [Green Version]
- Richardson, C.L.; Delehanty, L.L.; Bullock, G.C.; Rival, C.M.; Tung, K.S.; Kimpel, D.L.; Gardenghi, S.; Rivella, S.; Goldfarb, A.N. Isocitrate ameliorates anemia by suppressing the erythroid iron restriction response. J. Clin. Investig. 2013, 123, 3614–3623. [Google Scholar] [CrossRef] [Green Version]
- Savarese, G.; von Haehling, S.; Butler, J.; Cleland, J.G.F.; Ponikowski, P.; Anker, S.D. Iron deficiency and cardiovascular disease. Eur. Heart J. 2023, 44, 14–27. [Google Scholar] [CrossRef]
- Weiss, G.; Goodnough, L.T. Anemia of chronic disease. N. Engl. J. Med. 2005, 352, 1011–1023. [Google Scholar] [CrossRef] [Green Version]
- Webster, A.C.; Nagler, E.V.; Morton, R.L.; Masson, P. Chronic Kidney Disease. Lancet (Lond. Engl.) 2017, 389, 1238–1252. [Google Scholar] [CrossRef]
- Kindrat, I.; Tryndyak, V.; de Conti, A.; Shpyleva, S.; Mudalige, T.K.; Kobets, T.; Erstenyuk, A.M.; Beland, F.A.; Pogribny, I.P. MicroRNA-152-mediated dysregulation of hepatic transferrin receptor 1 in liver carcinogenesis. Oncotarget 2016, 7, 1276–1287. [Google Scholar] [CrossRef] [Green Version]
- Xue, X.; Ramakrishnan, S.K.; Weisz, K.; Triner, D.; Xie, L.; Attili, D.; Pant, A.; Győrffy, B.; Zhan, M.; Carter-Su, C.; et al. Iron Uptake via DMT1 Integrates Cell Cycle with JAK-STAT3 Signaling to Promote Colorectal Tumorigenesis. Cell Metab. 2016, 24, 447–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, D.; Zhou, C.X.; Shi, Y.B.; Lu, H.; He, X.Z. Decreased expression of ferroportin in prostate cancer. Oncol. Lett. 2015, 10, 913–916. [Google Scholar] [CrossRef] [Green Version]
- Pinnix, Z.K.; Miller, L.D.; Wang, W.; D’Agostino, R., Jr.; Kute, T.; Willingham, M.C.; Hatcher, H.; Tesfay, L.; Sui, G.; Di, X.; et al. Ferroportin and iron regulation in breast cancer progression and prognosis. Sci. Transl. Med. 2010, 2, 43ra56. [Google Scholar] [CrossRef] [PubMed]
- Pasricha, S.R.; Tye-Din, J.; Muckenthaler, M.U.; Swinkels, D.W. Iron deficiency. Lancet (Lond. Engl.) 2021, 397, 233–248. [Google Scholar] [CrossRef]
- Wang, W.; Jing, X.; Du, T.; Ren, J.; Liu, X.; Chen, F.; Shao, Y.; Sun, S.; Yang, G.; Cui, X. Iron overload promotes intervertebral disc degeneration via inducing oxidative stress and ferroptosis in endplate chondrocytes. Free Radic. Biol. Med. 2022, 190, 234–246. [Google Scholar] [CrossRef]
- Ramm, G.A.; Ruddell, R.G. Iron homeostasis, hepatocellular injury, and fibrogenesis in hemochromatosis: The role of inflammation in a noninflammatory liver disease. Semin. Liver Dis. 2010, 30, 271–287. [Google Scholar] [CrossRef]
- Dos Santos, L.; Bertoli, S.R.; Ávila, R.A.; Marques, V.B. Iron overload, oxidative stress and vascular dysfunction: Evidences from clinical studies and animal models. Biochim. Et Biophys. Acta Gen. Subj. 2022, 1866, 130172. [Google Scholar] [CrossRef]
- Mo, M.; Gao, Y.; Deng, L.; Liang, Y.; Xia, N.; Pan, L. Association Between Iron Metabolism and Acute Kidney Injury in Critically Ill Patients With Diabetes. Front. Endocrinol. 2022, 13, 892811. [Google Scholar] [CrossRef]
- Cook-Libin, S.; Sykes, E.M.E.; Kornelsen, V.; Kumar, A. Iron Acquisition Mechanisms and Their Role in the Virulence of Acinetobacter baumannii. Infect. Immun. 2022, 90, e0022322. [Google Scholar] [CrossRef]
- Yap, A.; Talasz, H.; Lindner, H.; Würzner, R.; Haas, H. Ambient Availability of Amino Acids, Proteins, and Iron Impacts Copper Resistance of Aspergillus fumigatus. Front. Cell. Infect. Microbiol. 2022, 12, 847846. [Google Scholar] [CrossRef]
- Valković, T.; Damić, M.S. Role of Iron and Iron Overload in the Pathogenesis of Invasive Fungal Infections in Patients with Hematological Malignancies. J. Clin. Med. 2022, 11, 4457. [Google Scholar] [CrossRef]
- Chhabra, R.; Saha, A.; Chamani, A.; Schneider, N.; Shah, R.; Nanjundan, M. Iron Pathways and Iron Chelation Approaches in Viral, Microbial, and Fungal Infections. Pharmaceuticals 2020, 13, 275. [Google Scholar] [CrossRef]
- Scozzi, D.; Liao, F.; Krupnick, A.S.; Kreisel, D.; Gelman, A.E. The role of neutrophil extracellular traps in acute lung injury. Front. Immunol. 2022, 13, 953195. [Google Scholar] [CrossRef]
- Torres Acosta, M.A.; Singer, B.D. Pathogenesis of COVID-19-induced ARDS: Implications for an ageing population. Eur. Respir. J. 2020, 56, 2002049. [Google Scholar] [CrossRef]
- Wiersinga, W.J.; Rhodes, A.; Cheng, A.C.; Peacock, S.J.; Prescott, H.C. Pathophysiology, Transmission, Diagnosis, and Treatment of Coronavirus Disease 2019 (COVID-19): A Review. JAMA 2020, 324, 782–793. [Google Scholar] [CrossRef]
- Habib, H.M.; Ibrahim, S.; Zaim, A.; Ibrahim, W.H. The role of iron in the pathogenesis of COVID-19 and possible treatment with lactoferrin and other iron chelators. Biomed. Pharmacother. Biomed. Pharmacother. 2021, 136, 111228. [Google Scholar] [CrossRef]
- El-Sayed, E.M.; Ibrahim, K.S. Ameliorating effects of probiotics on alterations in iron homeostasis and inflammation in COVID-19. Mol. Biol. Rep. 2022, 49, 5153–5163. [Google Scholar] [CrossRef]
- Ganz, T.; Nemeth, E. Iron homeostasis in host defence and inflammation. Nat. Rev Immunol. 2015, 15, 500–510. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Chen, P.; Zhai, B.; Zhang, M.; Xiang, Y.; Fang, J.; Xu, S.; Gao, Y.; Chen, X.; Sui, X.; et al. The emerging role of ferroptosis in inflammation. Biomed. Pharmacother. Biomed. Pharmacother. 2020, 127, 110108. [Google Scholar] [CrossRef]
- Phua, J.; Weng, L.; Ling, L.; Egi, M.; Lim, C.M.; Divatia, J.V.; Shrestha, B.R.; Arabi, Y.M.; Ng, J.; Gomersall, C.D.; et al. Intensive care management of coronavirus disease 2019 (COVID-19): Challenges and recommendations. Lancet Respir. Med. 2020, 8, 506–517. [Google Scholar] [CrossRef]
- Wang, M.P.; Joshua, B.; Jin, N.Y.; Du, S.W.; Li, C. Ferroptosis in viral infection: The unexplored possibility. Acta Pharmacol. Sin. 2022, 43, 1905–1915. [Google Scholar] [CrossRef] [PubMed]
- An, H.S.; Yoo, J.W.; Jeong, J.H.; Heo, M.; Hwang, S.H.; Jang, H.M.; Jeong, E.A.; Lee, J.; Shin, H.J.; Kim, K.E.; et al. Lipocalin-2 promotes acute lung inflammation and oxidative stress by enhancing macrophage iron accumulation. Int. J. Biol. Sci. 2023, 19, 1163–1177. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zeng, C.; Luo, G.; Sun, Y.; Zhang, J.; Xu, Z.; Guo, Y.; Ye, H.; Mao, J.; Chen, S.; et al. Macrophage ferroportin serves as a therapeutic target against bacteria-induced acute lung injury by promoting barrier restoration. iScience 2022, 25, 105698. [Google Scholar] [CrossRef] [PubMed]
- Cianciulli, P.; Trua, G.; Papa, G. Yersinia enterocolitica infection in patients with iron overload. Haematologica 1990, 75, 197–198. [Google Scholar]
- Abreu, R.; Quinn, F.; Giri, P.K. Role of the hepcidin-ferroportin axis in pathogen-mediated intracellular iron sequestration in human phagocytic cells. Blood Adv. 2018, 2, 1089–1100. [Google Scholar] [CrossRef]
- Han, N.; Li, H.; Li, G.; Shen, Y.; Fei, M.; Nan, Y. Effect of bovine lactoferrin as a novel therapeutic agent in a rat model of sepsis-induced acute lung injury. AMB Express 2019, 9, 177. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.; Datta, R. Leishmania infection triggers hepcidin-mediated proteasomal degradation of Nramp1 to increase phagolysosomal iron availability. Cell. Microbiol. 2020, 22, e13253. [Google Scholar] [CrossRef]
- Xu, Y.; Li, X.; Cheng, Y.; Yang, M.; Wang, R. Inhibition of ACSL4 attenuates ferroptotic damage after pulmonary ischemia-reperfusion. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 16262–16275. [Google Scholar] [CrossRef]
- Nakamura, T.; Naguro, I.; Ichijo, H. Iron homeostasis and iron-regulated ROS in cell death, senescence and human diseases. Biochim. Et Biophys. Acta Gen. Subj. 2019, 1863, 1398–1409. [Google Scholar] [CrossRef]
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef]
- Aloe, C.A.; Leong, T.L.; Wimaleswaran, H.; Papagianis, P.C.; McQualter, J.L.; McDonald, C.F.; Khor, Y.H.; Hoy, R.F.; Ingle, A.; Bansal, V.; et al. Excess iron promotes emergence of foamy macrophages that overexpress ferritin in the lungs of silicosis patients. Respirology (Carlton Vic.) 2022, 27, 427–436. [Google Scholar] [CrossRef]
- Zhang, V.; Ganz, T.; Nemeth, E.; Kim, A. Iron overload causes a mild and transient increase in acute lung injury. Physiol. Rep. 2020, 8, e14470. [Google Scholar] [CrossRef]
- Connelly, K.G.; Moss, M.; Parsons, P.E.; Moore, E.E.; Moore, F.A.; Giclas, P.C.; Seligman, P.A.; Repine, J.E. Serum ferritin as a predictor of the acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 1997, 155, 21–25. [Google Scholar] [CrossRef]
- Sharkey, R.A.; Donnelly, S.C.; Connelly, K.G.; Robertson, C.E.; Haslett, C.; Repine, J.E. Initial serum ferritin levels in patients with multiple trauma and the subsequent development of acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 1999, 159, 1506–1509. [Google Scholar] [CrossRef]
- Zheng, D.; Liu, J.; Piao, H.; Zhu, Z.; Wei, R.; Liu, K. ROS-triggered endothelial cell death mechanisms: Focus on pyroptosis, parthanatos, and ferroptosis. Front. Immunol. 2022, 13, 1039241. [Google Scholar] [CrossRef]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [Green Version]
- Friedmann Angeli, J.P.; Schneider, M.; Proneth, B.; Tyurina, Y.Y.; Tyurin, V.A.; Hammond, V.J.; Herbach, N.; Aichler, M.; Walch, A.; Eggenhofer, E.; et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 2014, 16, 1180–1191. [Google Scholar] [CrossRef] [Green Version]
- Mayr, L.; Grabherr, F.; Schwärzler, J.; Reitmeier, I.; Sommer, F.; Gehmacher, T.; Niederreiter, L.; He, G.W.; Ruder, B.; Kunz, K.T.R.; et al. Dietary lipids fuel GPX4-restricted enteritis resembling Crohn’s disease. Nat. Commun. 2020, 11, 1775. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Cao, L.m.; Zhang, X.j.; Chu, B. Targeting ferroptosis as a vulnerability in pulmonary diseases. Cell Death Dis. 2022, 13, 649. [Google Scholar] [CrossRef]
- Pan, Y.; Wang, X.; Liu, X.; Shen, L.; Chen, Q.; Shu, Q. Targeting Ferroptosis as a Promising Therapeutic Strategy for Ischemia-Reperfusion Injury. Antioxidants 2022, 11, 2196. [Google Scholar] [CrossRef] [PubMed]
- Qu, M.; Zhang, H.; Chen, Z.; Sun, X.; Zhu, S.; Nan, K.; Chen, W.; Miao, C. The Role of Ferroptosis in Acute Respiratory Distress Syndrome. Front. Med. 2021, 8, 651552. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Feng, Y.; Li, H.; Chen, X.; Wang, G.; Xu, S.; Li, Y.; Zhao, L. Ferrostatin-1 alleviates lipopolysaccharide-induced acute lung injury via inhibiting ferroptosis. Cell. Mol. Biol. Lett. 2020, 25, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Zheng, Y.; Wang, Y.; Wang, J.; Sang, A.; Song, X.; Li, X. YAP1 alleviates sepsis-induced acute lung injury via inhibiting ferritinophagy-mediated ferroptosis. Front. Immunol. 2022, 13, 884362. [Google Scholar] [CrossRef]
- Clemente, L.P.; Rabenau, M.; Tang, S.; Stanka, J.; Cors, E.; Stroh, J.; Culmsee, C.; von Karstedt, S. Dynasore Blocks Ferroptosis through Combined Modulation of Iron Uptake and Inhibition of Mitochondrial Respiration. Cells 2020, 9, 2259. [Google Scholar] [CrossRef]
- Kim, J.; Wessling-Resnick, M. The Role of Iron Metabolism in Lung Inflammation and Injury. J. Allergy Ther. 2012, 3, 004. [Google Scholar] [CrossRef]
- Preza, G.C.; Ruchala, P.; Pinon, R.; Ramos, E.; Qiao, B.; Peralta, M.A.; Sharma, S.; Waring, A.; Ganz, T.; Nemeth, E. Minihepcidins are rationally designed small peptides that mimic hepcidin activity in mice and may be useful for the treatment of iron overload. J. Clin. Investig. 2011, 121, 4880–4888. [Google Scholar] [CrossRef]
- Kaneko, Y.; Thoendel, M.; Olakanmi, O.; Britigan, B.E.; Singh, P.K. The transition metal gallium disrupts Pseudomonas aeruginosa iron metabolism and has antimicrobial and antibiofilm activity. J. Clin. Investig. 2007, 117, 877–888. [Google Scholar] [CrossRef]
- Itkonen, O.; Stenman, U.H.; Parkkinen, J.; Soliymani, R.; Baumann, M.; Hämäläinen, E. Binding of hepcidin to plasma proteins. Clin. Chem. 2012, 58, 1158–1160. [Google Scholar] [CrossRef] [Green Version]
- Ang, M.T.C.; Gumbau-Brisa, R.; Allan, D.S.; McDonald, R.; Ferguson, M.J.; Holbein, B.E.; Bierenstiel, M. DIBI, a 3-hydroxypyridin-4-one chelator iron-binding polymer with enhanced antimicrobial activity. MedChemComm 2018, 9, 1206–1212. [Google Scholar] [CrossRef]
- Parquet, M.D.C.; Savage, K.A.; Allan, D.S.; Davidson, R.J.; Holbein, B.E. Novel Iron-Chelator DIBI Inhibits Staphylococcus aureus Growth, Suppresses Experimental MRSA Infection in Mice and Enhances the Activities of Diverse Antibiotics in vitro. Front. Microbiol. 2018, 9, 1811. [Google Scholar] [CrossRef] [Green Version]
- Greenshields, A.L.; Power Coombs, M.R.; Fernando, W.; Holbein, B.E.; Hoskin, D.W. DIBI, a novel 3-hydroxypyridin-4-one chelator iron-binding polymer, inhibits breast cancer cell growth and functions as a chemosensitizer by promoting S-phase DNA damage. Biometals Int. J. Role Met. Ions Biol. Biochem. Med. 2019, 32, 909–921. [Google Scholar] [CrossRef]
- Nocera, F.P.; Iovane, G.; De Martino, L.; Holbein, B.E. Antimicrobial Activity of the Iron-Chelator, DIBI, against Multidrug-Resistant Canine Methicillin-Susceptible Staphylococcus pseudintermedius: A Preliminary Study of Four Clinical Strains. Pathogens 2022, 11, 656. [Google Scholar] [CrossRef]
- Islam, S.; Jarosch, S.; Zhou, J.; Parquet Mdel, C.; Toguri, J.T.; Colp, P.; Holbein, B.E.; Lehmann, C. Anti-inflammatory and anti-bacterial effects of iron chelation in experimental sepsis. J. Surg. Res. 2016, 200, 266–273. [Google Scholar] [CrossRef]
- Lehmann, C.; Alizadeh-Tabrizi, N.; Hall, S.; Faridi, S.; Euodia, I.; Holbein, B.; Zhou, J.; Chappe, V. Anti-Inflammatory Effects of the Iron Chelator, DIBI, in Experimental Acute Lung Injury. Molecules 2022, 27, 4036. [Google Scholar] [CrossRef]
- Shannahan, J.H.; Ghio, A.J.; Schladweiler, M.C.; McGee, J.K.; Richards, J.H.; Gavett, S.H.; Kodavanti, U.P. The role of iron in Libby amphibole-induced acute lung injury and inflammation. Inhal. Toxicol. 2011, 23, 313–323. [Google Scholar] [CrossRef]
- Hybertson, B.M.; Connelly, K.G.; Buser, R.T.; Repine, J.E. Ferritin and desferrioxamine attenuate xanthine oxidase-dependent leak in isolated perfused rat lungs. Inflammation 2002, 26, 153–159. [Google Scholar] [CrossRef]
- Kostopanagiotou, G.G.; Kalimeris, K.A.; Arkadopoulos, N.P.; Pafiti, A.; Panagopoulos, D.; Smyrniotis, V.; Vlahakos, D.; Routsi, C.; Lekka, M.E.; Nakos, G. Desferrioxamine attenuates minor lung injury following surgical acute liver failure. Eur. Respir. J. 2009, 33, 1429–1436. [Google Scholar] [CrossRef] [Green Version]
- Ritter, C.; da Cunha, A.A.; Echer, I.C.; Andrades, M.; Reinke, A.; Lucchiari, N.; Rocha, J.; Streck, E.L.; Menna-Barreto, S.; Moreira, J.C.; et al. Effects of N-acetylcysteine plus deferoxamine in lipopolysaccharide-induced acute lung injury in the rat. Crit. Care Med. 2006, 34, 471–477. [Google Scholar] [CrossRef]
- Baldwin, S.R.; Simon, R.H.; Boxer, L.A.; Till, G.O.; Kunkel, R.G. Attenuation by 2,3-dihydroxybenzoic acid of acute lung injury induced by cobra venom factor in the rat. Am. Rev. Respir. Dis. 1985, 132, 1288–1293. [Google Scholar] [CrossRef]
- Sharpe, M.D.; Mustard, R.A.; Finley, R.R.; Rutledge, F.S.; Sibbald, W.J. Failure of therapy with 2,3-dihydroxybenzoic acid to modify the course of sepsis-induced lung injury. J. Appl. Physiol. (Bethesda MD 1985) 1990, 69, 1893–1902. [Google Scholar] [CrossRef] [PubMed]
- Kono, M.; Matsuhiroya, S.; Obuchi, A.; Takahashi, T.; Imoto, S.; Kawano, S.; Saigo, K. Deferasirox, an iron-chelating agent, alleviates acute lung inflammation by inhibiting neutrophil activation and extracellular trap formation. J. Int. Med. Res. 2020, 48, 300060520951015. [Google Scholar] [CrossRef] [PubMed]
- Kono, M.; Saigo, K.; Yamamoto, S.; Shirai, K.; Iwamoto, S.; Uematsu, T.; Takahashi, T.; Imoto, S.; Hashimoto, M.; Minami, Y.; et al. Iron-chelating agent, deferasirox, inhibits neutrophil activation and extracellular trap formation. Clin. Exp. Pharmacol. Physiol. 2016, 43, 915–920. [Google Scholar] [CrossRef] [PubMed]
- Frank, L.; McLaughlin, G.E. Protection against acute and chronic hyperoxic inhibition of neonatal rat lung development with the 21-aminosteroid drug U74389F. Pediatr. Res. 1993, 33, 632–638. [Google Scholar] [CrossRef] [Green Version]
- Dallessio, J.J.; McLaughlin, G.E.; Frank, L. Reduction of bleomycin-induced acute DNA injury in the rat lung by the 21-aminosteroid, U-74389G. Crit. Care Med. 1997, 25, 652–656. [Google Scholar] [CrossRef]
- Crichton, R.R.; Ward, R.J.; Hider, R.C. The Efficacy of Iron Chelators for Removing Iron from Specific Brain Regions and the Pituitary-Ironing out the Brain. Pharmaceuticals 2019, 12, 138. [Google Scholar] [CrossRef] [Green Version]
- Bareggi, S.R.; Cornelli, U. Clioquinol: Review of its mechanisms of action and clinical uses in neurodegenerative disorders. CNS Neurosci. Ther. 2012, 18, 41–46. [Google Scholar] [CrossRef]
- Liu, Z.; Purro, M.; Qiao, J.; Xiong, M.P. Multifunctional Polymeric Micelles for Combining Chelation and Detection of Iron in Living Cells. Adv. Healthc. Mater. 2017, 6, 1700162. [Google Scholar] [CrossRef] [Green Version]
- You, L.; Wang, J.; Liu, T.; Zhang, Y.; Han, X.; Wang, T.; Guo, S.; Dong, T.; Xu, J.; Anderson, G.J.; et al. Targeted Brain Delivery of Rabies Virus Glycoprotein 29-Modified Deferoxamine-Loaded Nanoparticles Reverses Functional Deficits in Parkinsonian Mice. ACS Nano 2018, 12, 4123–4139. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Model | Iron Chelators | Readout | Outcome | Refs |
|---|---|---|---|---|
| RAW264.7 cells | DFP | Lipocalin-2 | IL-6, TNF-α, iNOS ↓ Oxidative stress ↓ | [62] |
| Mice | DIBI | Leukocyte adhesion Inflammatory mediators | Adhering leukocytes ↓ CXCL-2, IL-6 ↓ | [95] |
| BEAS-2B cells | DFO | ROS | ROS generation ↓ | [96] |
| Rats | DFO | TBARs | Lipid peroxidation ↓ | [97] |
| Pigs | DFO | PLA2 activity | Alveolar collapse ↓ | [98] |
| Rats | DFO | Oxidative stress | Oxidative damage ↓ | [99] |
| Rats | 2,3-DHB | Phospholipids | Peroxidation ↓ | [100] |
| Sheep | 2,3-DHB | Pulmonary response | Pulmonary arterial pressures ↓ | [101] |
| Mice | DFX | Neutrophil activation | Neutrophil invasion ↓ | [102] |
| Neutrophils | DFX | Neutrophils | ROS production ↓ NET formation ↓ | [103] |
| Rats | 21-aminosteroid | Elastin deposition DNA | Restructuring of newborn lung DNA injury ↓ | [104] [105] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Zhou, J.; Holbein, B.E.; Lehmann, C. Iron Chelation as a Potential Therapeutic Approach in Acute Lung Injury. Life 2023, 13, 1659. https://doi.org/10.3390/life13081659
Zhang X, Zhou J, Holbein BE, Lehmann C. Iron Chelation as a Potential Therapeutic Approach in Acute Lung Injury. Life. 2023; 13(8):1659. https://doi.org/10.3390/life13081659
Chicago/Turabian StyleZhang, Xiyang, Juan Zhou, Bruce E. Holbein, and Christian Lehmann. 2023. "Iron Chelation as a Potential Therapeutic Approach in Acute Lung Injury" Life 13, no. 8: 1659. https://doi.org/10.3390/life13081659