
Clonal Hematopoiesis of Indeterminate Potential and Cardiovascular Risk in Patients with Chronic Kidney Disease without Previous Cardiac Pathology
 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
3. Results
3.1. Description of the Sample and Clinical Events
3.2. CHIP’s Relationship with Coronary Calcification
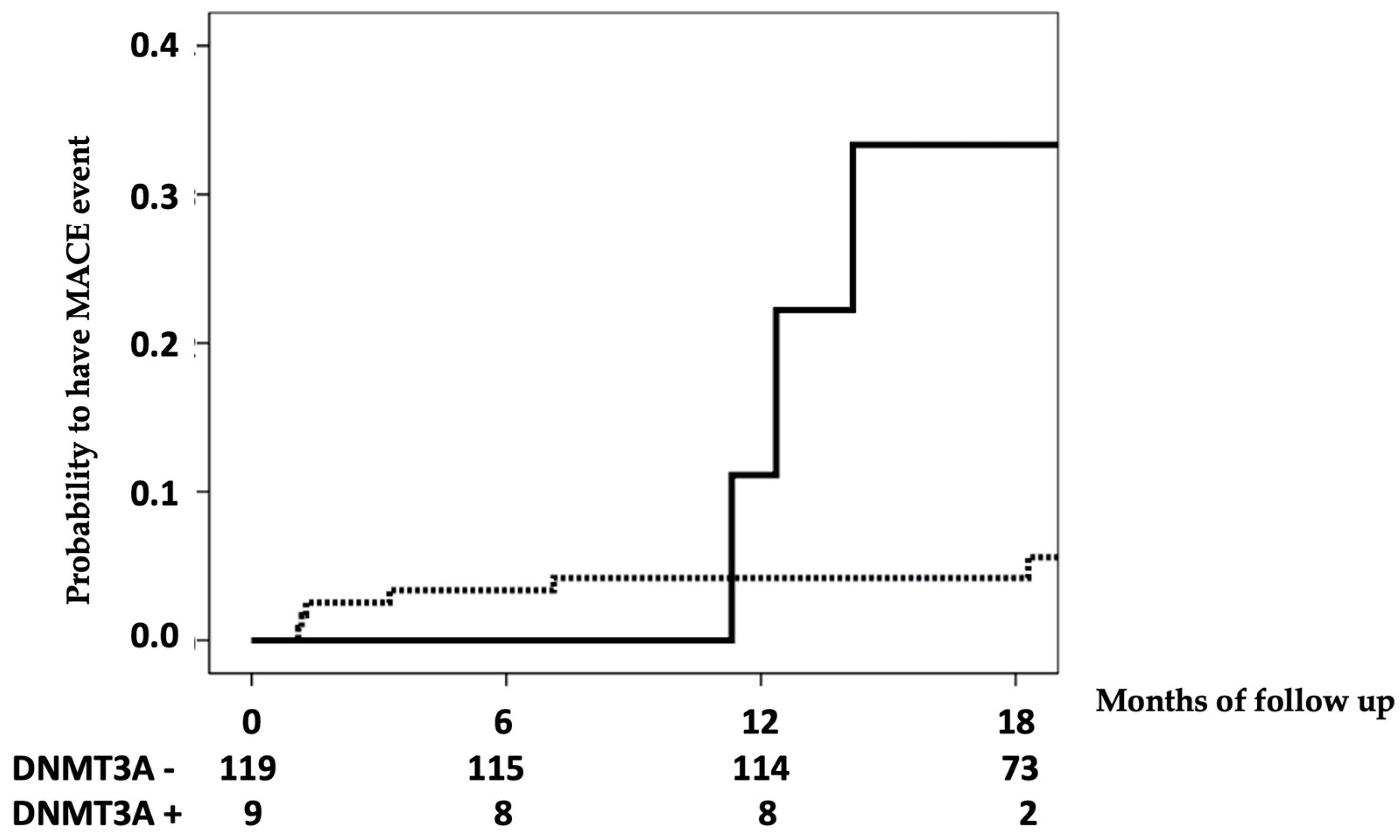
3.3. CHIP’s Relationship with Cardiovascular Events
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- GBD Chronic Kidney Disease Collaboration. Global, regional, and national burden of chronic kidney disease, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2020, 395, 709–733. [Google Scholar] [CrossRef]
- Díez, J.; Ortiz, A. The need for a cardionephrology subspecialty. Clin. Kidney J. 2021, 14, 1491–1494. [Google Scholar] [CrossRef] [PubMed]
- Sarnak, M.J.; Amann, K.; Bangalore, S.; Cavalcante, J.L.; Charytan, D.M.; Craig, J.C.; Gill, J.S.; Hlatky, M.A.; Jardine, A.G.; Landmesser, U.; et al. Conference Participants. Chronic Kidney Disease and Coronary Artery Disease: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2019, 74, 1823–1838. [Google Scholar] [CrossRef] [PubMed]
- Zawada, A.M.; Rogacev, K.S.; Heine, G.H. Clinical relevance of epigenetic dysregulation in chronic kidney disease-associated cardiovascular disease. Nephrol. Dial. Transplant. 2013, 28, 1663–1671. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.C.; Lu, C.L.; Yuan, T.H.; Liao, M.T.; Chao, C.T.; Lu, K.C. The Epigenetic Landscape of Vascular Calcification: An Integrative Perspective. Int. J. Mol. Sci. 2020, 21, 980. [Google Scholar] [CrossRef]
- Jaiswal, S.; Libby, P. Clonal haematopoiesis: Connecting ageing and inflammation in cardiovascular disease. Nat. Rev. Cardiol. 2020, 17, 137–144. [Google Scholar] [CrossRef]
- Asada, S.; Kitamura, T. Clonal hematopoiesis and associated diseases: A review of recent findings. Cancer Sci. 2021, 112, 3962–3971. [Google Scholar] [CrossRef]
- Florez, M.A.; Tran, B.T.; Wathan, T.K.; De Gregori, J.; Pietras, E.M.; King, K.Y. Clonal hematopoiesis: Mutation-specific adaptation to environmental change. Cell Stem Cell 2022, 29, 882–904. [Google Scholar] [CrossRef]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef]
- Dawoud, A.A.Z.; Gilbert, R.D.; Tapper, W.J.; Cross, N.C.P. Clonal myelopoiesis promotes adverse outcomes in chronic kidney disease. Leukemia 2022, 36, 507–515. [Google Scholar] [CrossRef]
- Vlasschaert, C.; McNaughton, A.J.M.; Chong, M.; Cook, E.K.; Hopman, W.; Kestenbaum, B.; Robinson-Cohen, C.; Garland, J.; Moran, S.M.; Paré, G.; et al. Association of Clonal Hematopoiesis of Indeterminate Potential with Worse Kidney Function and Anemia in Two Cohorts of Patients with Advanced Chronic Kidney Disease. J. Am. Soc. Nephrol. 2022, 33, 985–995. [Google Scholar] [CrossRef] [PubMed]
- Denicolò, S.; Vogi, V.; Keller, F.; Thöni, S.; Eder, S.; Heerspink, H.J.L.; Rosivall, L.; Wiecek, A.; Mark, P.B.; Perco, P.; et al. Clonal Hematopoiesis of Indeterminate Potential and Diabetic Kidney Disease: A Nested Case-Control Study. Kidney Int. Rep. 2022, 7, 876–888. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Vervloet, M.; Cozzolino, M. Vascular calcification in chronic kidney disease: Different bricks in the wall? Kidney Int. 2017, 91, 808–817. [Google Scholar] [CrossRef] [PubMed]
- Budoff, M.J.; Young, R.; Burke, G.; Jeffrey Carr, J.; Detrano, R.C.; Folsom, A.R.; Kronmal, R.; Lima, J.A.C.; Liu, K.J.; McClelland, R.L.; et al. Ten-year association of coronary artery calcium with atherosclerotic cardiovascular disease (ASCVD) events: The multi-ethnic study of atherosclerosis (MESA). Eur. Heart J. 2018, 39, 2401–2408. [Google Scholar] [CrossRef] [PubMed]
- Peng, A.W.; Mirbolouk, M.; Orimoloye, O.A.; Osei, A.D.; Dardari, Z.; Dzaye, O.; Budoff, M.J.; Shaw, L.; Miedema, M.D.; Rumberger, J.; et al. Long-Term All-Cause and Cause-Specific Mortality in Asymptomatic Patients With CAC ≥1,000: Results From the CAC Consortium. JACC Cardiovasc. Imaging 2020, 13, 83–93. [Google Scholar] [CrossRef]
- Abuzaid, A.; Saad, M.; Addoumieh, A.; Ha, L.D.; Elbadawi, A.; Mahmoud, A.N.; Elgendy, A.; Abdelaziz, H.K.; Barakat, A.F.; Mentias, A.; et al. Coronary artery calcium score and risk of cardiovascular events without established coronary artery disease: A systemic review and meta-analysis. Coron. Artery Dis. 2021, 32, 317–328. [Google Scholar] [CrossRef]
- Lo-Kioeng-Shioe, M.S.; Rijlaarsdam-Hermsen, D.; van Domburg, R.T.; Hadamitzky, M.; Lima, J.A.C.; Hoeks, S.E.; Deckers, J.W. Prognostic value of coronary artery calcium score in symptomatic individuals: A meta-analysis of 34,000 subjects. Int. J. Cardiol. 2020, 299, 56–62. [Google Scholar] [CrossRef]
- Peters, S.A.; den Ruijter, H.M.; Bots, M.L.; Moons, K.G. Improvements in risk stratification for the occurrence of cardiovascular disease by imaging subclinical atherosclerosis: A systematic review. Heart 2012, 98, 177–184. [Google Scholar] [CrossRef]
- Lamarche, M.C.; Hopman, W.M.; Garland, J.S.; White, C.A.; Holden, R.M. Relationship of coronary artery calcification with renal function decline and mortality in predialysis chronic kidney disease patients. Nephrol. Dial. Transplant. 2019, 34, 1715–1722. [Google Scholar] [CrossRef]
- Kouis, P.; Kousios, A.; Kanari, A.; Kleopa, D.; Papatheodorou, S.I.; Panayiotou, A.G. Association of non-invasive measures of subclinical atherosclerosis and arterial stiffness with mortality and major cardiovascular events in chronic kidney disease: Systematic review and meta-analysis of cohort studies. Clin. Kidney J. 2019, 13, 842–854. [Google Scholar] [CrossRef] [PubMed]
- Go, A.S.; Chertow, G.M.; Fan, D.; McCulloch, C.E.; Hsu, C.Y. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N. Engl. J. Med. 2004, 351, 1296–1305. [Google Scholar] [CrossRef] [PubMed]
- Ford, I.; Shah, A.S.; Zhang, R.; McAllister, D.A.; Strachan, F.E.; Caslake, M.; Newby, D.E.; Packard, C.J.; Mills, N.L. High-Sensitivity Cardiac Troponin, Statin Therapy, and Risk of Coronary Heart Disease. J. Am. Coll. Cardiol. 2016, 68, 2719–2728. [Google Scholar] [CrossRef] [PubMed]
- Kistorp, C.; Raymond, I.; Pedersen, F.; Gustafsson, F.; Faber, J.; Hildebrandt, P. N-terminal pro-brain natriuretic peptide, C-reactive protein, and urinary albumin levels as predictors of mortality and cardiovascular events in older adults. JAMA 2005, 293, 1609–1616. [Google Scholar] [CrossRef]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef]
- David, C.; Duployez, N.; Eloy, P.; Belhadi, D.; Chezel, J.; Le Guern, V.; Laouénan, C.; Fenwarth, L.; Rouzaud, D.; Mathian, A.; et al. Clonal hematopoiesis of indeterminate potential and cardiovascular events in systemic lupus erythematosus (HEMATOPLUS study). Rheumatology 2022, 61, 4355–4363. [Google Scholar] [CrossRef]
- Marco, M.P.; Craver, L.; Betriu, A.; Belart, M.; Fibla, J.; Fernández, E. Higher impact of mineral metabolism on cardiovascular mortality in a European hemodialysis population. Kidney Int. 2003, 63, S111–S114. [Google Scholar] [CrossRef]
- Björkegren, J.L.M.; Lusis, A.J. Atherosclerosis: Recent developments. Cell 2022, 185, 1630–1645. [Google Scholar] [CrossRef]
- Fuster, J.J.; MacLauchlan, S.; Zuriaga, M.A.; Polackal, M.N.; Ostriker, A.C.; Chakraborty, R.; Wu, C.L.; Sano, S.; Muralidharan, S.; Rius, C.; et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 2017, 355, 842–847. [Google Scholar] [CrossRef]
- Sano, S.; Oshima, K.; Wang, Y.; Katanasaka, Y.; Sano, M.; Walsh, K. CRISPR-Mediated Gene Editing to Assess the Roles of Tet2 and Dnmt3a in Clonal Hematopoiesis and Cardiovascular Disease. Circ. Res. 2018, 123, 335–341. [Google Scholar] [CrossRef]
- Cobo, I.; Tanaka, T.; Glass, C.K.; Yeang, C. Clonal hematopoiesis driven by DNMT3A and TET2 mutations: Role in monocyte and macrophage biology and atherosclerotic cardiovascular disease. Curr. Opin. Hematol. 2022, 29, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Khetarpal, S.A.; Qamar, A.; Bick, A.G.; Fuster, J.J.; Kathiresan, S.; Jaiswal, S.; Natarajan, P. Clonal Hematopoiesis of Indeterminate Potential Reshapes Age-Related CVD: JACC Review Topic of the Week. J. Am. Coll. Cardiol. 2019, 74, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Uddin, M.D.M.; Nguyen, N.Q.H.; Yu, B.; Brody, J.A.; Pampana, A.; Nakao, T.; Fornage, M.; Bressler, J.; Sotoodehnia, N.; Weinstock, J.S.; et al. Clonal hematopoiesis of indeterminate potential, DNA methylation, and risk for coronary artery disease. Nat. Commun. 2022, 13, 5350. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Total | No CHIP (n = 81) | CHIP (n = 47) | p | |
|---|---|---|---|---|
| Age (years) | 61 ± 7 | 61 ± 7 | 61 ± 7 | 0.949 |
| Sex—male | 54.7% | 53.8% | 55.3% | 0.874 |
| BMI (kg/m2) | 28.3 ± 5.3 | 28.5 ± 5.5 | 28.0 ± 5.1 | 0.586 |
| CKD stage | 0.565 | |||
| 1 | 12.5% | 9.9% | 17.0% | |
| 2 | 13.3% | 12.3% | 14.9% | |
| 3 | 21.9% | 21.0% | 23.4% | |
| 4 | 21.1% | 25.9% | 12.8% | |
| 5 | 31.3% | 30.9% | 31.9% | |
| GFR (mL/min/1.73 m2) | 38.1 ± 30.4 | 35.1 ± 29.2 | 43.2 ± 32.1 | 0.145 |
| Etiology of CKD | 0.425 | |||
| Glomerular | 22.7% | 21.0% | 25.5% | |
| Diabetic | 17.2% | 18.5% | 14.9% | |
| Vascular | 14.8% | 13.6% | 17.0% | |
| Hereditary | 9.4% | 9.9% | 8.5% | |
| Interstitial | 8.6% | 7.4% | 10.6% | |
| Systemic | 6.3% | 3.7% | 10.6% | |
| Unknown | 21.0% | 25.9% | 12.8% | |
| Diabetes mellitus | 36.7% | 43.2% | 25.5% | 0.045 |
| Nonsmoker | 31.3% | 32.1% | 29.8% | 0.786 |
| Troponin (ng/L) | 15.9 ± 26.1 | 18.3 ± 31.2 | 11.7 ± 13.0 | 0.098 |
| NT-ProBNP (pg/mL) | 2306.8 ± 6340.2 | 2793 ± 7245 | 1468 ± 2306 | 0.197 |
| LDL cholesterol (mg/dl) | 93.7 ± 38.6 | 92.5 ± 35.2 | 95.8 ± 44.2 | 0.645 |
| HDL cholesterol (mg/dl) | 48.3 ± 16.4 | 45.9 ± 12.3 | 52.5 ± 21.2 | 0.057 |
| Lipoprotein A (mg/dl) | 40.2 ± 50.3 | 43.0 ± 54.1 | 35.6 ± 43.3 | 0.427 |
| Calcium (mg/dl) | 9.1 ± 0.7 | 9.0 ± 0.7 | 9.1 ± 0.6 | 0.442 |
| Phosphate (mg/dl) | 4.1 ± 1.3 | 4.1 ± 1.3 | 4.0 ± 1.3 | 0.603 |
| PTH (pg/mL) | 216.6 ± 255.7 | 238.3 ± 275.0 | 179.3 ± 216.0 | 0.210 |
| Vitamin D (ng/mL) | 20.8 ± 10.3 | 19.7 ± 10.4 | 22.7 ± 10.0 | 0.120 |
| Calcification of anterior descendent artery * | 222.7 ± 382.8 | 270.8 ± 427.7 | 140.6 ± 276.2 | 0.039 |
| Global coronary calcification * | 543.6 ± 1250.6 | 695.3 ± 1451.4 | 285.2 ± 637.8 | 0.033 |
| Agatston ≥ 400 * | 30.7% | 38.8% | 17.0% | 0.010 |
| MACE | 7% | 7.4% | 6.4% | 0.827 |
| Global mortality | 7% | 8.6% | 4.3% | 0.349 |
| CV mortality | 2.3% | 3.7% | 0.0% | 0.182 |
| CHIP | 36.7% | - | - | - |
| One gene mutation | - | - | - | |
| TET2 | 21.1% | |||
| DNMT3A | 7.0% | |||
| ASXL1 | 0.8% | |||
| JAK2 | 1.6% | |||
| MPL | 12.5% | |||
| Two gene mutations | 6.3% | - | - | - |
| Agatston < 400 (n = 88) | Agatston ≥ 400 (n = 39) | p | |
|---|---|---|---|
| Age (years) | 60 ± 7 | 62 ± 7 | 0.071 |
| Sex—male | 64.1% | 50.0% | 0.455 |
| BMI (kg/m2) | 28.0 ± 5.4 | 29.0 ± 5.3 | 0.351 |
| CKD stage 5 | 25.0% | 43.6% | 0.036 |
| GFR (mL/min/1.73 m2) | 42.8 ± 31.1 | 28.2 ± 26.4 | 0.008 |
| Diabetes mellitus | 23.9% | 66.7% | <0.001 |
| Smoker | 27.3% | 41.0% | 0.124 |
| Troponin (ng/l) | 12.0 ± 19.3 | 25.0 ± 36.1 | 0.040 |
| NT-ProBNP (pg/mL) | 1049.7 ± 2110.6 | 5157.2 ± 10,589.7 | 0.021 |
| LDL cholesterol (mg/dl) | 99.3 ± 41.4 | 81.5 ± 28.7 | 0.006 |
| HDL cholesterol (mg/dl) | 50.0 ± 17.4 | 44.6 ± 13.3 | 0.060 |
| Lipoprotein A (mg/dl) | 40.1 ± 53.6 | 41.1 ± 43.3 | 0.913 |
| Calcium (mg/dl) | 9.1 ± 0.7 | 9.1 ± 0.7 | 0.755 |
| Phosphate (mg/dl) | 3.8 ± 1.0 | 4.7 ± 1.7 | 0.004 |
| PTH (pg/mL) | 187.1 ± 187 | 258.6 ± 332.5 | 0.125 |
| Vitamin D (ng/mL) | 21.8 ± 9.4 | 18.9 ± 12.1 | 0.144 |
| CHIP | 44.3% | 20.5% | 0.010 |
| Gene mutation | |||
| TET2 | 27.3% | 7.7% | 0.013 |
| DNMT3A | 6.8% | 7.7% | 0.859 |
| MPL | 15.9% | 5.1% | 0.091 |
| HR | 95%IC | p | |
|---|---|---|---|
| Age (years) | 1.051 | 0.950–1.163 | 0.335 |
| Sex—male | 0.216 | 0.045–1.043 | 0.056 |
| BMI (kg/m2) | 1.075 | 0.949–1.217 | 0.254 |
| CKD stage 5 | 8.469 | 1.756–40.834 | 0.008 |
| GFR (mL/min/1.73 m2) | 0.916 | 0.842–0.997 | 0.043 |
| Diabetes mellitus | 3.532 | 0.882–14.143 | 0.075 |
| Smoker | 2.796 | 0.750–10.415 | 0.125 |
| Troponin (ng/l) | 1.000 | 0.977–1.024 | 0.977 |
| NT-ProBNP (pg/mL) | 1.090 | 1.050–1.132 | <0.001 |
| LDL cholesterol (mg/dl) | 0.989 | 0.969–1.010 | 0.311 |
| HDL cholesterol (mg/dl) | 0.908 | 0.836–0.987 | 0.024 |
| Lipoprotein A (mg/dl) | 1.002 | 0.991–1.014 | 0.717 |
| Calcium (mg/dl) | 0.451 | 0.194–1.048 | 0.064 |
| Phosphate (mg/dl) | 1.741 | 1.284–2.361 | <0.001 |
| PTH (pg/mL) | 1.002 | 1.000–1.003 | 0.014 |
| Vitamin D (ng/mL) | 0.961 | 0.893–1.034 | 0.290 |
| Calcification of anterior descendent artery * | 1.002 | 1.001–1.003 | <0.001 |
| Global coronary calcification * | 1.001 | 1.001–1.001 | <0.001 |
| Agatston ≥ 400 * | 8.223 | 1.012–66.842 | 0.049 |
| CHIP | 0.848 | 0.212–3.393 | 0.816 |
| Gene mutation | |||
| TET2 | 0.034 | 0.000–30.509 | 0.330 |
| DNMT3A | 7.343 | 1.817–29.673 | 0.005 |
| MPL | 0.040 | 0.000–202.888 | 0.460 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kislikova, M.; Lopez, M.A.B.; Salinas, F.J.F.; Blanco, J.A.P.; Molina, M.P.G.-B.; Fernandez, A.A.; Haces, V.C.P.; Unzueta, M.T.G.; Hernández, A.B.; Millan, J.C.R.S.; et al. Clonal Hematopoiesis of Indeterminate Potential and Cardiovascular Risk in Patients with Chronic Kidney Disease without Previous Cardiac Pathology. Life 2023, 13, 1801. https://doi.org/10.3390/life13091801
Kislikova M, Lopez MAB, Salinas FJF, Blanco JAP, Molina MPG-B, Fernandez AA, Haces VCP, Unzueta MTG, Hernández AB, Millan JCRS, et al. Clonal Hematopoiesis of Indeterminate Potential and Cardiovascular Risk in Patients with Chronic Kidney Disease without Previous Cardiac Pathology. Life. 2023; 13(9):1801. https://doi.org/10.3390/life13091801
Chicago/Turabian StyleKislikova, Maria, Maria Ana Batlle Lopez, Francisco Javier Freire Salinas, José Antonio Parra Blanco, Maria Pilar García-Berbel Molina, Alejandro Aguilera Fernandez, Vicente Celestino Piñera Haces, Maria Teresa García Unzueta, Adalberto Benito Hernández, Juan Carlos Ruiz San Millan, and et al. 2023. "Clonal Hematopoiesis of Indeterminate Potential and Cardiovascular Risk in Patients with Chronic Kidney Disease without Previous Cardiac Pathology" Life 13, no. 9: 1801. https://doi.org/10.3390/life13091801