
Prediction of Amino Acid Substitutions in ABL1 Protein Leading to Tumor Drug Resistance Based on “Structure-Property” Relationship Classification Models

Abstract
:1. Introduction
2. Materials and Methods
2.1. ABL1 Protein
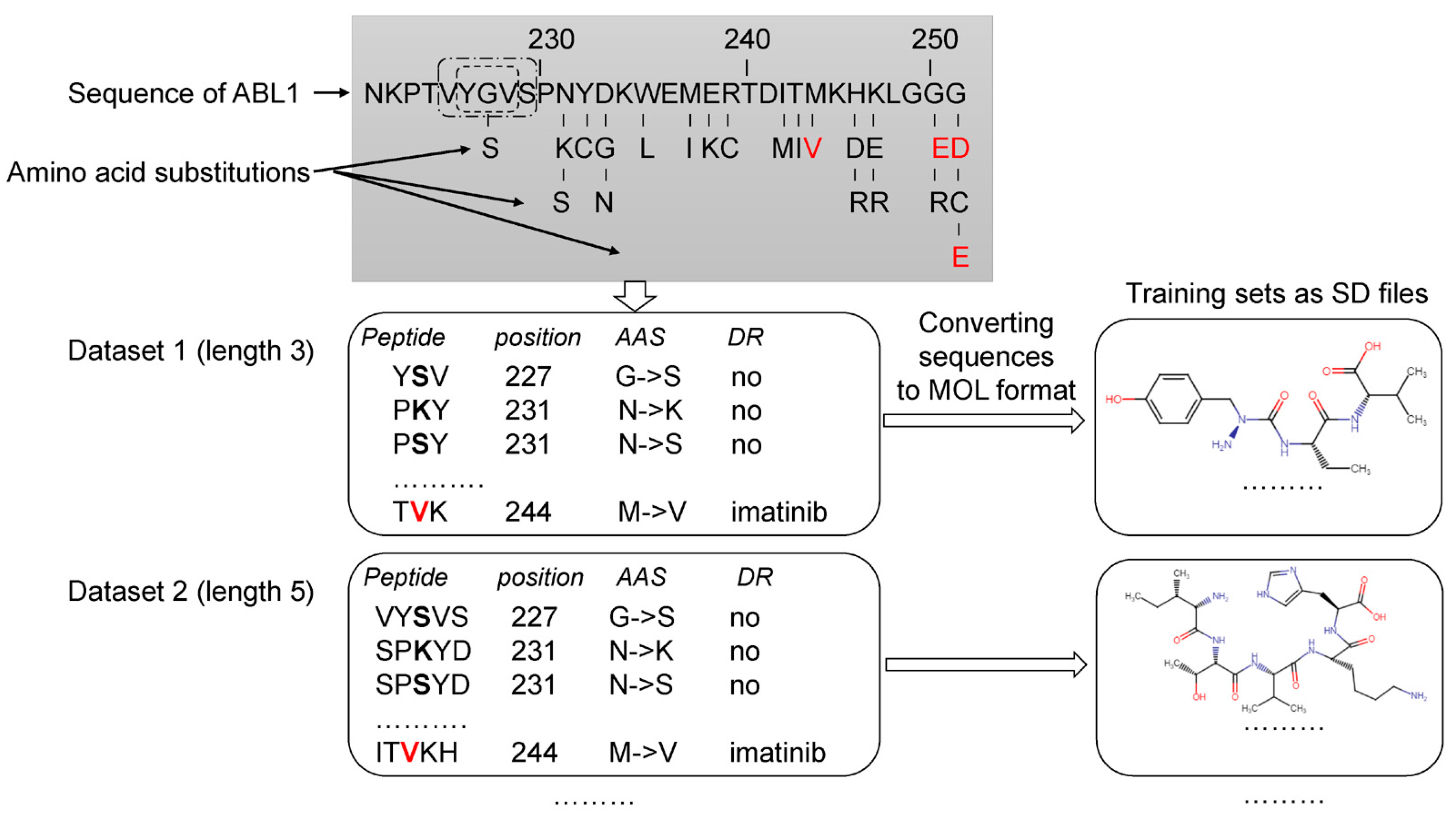
2.2. Datasets and Data Preparation
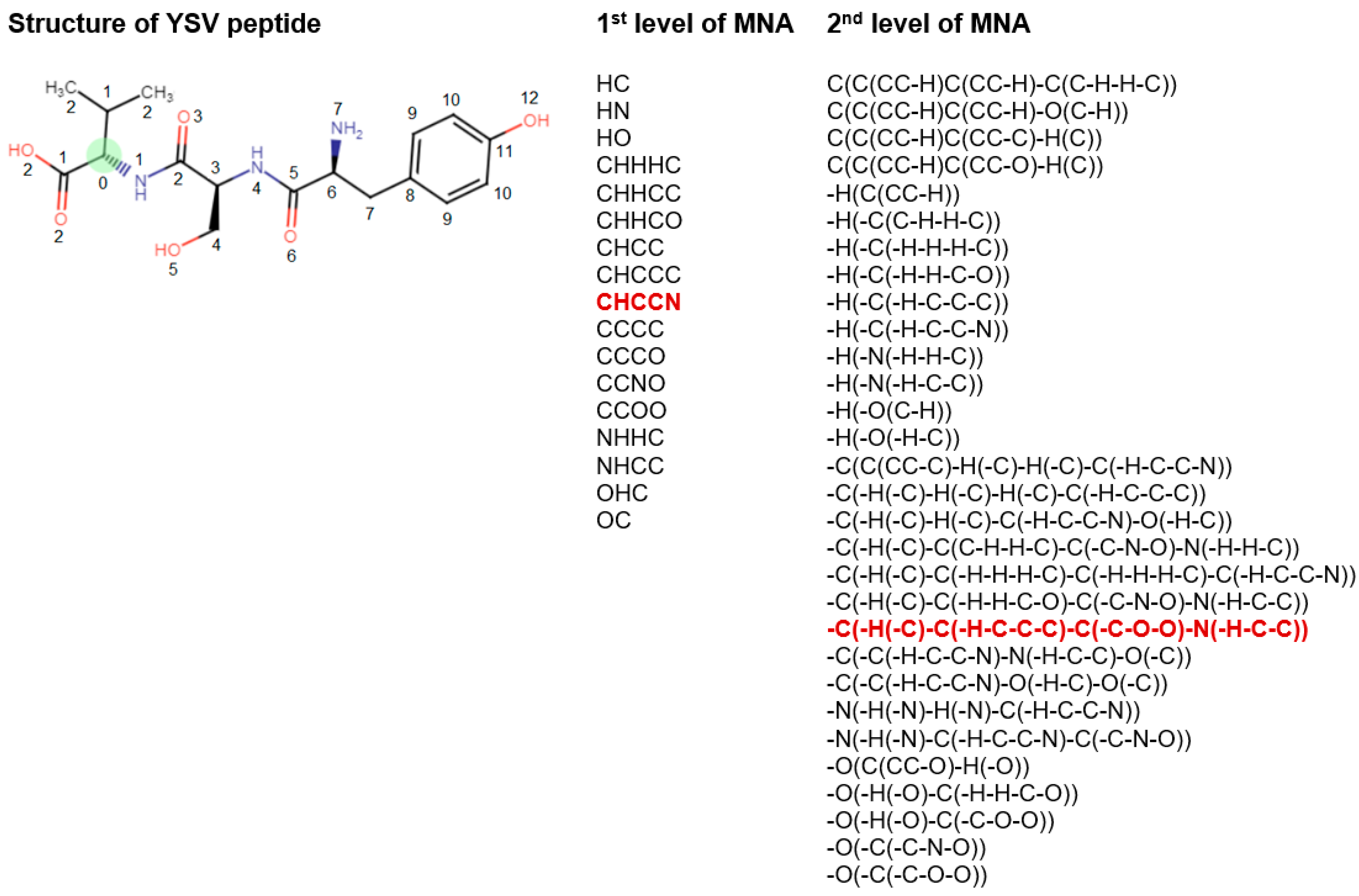
2.3. PASS Software and MNA Descriptors
- Zero-level MNA descriptor for each atom is the mark A of the atom itself;
- Any next-level MNA descriptor for the atom is the sub-structure notation A(D1D2…Di…), where Di is the previous-level MNA descriptor for i–th immediate neighbor of the atom A.
2.4. Approach Estimation
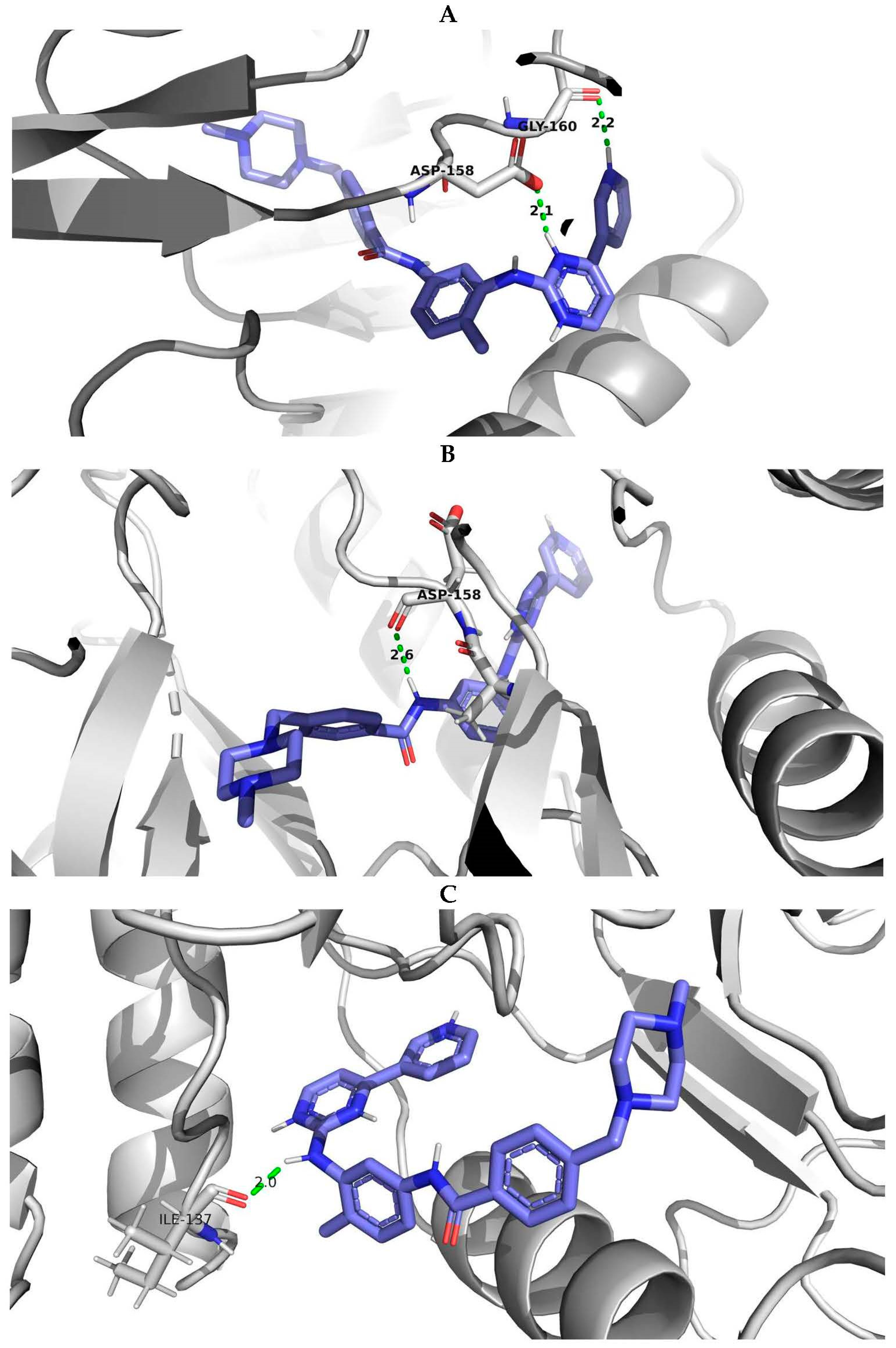
2.5. Molecular Modeling and Docking Procedure
- The structure exhibited high validation scores according to the wwPDB validation data, surpassing the parameters of most similar structures in the PDB database, indicating the quality of the X-ray diffraction investigation.
- The structure consisted of a single chain, facilitating the preparation process of the three-dimensional structure for docking procedures.
- The examined structure of the wild type of human ABL1 possessed a high crystallographic resolution of 1.53 Å ensuring enhanced precision during the docking simulations.
- The presence of the CHMFL-074 inhibitor within the model was situated in the same region of the protein as in other examples of ABL1 protein structures from the PDB, where the inhibitor imatinib was located. This enabled us to utilize the ligand coordinates of CHMFL-074 for modeling the docking of ABL1 with imatinib.”
3. Results
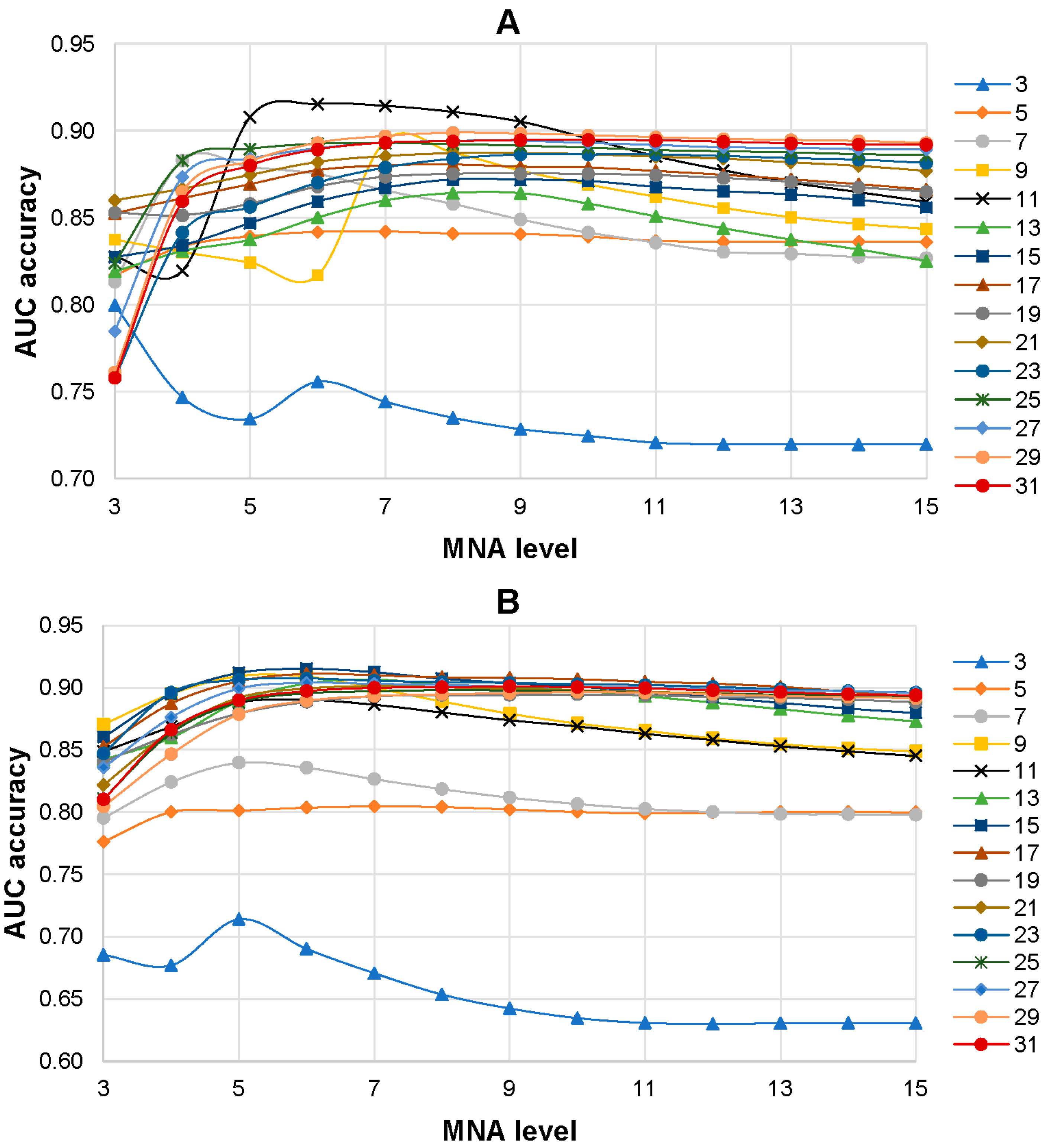
3.1. Selection of the Best SPR Models
3.2. Comparison Prediction Results of MultiPASS and SUSPECT-ABL
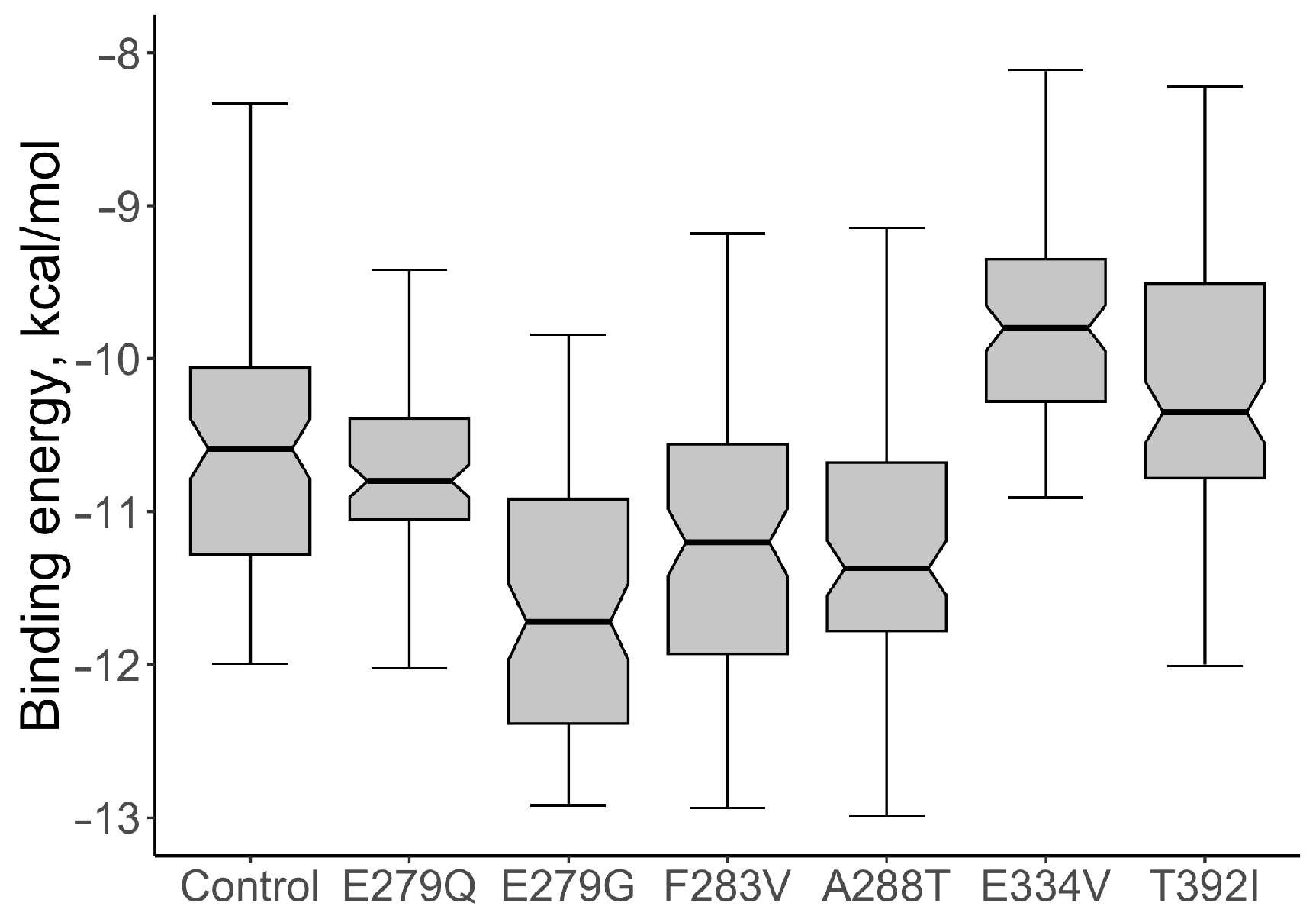
3.3. Estimation of Drug-Resistant AASs from gnomAD and dbSNP Databases
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Housman, G.; Byler, S.; Heerbogh, S.; Lapinska, K.; Longacre, M.; Snyder, N.; Sarkar, S. Drug Resistance in Cancer: An Overview. Cancers 2014, 6, 1769–1792. [Google Scholar] [CrossRef] [PubMed]
- Nussinov, R.; Tsai, C.J.; Jang, H. Anticancer drug resistance: An update and perspective. Drug Resist. Updates 2021, 59, 371–385. [Google Scholar] [CrossRef] [PubMed]
- Libbrecht, M.W.; Noble, W.S. Machine learning applications in genetics and genomics. Nat. Rev. Genet. 2015, 16, 321–332. [Google Scholar] [CrossRef]
- Cao, Z.W.; Han, L.Y.; Zheng, C.J.; Ji, Z.L.; Chen, X.; Lin, H.H.; Chen, Y.Z. Computer prediction of drug resistance mutations in proteins. Drug Discov. Today 2005, 10, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Park, H.; Yang, H.J.; Lee, S.; Lee, K.Y.; Kim, T.S.; Jung, J.; Shin, J.M. Cancer Drug Response Profile scan (CDRscan): A Deep Learning Model that Predicts Drug Effectiveness from Cancer Genomic Signature. Sci. Rep. 2018, 8, 3367–3373. [Google Scholar] [CrossRef]
- Choi, M.; Shi, J.; Zhu, Y.; Yang, R.; Cho, K.-H. Network dynamics-based cancer panel stratification for systemic prediction of an-ticancer drug response. Nat. Commun. 2017, 8, 1940. [Google Scholar] [CrossRef]
- Lin, J.J.; Choudhury, N.J.; Yoda, S.; Zhu, V.W.; Johnson, T.W.; Sakhtemani, R.; Dagogo-Jack, I.; Digumarthy, S.R.; Lee, C.; Do, A.; et al. Spectrum of Mechanisms of Resistance to Crizotinib and Lorlatinib in ROS1 Fusion-Positive Lung Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 2899–2909. [Google Scholar] [CrossRef]
- Wu, C.; Chen, X.; Chen, D.; Xia, Q.; Liu, Z.; Li, F.; Yan, Y.; Cai, Y. Insight into ponatinib resistance mechanisms in rhabdomyosarcoma caused by the mutations in FGFR4 tyrosine kinase using molecular modeling strategies. Int. J. Biol. Macromol. 2019, 135, 294–302. [Google Scholar] [CrossRef]
- Kim, P.; Li, H.; Wang, J.; Zhao, Z. Landscape of drug-resistance mutations in kinase regulatory hotspots. Brief. Bioinform. 2021, 22, bbaa108. [Google Scholar] [CrossRef]
- Hauser, K.; Negron, C.; Albanese, S.K.; Ray, S.; Steinbrecher, T.; Abel, R.; Chodera, J.D.; Wang, L. Predicting resistance of clinical Abl mutations to targeted kinase inhibitors using alchemical free-energy calculations. Commun. Biol. 2018, 1, 70. [Google Scholar] [CrossRef]
- Liu, J.; Pei, J.; Lai, L. A combined computational and experimental strategy identifies mutations conferring resistance to drugs targeting the BCR-ABL fusion protein. Commun. Biol. 2020, 3, 18. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Chen, Y.; Wen, Y.; Zhu, Z.; Li, M. PremPLI: A machine learning model for predicting the effects of missense mutations on protein-ligand interactions. Commun. Biol. 2021, 4, 1311. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Portelli, S.; Pat, M.; Rodrigues, C.H.M.; Nguyen, T.B.; Pires, D.E.V.; Ascher, D.B. Structure-guided machine learning prediction of drug resistance mutations in Abelson 1 kinase. Comput. Struct. Biotechnol. J. 2021, 19, 5381–5391. [Google Scholar] [CrossRef] [PubMed]
- Aldeghi, M.; Gapsys, V.; de Groot, B.L. Predicting Kinase Inhibitor Resistance: Physics-Based and Data-Driven Approaches. ACS Cent. Sci. 2019, 5, 1468–1474. [Google Scholar] [CrossRef]
- Karasev, D.A.; Savosina, P.I.; Sobolev, B.N.; Filimonov, D.A.; Lagunin, A.A. Application of molecular descriptors for recognition of phosphorylation sites in amino acid sequences. Biomeditsinskaia Khimiia 2017, 63, 423–427. [Google Scholar] [CrossRef]
- Zadorozhny, A.; Smirnov, A.; Filimonov, D.; Lagunin, A. Prediction of pathogenic single amino acid substitutions using molecular fragment descriptors. Bioinformatics 2023, 39, btad484. [Google Scholar] [CrossRef]
- Zadorozhny, A.D.; Rudik, A.V.; Filimonov, D.A.; Lagunin, A.A. SAV-Pred: A Freely Available Web Application for the Prediction of Pathogenic Amino Acid Substitutions for Monogenic Hereditary Diseases Studied in Newborn Screening. Int. J. Mol. Sci. 2023, 24, 2463. [Google Scholar] [CrossRef]
- Smirnov, A.S.; Rudik, A.V.; Filimonov, D.A.; Lagunin, A.A. TCR-Pred: A new web-application for prediction of epitope and MHC specificity for CDR3 TCR sequences using molecular fragment descriptors. Immunology 2023, 169, 447–453. [Google Scholar] [CrossRef]
- Jabbour, E.; Kantarjian, H. Chronic myeloid leukemia: 2020 update on diagnosis, therapy and monitoring. Am. J. Hematol. 2020, 95, 691–709. [Google Scholar] [CrossRef]
- Heisterkamp, N.; Stephenson, J.R.; Groffen, J.; Hansen, P.F.; de Klein, A.; Bartram, C.R.; Grosveld, G. Localization of the c-abl oncogene adjacent to a translocation break point in chronic myelocytic leukaemia. Nature 1983, 306, 239–242. [Google Scholar] [CrossRef]
- Baykal-Köse, S.; Acikgoz, E.; Yavuz, A.S.; Gönül Geyik, Ö.; Ateş, H.; Sezerman, O.U.; Özsan, G.H.; Yüce, Z. Adaptive phenotypic modulations lead to therapy resistance in chronic myeloid leukemia cells. PLoS ONE 2020, 15, e0229104. [Google Scholar] [CrossRef]
- Braun, T.P.; Eide, C.A.; Druker, B.J. Response and Resistance to BCR-ABL1-Targeted Therapies. Cancer Cell 2020, 37, 530–542. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.W.; Pritchard, J.R.; Leighow, S.M.; Aminov, B.I.; Beppu, L.; Kim, D.S.; Hodgson, J.G.; Rivera, V.M.; Loeb, L.A.; Radich, J.P. Single-Molecule Sequencing Reveals Patterns of Preexisting Drug Resistance That Suggest Treatment Strategies in Philadelphia-Positive Leukemias. Clin. Cancer Res. 2018, 24, 5321–5334. [Google Scholar] [CrossRef]
- COSMIC Database. Available online: https://cancer.sanger.ac.uk/cosmic (accessed on 19 September 2022).
- Lagunin, A.; Stepanchikova, A.; Filimonov, D.; Poroikov, V. PASS: Prediction of activity spectra for biologically active substances. Bioinformatics 2000, 16, 747–748. [Google Scholar] [CrossRef] [PubMed]
- Lagunin, A.; Ivanov, S.; Rudik, A.; Filimonov, D.; Poroikov, V. DIGEP-Pred: Web service for in silico pre-diction of drug-induced gene expression profiles based on structural formula. Bioinformatics 2013, 29, 2062–2063. [Google Scholar] [CrossRef]
- Rudik, A.; Dmitriev, A.; Lagunin, A.; Filimonov, D.; Poroikov, V. SOMP: Web server for in silico prediction of sites of metabolism for drug-like compounds. Bioinformatics 2015, 31, 2046–2048. [Google Scholar] [CrossRef]
- Lagunin, A.; Rudik, A.; Druzhilovsky, D.; Filimonov, D.; Poroikov, V.; Wren, J. ROSC-Pred: Web-service for rodent organ-specific carcinogenicity prediction. Bioinformatics 2018, 34, 710–712. [Google Scholar] [CrossRef] [PubMed]
- Lagunin, A.A.; Rudik, A.V.; Pogodin, P.V.; Savosina, P.I.; Tarasova, O.A.; Dmitriev, A.V.; Ivanov, S.M.; Biziukova, N.Y.; Druzhilovskiy, D.S.; Filimonov, D.A.; et al. CLC-Pred 2.0: A Freely Available Web Application for in Silico Prediction of Human Cell Line Cytotoxicity and Molecular Mechanisms of Action for Druglike Compounds. Int. J. Mol. Sci. 2023, 24, 1689. [Google Scholar] [CrossRef]
- Poroikov, V.V.; Filimonov, D.A.; Borodina, Y.V.; Lagunin, A.A.; Kos, A. Robustness of biological activity spectra predicting by computer program PASS for non-congeneric sets of chemical compounds. J. Chem. Inf. Comput. Sci. 2000, 40, 1349–1355. [Google Scholar] [CrossRef]
- Filimonov, D.A.; Lagunin, A.A.; Gloriozova, T.A.; Rudik, A.V.; Druzhilovskii, D.S.; Pogodin, P.V.; Poroikov, V.V. Prediction of the Biological Activity Spectra of Organic Compounds Using the Pass Online Web Resource. Chem. Heterocycl. Comp. 2014, 50, 444–457. [Google Scholar] [CrossRef]
- Song, Y.; DiMaio, F.; Wang, R.Y.; Kim, D.; Miles, C.; Brunette, T.; Thompson, J.; Baker, D. High-resolution comparative modeling with RosettaCM. Structure 2013, 21, 1735–1742. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Allaire, J.; Cheng, J.; Russell, K.; Schramm, S. RStudio: Integrated Development Environment for R. RStudio, PBC, Boston, MA. 2021. Available online: https://www.kdnuggets.com/2011/03/rstudio-ide-for-r.html (accessed on 20 April 2023).
- Soverini, S.; Mancini, M.; Bavaro, L.; Cavo, M.; Martinelli, G. Chronic myeloid leukemia: The paradigm of targeting oncogenic tyrosine kinase signaling and counteracting resistance for successful cancer therapy. Mol. Cancer 2018, 17, 49. [Google Scholar] [CrossRef] [PubMed]
- Reddy, E.P.; Aggarwal, A.K. The ins and outs of bcr-abl inhibition. Genes Cancer 2012, 3, 447–454. [Google Scholar] [CrossRef]
- Hochhaus, A.; Baccarani, M.; Silver, R.T.; Schiffer, C.; Apperley, J.F.; Cervantes, F.; Clark, R.E.; Cortes, J.E.; Deininger, M.W.; Guilhot, F.; et al. European LeukemiaNet 2020 recommendations for treating chronic myeloid leukemia. Leukemia 2020, 34, 966–984. [Google Scholar] [CrossRef]
- Barouch-Bentov, R.; Sauer, K. Mechanisms of drug resistance in kinases. Expert Opin. Investig. Drugs 2011, 20, 153–208. [Google Scholar] [CrossRef]
- Zhang, B.; Chen, Y.; Dai, P.; Yu, H.; Ma, J.; Chen, C.; Zhang, Y.; Guan, Y.; Chen, R.; Liu, T.; et al. Oncogenic mutations within the β3-αC loop of EGFR/ERBB2/BRAF/MAP2K1 predict response to therapies. Mol. Genet. Genom. Med. 2020, 8, e1395. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Drug Resistance AASs | Number of Not Drug Resistance AASs | Drug Resistance to | AUCLOO CV | AUC20-F CV |
|---|---|---|---|---|
| ABL1, isoform 1 | ||||
| 84 | 576 | imatinib | 0.851 | 0.846 |
| 3 | 657 | dasatinib | 0.980 | 0.980 |
| ABL1, isoform 2 | ||||
| 30 | 670 | imatinib | 0.909 | 0.907 |
| Software | TP | FP | TN | FN | Sen. | Sp. | Bal. Acc. |
|---|---|---|---|---|---|---|---|
| SUSPECT-ABL | 5 | 8 | 181 | 77 | 0.061 | 0.958 | 0.509 |
| MultiPASS, Pa > Pi | 78 | 102 | 87 | 4 | 0.951 | 0.460 | 0.706 |
| MultiPASS, Pa > 0.3 | 77 | 87 | 102 | 5 | 0.939 | 0.540 | 0.739 |
| MultiPASS, Pa > 0.4 | 76 | 64 | 125 | 6 | 0.927 | 0.661 | 0.794 |
| MultiPASS, Pa > 0.5 | 71 | 42 | 147 | 11 | 0.866 | 0.778 | 0.822 |
| MultiPASS, Pa > 0.6 | 66 | 32 | 157 | 16 | 0.805 | 0.831 | 0.818 |
| MultiPASS, Pa > 0.7 | 60 | 26 | 163 | 22 | 0.732 | 0.862 | 0.797 |
| MultiPASS, Pa > 0.8 | 42 | 18 | 171 | 40 | 0.512 | 0.905 | 0.708 |
| MultiPASS, Pa > 0.9 | 24 | 6 | 183 | 58 | 0.293 | 0.968 | 0.630 |
| AAS | Number of MNA Descriptors | Number of New MNA Descriptors | Pa | Pi | Drug Resistance |
|---|---|---|---|---|---|
| E279G | 839 | 21 | 0.743 | 0.042 | Imatinib |
| E279Q | 778 | 11 | 0.742 | 0.043 | Imatinib |
| T392I | 1002 | 17 | 0.671 | 0.055 | Imatinib |
| F283V | 635 | 1 | 0.671 | 0.055 | Imatinib |
| E334V | 693 | 1 | 0.603 | 0.064 | Imatinib |
| A288T | 674 | 10 | 0.575 | 0.068 | Imatinib |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhuravleva, S.I.; Zadorozhny, A.D.; Shilov, B.V.; Lagunin, A.A. Prediction of Amino Acid Substitutions in ABL1 Protein Leading to Tumor Drug Resistance Based on “Structure-Property” Relationship Classification Models. Life 2023, 13, 1807. https://doi.org/10.3390/life13091807
Zhuravleva SI, Zadorozhny AD, Shilov BV, Lagunin AA. Prediction of Amino Acid Substitutions in ABL1 Protein Leading to Tumor Drug Resistance Based on “Structure-Property” Relationship Classification Models. Life. 2023; 13(9):1807. https://doi.org/10.3390/life13091807
Chicago/Turabian StyleZhuravleva, Svetlana I., Anton D. Zadorozhny, Boris V. Shilov, and Alexey A. Lagunin. 2023. "Prediction of Amino Acid Substitutions in ABL1 Protein Leading to Tumor Drug Resistance Based on “Structure-Property” Relationship Classification Models" Life 13, no. 9: 1807. https://doi.org/10.3390/life13091807