
Simple and Effective Squash-PCR for Rapid Genotyping of Industrial Microalgae
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains
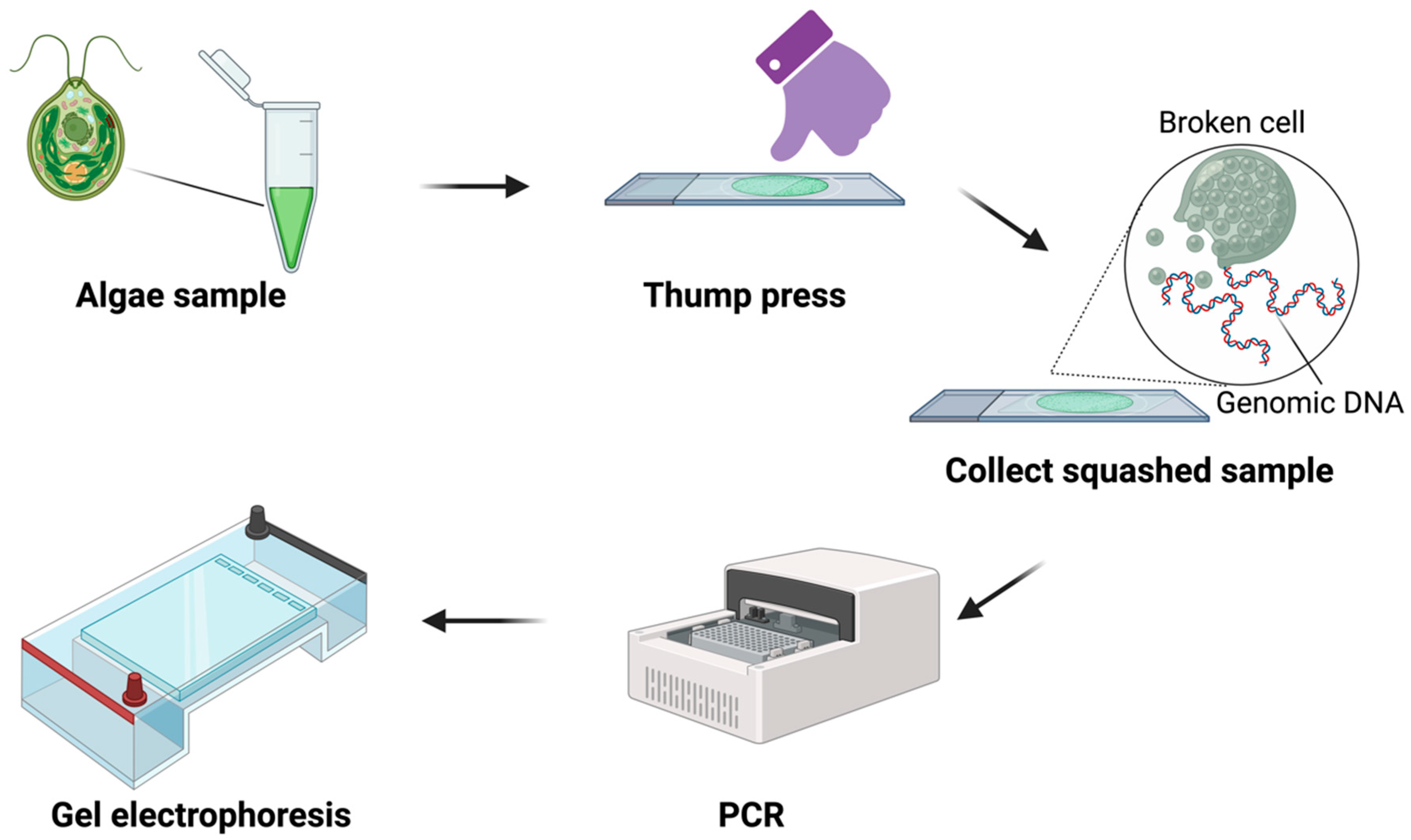
2.2. Squash Preparation
2.3. Genomic DNA Extraction
2.4. PCR Procedure
2.5. Primers for PCR
2.6. Microscopy
2.7. Cell Counting
3. Results
3.1. DNA Template Preparation via Squash Technique
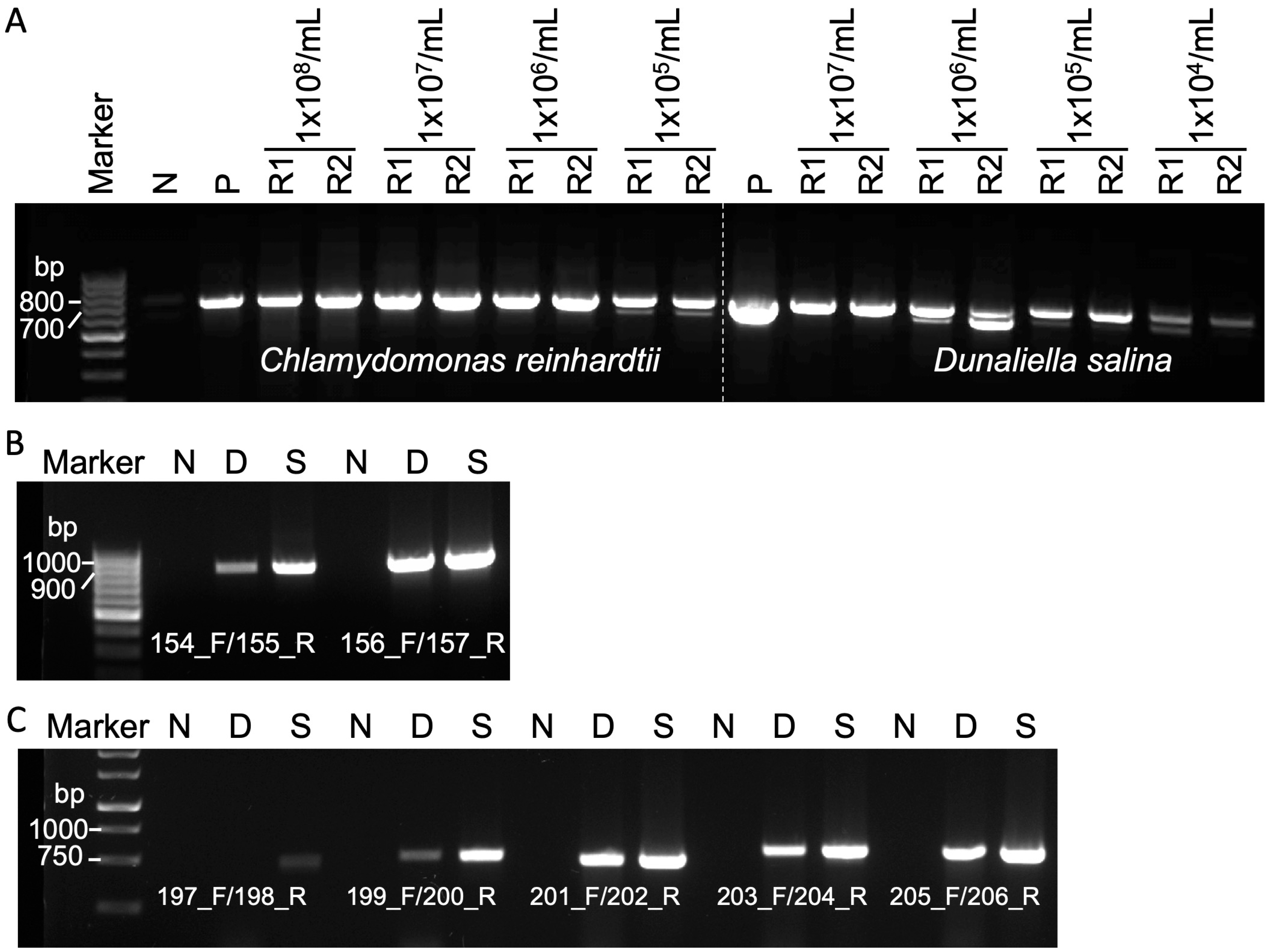
3.2. Efficacy of Squash-PCR in Diverse Microalgae Strains
3.3. Evaluation of Squash-PCR in Microalgae Using Different Cell Numbers and Primer Pairs
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dolganyuk, V.; Belova, D.; Babich, O.; Prosekov, A.; Ivanova, S.; Katserov, D.; Patyukov, N.; Sukhikh, S. Microalgae: A Promising Source of Valuable Bioproducts. Biomolecules 2020, 10, 1153. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Wang, Q.; Shen, Q.; Zhan, J.; Zhao, Y. Genetic engineering of microorganisms for biodiesel production. Bioengineered 2013, 4, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Radakovits, R.; Jinkerson, R.E.; Darzins, A.; Posewitz, M.C. Genetic engineering of algae for enhanced biofuel production. Eukaryot Cell 2010, 9, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Nouemssi, S.B.; Ghribi, M.; Beauchemin, R.; Meddeb-Mouelhi, F.; Germain, H.; Desgagné-Penix, I. Rapid and Efficient Colony-PCR for High Throughput Screening of Genetically Transformed Chlamydomonas reinhardtii. Life 2020, 10, 186. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Gerken, H.; Li, Y. Single-tube colony PCR for DNA amplification and transformant screening of oleaginous microalgae. J. Appl. Phycol. 2014, 26, 1719–1726. [Google Scholar] [CrossRef]
- Tear, C.J.; Lim, C.; Wu, J.; Zhao, H. Accumulated lipids rather than the rigid cell walls impede the extraction of genetic materials for effective colony PCRs in Chlorella vulgaris. Microb. Cell Fact. 2013, 12, 106. [Google Scholar] [CrossRef]
- Yuan, G.; Czajka, J.J.; Dai, Z.; Hu, D.; Pomraning, K.R.; Hofstad, B.A.; Kim, J.; Robles, A.L.; Deng, S.; Magnuson, J.K. Rapid and robust squashed spore/colony PCR of industrially important fungi. Fungal Biol. Biotechnol. 2023, 10, 15. [Google Scholar] [CrossRef]
- Kasajima, I.; Ide, Y.; Ohkama-Ohtsu, N.; Hayashi, H.; Yoneyama, T.; Fujiwara, T. A protocol for rapid DNA extraction from Arabidopsis thaliana for PCR analysis. Plant Mol. Biol. Report. 2004, 22, 49–52. [Google Scholar] [CrossRef]
- Thesseling, F.A.; Bircham, P.W.; Mertens, S.; Voordeckers, K.; Verstrepen, K.J. A Hands-On Guide to Brewing and Analyzing Beer in the Laboratory. Curr. Protoc. Microbiol. 2019, 54, e91. [Google Scholar] [CrossRef] [PubMed]
- Radha, S.; Fathima, A.A.; Iyappan, S.; Ramya, M. Direct colony PCR for rapid identification of varied microalgae from freshwater environment. J. Appl. Phycol. 2013, 25, 609–613. [Google Scholar] [CrossRef]
- Chen, S.; Yao, H.; Han, J.; Liu, C.; Song, J.; Shi, L.; Zhu, Y.; Ma, X.; Gao, T.; Pang, X.; et al. Validation of the ITS2 Region as a Novel DNA Barcode for Identifying Medicinal Plant Species. PLoS ONE 2010, 5, e8613. [Google Scholar] [CrossRef] [PubMed]
- Kira, N.; Ohnishi, K.; Miyagawa-Yamaguchi, A.; Kadono, T.; Adachi, M. Nuclear transformation of the diatom Phaeodactylum tricornutum using PCR-amplified DNA fragments by microparticle bombardment. Mar. Genom. 2016, 25, 49–56. [Google Scholar] [CrossRef]
- Fawley, M.W.; Fawley, K.P. Identification of Eukaryotic Microalgal Strains. J. Appl. Phycol. 2020, 32, 2699–2709. [Google Scholar] [CrossRef] [PubMed]
- Wan, M.; Rosenberg, J.N.; Faruq, J.; Betenbaugh, M.J.; Xia, J. An improved colony PCR procedure for genetic screening of Chlorella and related microalgae. Biotechnol. Lett. 2011, 33, 1615–1619. [Google Scholar] [CrossRef]
- Godhe, A.; Anderson, D.M.; Rehnstam-Holm, A.-S. PCR amplification of microalgal DNA for sequencing and species identification: Studies on fixatives and algal growth stages. Harmful Algae 2002, 1, 375–382. [Google Scholar] [CrossRef]
- Penna, A.; Galluzzi, L. The quantitative real-time PCR applications in the monitoring of marine harmful algal bloom (HAB) species. Environ. Sci. Pollut. Res. Int. 2013, 20, 6851–6862. [Google Scholar] [CrossRef]
- Duong, V.T.; Li, Y.; Nowak, E.; Schenk, P.M. Microalgae Isolation and Selection for Prospective Biodiesel Production. Energies 2012, 5, 1835–1849. [Google Scholar] [CrossRef]
- Jagielski, T.; Gawor, J.; Bakuła, Z.; Zuchniewicz, K.; Żak, I.; Gromadka, R. An optimized method for high quality DNA extraction from microalga Prototheca wickerhamii for genome sequencing. Plant Methods 2017, 13, 77. [Google Scholar] [CrossRef]
- Stark, J.R.; Cardon, Z.G.; Peredo, E.L. Extraction of high-quality, high-molecular-weight DNA depends heavily on cell homogenization methods in green microalgae. Appl. Plant Sci. 2020, 8, e11333. [Google Scholar] [CrossRef]
- Cao, M.; Fu, Y.; Guo, Y.; Pan, J. Chlamydomonas (Chlorophyceae) colony PCR. Protoplasma 2009, 235, 107–110. [Google Scholar] [CrossRef]
- Packeiser, H.; Lim, C.; Balagurunathan, B.; Wu, J.; Zhao, H. An extremely simple and effective colony PCR procedure for bacteria, yeasts, and microalgae. Appl. Biochem. Biotechnol. 2013, 169, 695–700. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-B.; Hu, Q.; Sommerfeld, M.; Chen, F. Cell wall proteomics of the green alga Haematococcus pluvialis (Chlorophyceae). Proteomics 2004, 4, 692–708. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Microalgae Species | 1 Direct-PCR | Squash-PCR |
|---|---|---|---|
| 1 | Monoraphidium minutum 26B-AM | + | ++ |
| 2 | Chlorella sorokiniana DOE1412 | + * | ++ |
| 3 | Tetraselmis striata LANL1001 | − | ++ |
| 4 | Picochlorum celeri TG2 | + * | ++ |
| 5 | Scenedesmus obliquus UTEX 393 | + | ++ |
| 6 | Chlamydomonas reinhardtii UTEX 89 | + | ++ |
| 7 | Scenedesmus sp. UTEX 1589 | + * | ++ |
| 8 | Haematococcus pluvialis UTEX 2505 | + | ++ |
| 9 | Chlorella vulgaris UTEX 395 | + | ++ |
| 10 | Chlamydomonas reinhardtii UTEX 90 | + | ++ |
| 11 | Botryococcus braunii UTEX 572 | + | ++ |
| 12 | Dunaliella salina UTEX LB 200 | − | ++ |
| Primer | Sequence | Microalgae Species | Gene | Size (bp) |
|---|---|---|---|---|
| 102_F | AGGAGAAGTCGTAACAAGGT | Strains 1 to 12 | ITS2 | 700–900 |
| 103_R | TCCTCCGCTTATTGATATGC | |||
| 154_F | TTGCACACAAGAACGCATGA | Chlamydomonas reinhardtii | FTSY | 815 |
| 155_R | GGCATTGGAGTAAACGACCC | |||
| 156_F | CTACGTCGCCTTACTGTGTG | Chlamydomonas reinhardtii | ZEP | 809 |
| 157_R | GATTGCCTACTCACCACTCG | |||
| 197_F | ATGGTTCCACAAACTGAAACG | Dunaliella salina | rbcL | 480 |
| 198_R | GTCACGCTCAACTTGAATACC | |||
| 199_F | CGTAGACTGTGTAGAAGCTG | Dunaliella salina | orf121 | 518 |
| 200_R | GTGGAACTACACCTTCAGGT | |||
| 201_F | ATGCCAATTGGTGTTCCAC | Dunaliella salina | clpP | 496 |
| 202_R | GTGCATTAAAATCGTAAGGCG | |||
| 203_F | CAATGCGCACACCAGAAG | Dunaliella salina | atpA | 551 |
| 204_R | GCATATAGTTTCCTGCGTCC | |||
| 205_F | ATGGCACGTGCTAAATTTGA | Dunaliella salina | tufA | 524 |
| 206_R | GCAGAACCTGAAACGATAGG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, G.; Gao, S.; Czajka, J.J.; Dai, Z.; Pomraning, K.R.; Duong, R.D.; Hofstad, B.A.; Deng, S. Simple and Effective Squash-PCR for Rapid Genotyping of Industrial Microalgae. Life 2024, 14, 115. https://doi.org/10.3390/life14010115
Yuan G, Gao S, Czajka JJ, Dai Z, Pomraning KR, Duong RD, Hofstad BA, Deng S. Simple and Effective Squash-PCR for Rapid Genotyping of Industrial Microalgae. Life. 2024; 14(1):115. https://doi.org/10.3390/life14010115
Chicago/Turabian StyleYuan, Guoliang, Song Gao, Jeffrey J. Czajka, Ziyu Dai, Kyle R. Pomraning, Rylan D. Duong, Beth A. Hofstad, and Shuang Deng. 2024. "Simple and Effective Squash-PCR for Rapid Genotyping of Industrial Microalgae" Life 14, no. 1: 115. https://doi.org/10.3390/life14010115