

Nilotinib as a Prospective Treatment for Alzheimer’s Disease: Effect on Proteins Involved in Neurodegeneration and Neuronal Homeostasis

 ,
,  ,
,  ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Nilotinib Treatment
2.2. Real-Time PCR
2.3. Western Blotting
2.4. Transmission Electron Microscopy
2.5. MitoTracker Staining
2.6. Statistical Analysis
3. Results
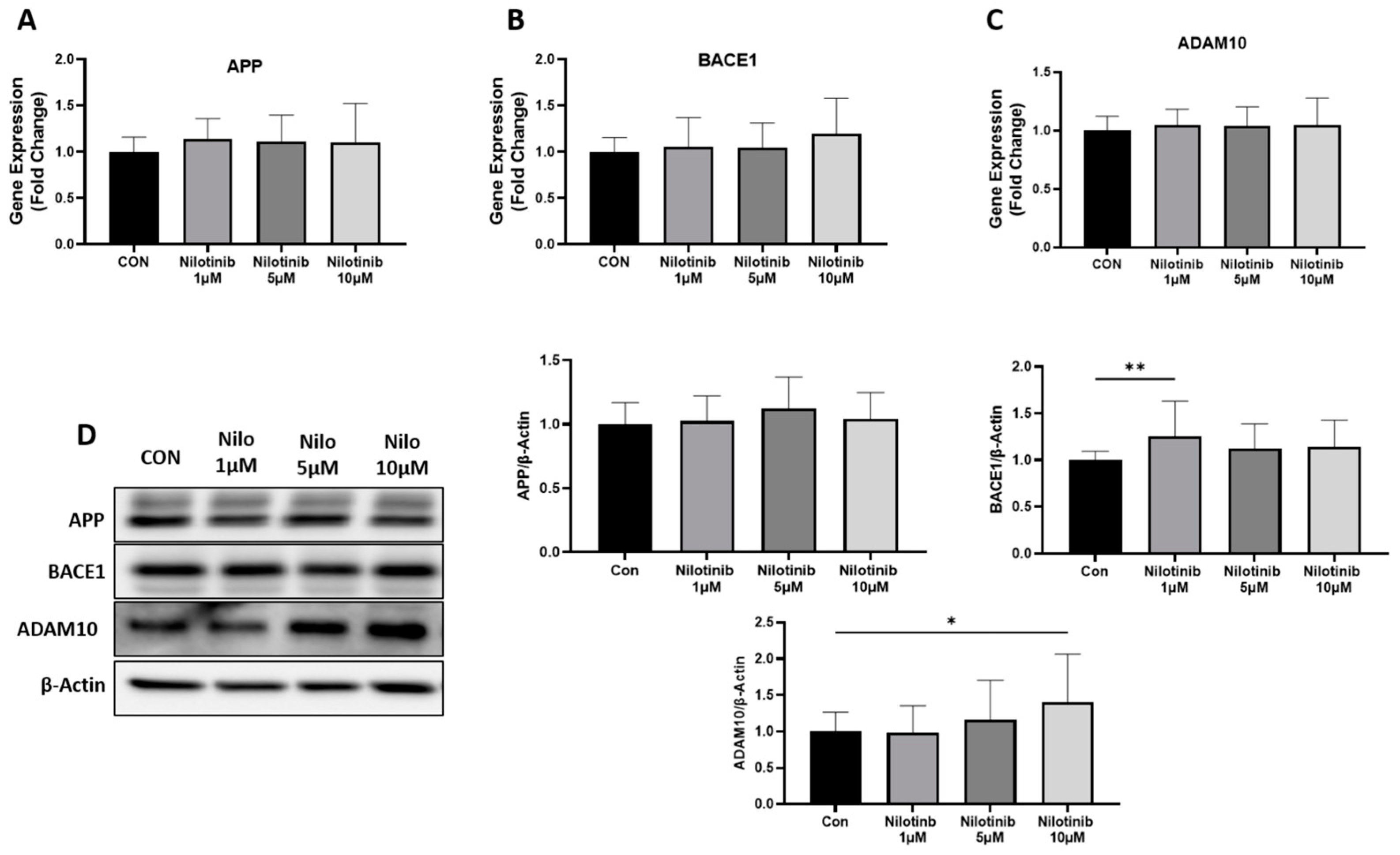
3.1. Effect of Nilotinib on Genes Regulating Amyloid-β Formation in SH-SY5Y Cells
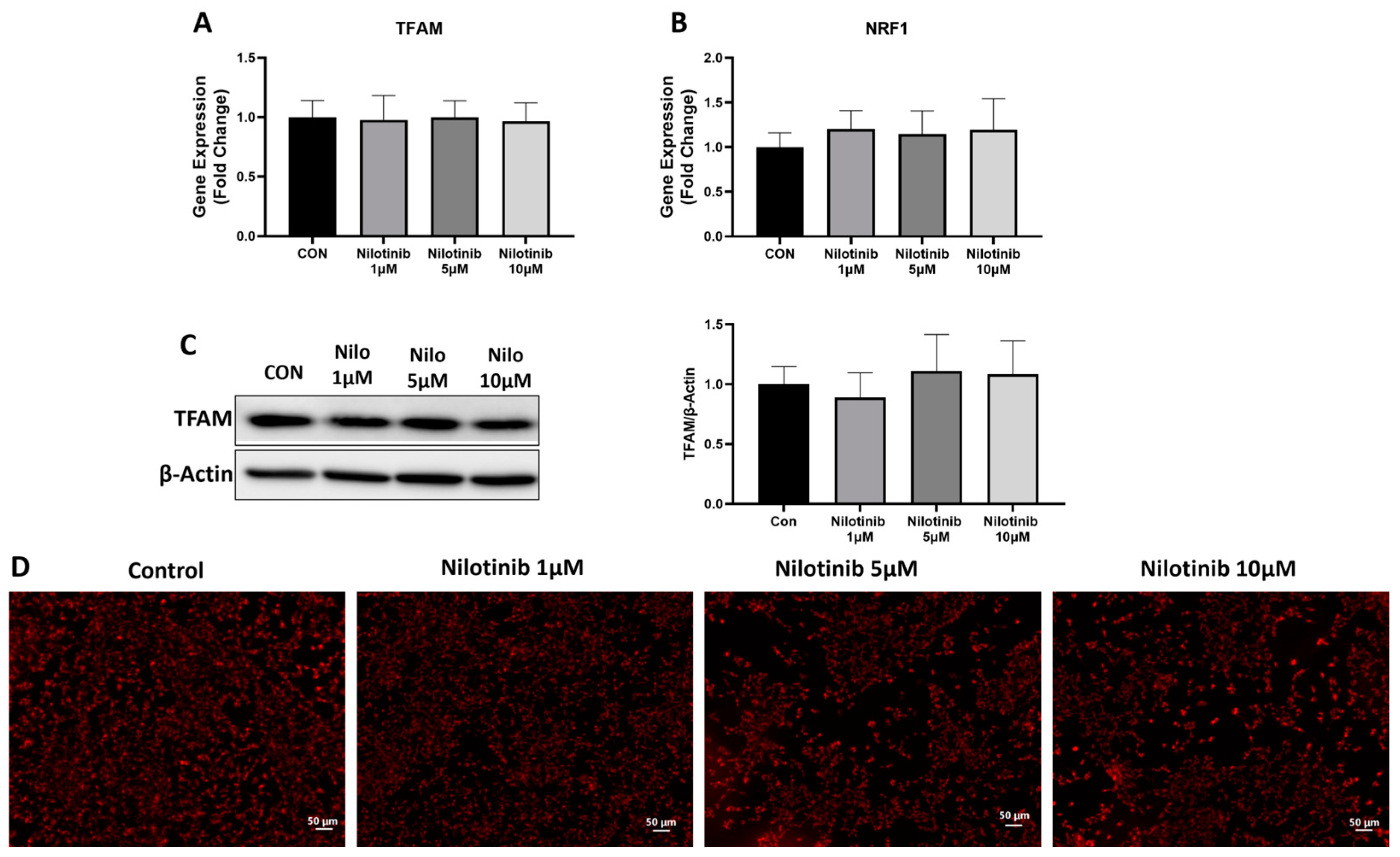
3.2. Effect of Nilotinib on Genes Associated with Neuronal Health in SH-SY5Y Cells
3.3. Effect of Nilotinib on Mitochondrial Functioning in SH-SY5Y Cells
3.4. Effect of Nilotinib on Mitochondrial Morphology in SH-SY5Y Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, X.; Hu, J.; Xie, S.; Li, W.; Zhang, H.; Huang, L.; Qian, Z.; Zhao, C.; Zhang, L. Hidden role of microglia during neurodegenerative disorders and neurocritical care: A mitochondrial perspective. Int. Immunopharmacol. 2024, 142, 113024. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Kril, J.J. Alzheimer disease: Alzheimer disease neuropathology in the oldest old. Nat. Rev. Neurol. 2009, 5, 411–412. [Google Scholar] [CrossRef]
- Nelson, L.; Tabet, N. Slowing the progression of Alzheimer’s disease; what works? Ageing Res. Rev. 2015, 23, 193–209. [Google Scholar] [CrossRef]
- Liu, P.P.; Xie, Y.; Meng, X.Y.; Kang, J.S. History and progress of hypotheses and clinical trials for Alzheimer’s disease. Signal Transduct. Target. Ther. 2019, 4, 29. [Google Scholar] [CrossRef] [PubMed]
- Reiss, A.B.; Montufar, N.; DeLeon, J.; Pinkhasov, A.; Gomolin, I.H.; Glass, A.D.; Arain, H.A.; Stecker, M.M. Alzheimer Disease Clinical Trials Targeting Amyloid: Lessons Learned From Success in Mice and Failure in Humans. Neurologist 2021, 26, 52–61. [Google Scholar] [CrossRef]
- Suzuki, N.; Hatta, T.; Ito, M.; Kusakabe, K.I. Anti-Amyloid-Beta Antibodies and Anti-Tau Therapies for Alzheimer’s Disease: Recent Advances and Perspectives. Chem. Pharm. Bull. 2024, 72, 602–609. [Google Scholar] [CrossRef]
- Volloch, V.; Rits-Volloch, S. Principles of Design of Clinical Trials for Prevention and Treatment of Alzheimer’s Disease and Aging-Associated Cognitive Decline in the ACH2.0 Perspective: Potential Outcomes, Challenges, and Solutions. J. Alzheimers Dis. Rep. 2023, 7, 921–955. [Google Scholar] [CrossRef]
- Fowler, A.J.; Hebron, M.; Missner, A.A.; Wang, R.; Gao, X.; Kurd-Misto, B.T.; Liu, X.; Moussa, C.E. Multikinase Abl/DDR/Src Inhibition Produces Optimal Effects for Tyrosine Kinase Inhibition in Neurodegeneration. Drugs R D 2019, 19, 149–166. [Google Scholar] [CrossRef]
- Stevenson, M.; Varghese, R.; Hebron, M.L.; Liu, X.; Ratliff, N.; Smith, A.; Turner, R.S.; Moussa, C. Inhibition of discoidin domain receptor (DDR)-1 with nilotinib alters CSF miRNAs and is associated with reduced inflammation and vascular fibrosis in Alzheimer’s disease. J. Neuroinflammation 2023, 20, 116. [Google Scholar] [CrossRef]
- Turner, R.S.; Hebron, M.L.; Lawler, A.; Mundel, E.E.; Yusuf, N.; Starr, J.N.; Anjum, M.; Pagan, F.; Torres-Yaghi, Y.; Shi, W.; et al. Nilotinib Effects on Safety, Tolerability, and Biomarkers in Alzheimer’s Disease. Ann. Neurol. 2020, 88, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Fagiani, F.; Lanni, C.; Racchi, M.; Govoni, S. Targeting dementias through cancer kinases inhibition. Alzheimers Dement. 2020, 6, e12044. [Google Scholar] [CrossRef]
- Franceschi, R.T.; Hallett, S.A.; Ge, C. Discoidin domain receptors; an ancient family of collagen receptors has major roles in bone development, regeneration and metabolism. Front. Dent. Med. 2023, 4, 1181817. [Google Scholar] [CrossRef]
- Deremer, D.L.; Ustun, C.; Natarajan, K. Nilotinib: A second-generation tyrosine kinase inhibitor for the treatment of chronic myelogenous leukemia. Clin. Ther. 2008, 30, 1956–1975. [Google Scholar] [CrossRef] [PubMed]
- Rosti, G.; Castagnetti, F.; Gugliotta, G.; Baccarani, M. Tyrosine kinase inhibitors in chronic myeloid leukaemia: Which, when, for whom? Nat. Rev. Clin. Oncol. 2017, 14, 141–154. [Google Scholar] [CrossRef]
- Day, E.; Waters, B.; Spiegel, K.; Alnadaf, T.; Manley, P.W.; Buchdunger, E.; Walker, C.; Jarai, G. Inhibition of collagen-induced discoidin domain receptor 1 and 2 activation by imatinib, nilotinib and dasatinib. Eur. J. Pharmacol. 2008, 599, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Fowler, A.J.; Hebron, M.; Balaraman, K.; Shi, W.; Missner, A.A.; Greenzaid, J.D.; Chiu, T.L.; Ullman, C.; Weatherdon, E.; Duka, V.; et al. Discoidin Domain Receptor 1 is a therapeutic target for neurodegenerative diseases. Hum. Mol. Genet. 2020, 29, 2882–2898. [Google Scholar] [CrossRef]
- Imbimbo, B.P.; Ippati, S.; Watling, M. Should drug discovery scientists still embrace the amyloid hypothesis for Alzheimer’s disease or should they be looking elsewhere? Expert. Opin. Drug Discov. 2020, 15, 1241–1251. [Google Scholar] [CrossRef]
- Schlatterer, S.D.; Tremblay, M.A.; Acker, C.M.; Davies, P. Neuronal c-Abl overexpression leads to neuronal loss and neuroinflammation in the mouse forebrain. J. Alzheimers Dis. 2011, 25, 119–133. [Google Scholar] [CrossRef]
- Karuppagounder, S.S.; Brahmachari, S.; Lee, Y.; Dawson, V.L.; Dawson, T.M.; Ko, H.S. The c-Abl inhibitor, nilotinib, protects dopaminergic neurons in a preclinical animal model of Parkinson’s disease. Sci. Rep. 2014, 4, 4874. [Google Scholar] [CrossRef]
- Wu, J.; Xu, X.; Zheng, L.; Mo, J.; Jin, X.; Bao, Y. Nilotinib inhibits microglia-mediated neuroinflammation to protect against dopaminergic neuronal death in Parkinson’s disease models. Int. Immunopharmacol. 2021, 99, 108025. [Google Scholar] [CrossRef] [PubMed]
- Simuni, T.; Fiske, B.; Merchant, K.; Coffey, C.S.; Klingner, E.; Caspell-Garcia, C.; Lafontant, D.E.; Matthews, H.; Wyse, R.K.; Brundin, P.; et al. Efficacy of Nilotinib in Patients With Moderately Advanced Parkinson Disease: A Randomized Clinical Trial. JAMA Neurol. 2021, 78, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Werner, M.H.; Olanow, C.W. Parkinson’s Disease Modification Through Abl Kinase Inhibition: An Opportunity. Mov. Disord. 2022, 37, 6–15. [Google Scholar] [CrossRef]
- Pagan, F.L.; Wilmarth, B.; Torres-Yaghi, Y.; Hebron, M.L.; Mulki, S.; Ferrante, D.; Matar, S.; Ahn, J.; Moussa, C. Long-Term Safety and Clinical Effects of Nilotinib in Parkinson’s Disease. Mov. Disord. 2021, 36, 740–749. [Google Scholar] [CrossRef]
- Lonskaya, I.; Hebron, M.L.; Desforges, N.M.; Franjie, A.; Moussa, C.E. Tyrosine kinase inhibition increases functional parkin-Beclin-1 interaction and enhances amyloid clearance and cognitive performance. EMBO Mol. Med. 2013, 5, 1247–1262. [Google Scholar] [CrossRef]
- Burns, M.P.; Zhang, L.; Rebeck, G.W.; Querfurth, H.W.; Moussa, C.E. Parkin promotes intracellular Abeta1-42 clearance. Hum. Mol. Genet. 2009, 18, 3206–3216. [Google Scholar] [CrossRef] [PubMed]
- Lonskaya, I.; Hebron, M.L.; Desforges, N.M.; Schachter, J.B.; Moussa, C.E. Nilotinib-induced autophagic changes increase endogenous parkin level and ubiquitination, leading to amyloid clearance. J. Mol. Med. 2014, 92, 373–386. [Google Scholar] [CrossRef]
- Lonskaya, I.; Hebron, M.; Chen, W.; Schachter, J.; Moussa, C. Tau deletion impairs intracellular beta-amyloid-42 clearance and leads to more extracellular plaque deposition in gene transfer models. Mol. Neurodegener. 2014, 9, 46. [Google Scholar] [CrossRef]
- Ali, M.; Wani, S.U.D.; Dey, T.; Sridhar, S.B.; Qadrie, Z.L. A common molecular and cellular pathway in developing Alzheimer and cancer. Biochem. Biophys. Rep. 2024, 37, 101625. [Google Scholar] [CrossRef]
- Motaln, H.; Rogelj, B. The Role of c-Abl Tyrosine Kinase in Brain and Its Pathologies. Cells 2023, 12, 2041. [Google Scholar] [CrossRef]
- Jing, Z.; Caltagarone, J.; Bowser, R. Altered subcellular distribution of c-Abl in Alzheimer’s disease. J. Alzheimers Dis. 2009, 17, 409–422. [Google Scholar] [CrossRef] [PubMed]
- Koga, S.; Sekiya, H.; Kondru, N.; Ross, O.A.; Dickson, D.W. Neuropathology and molecular diagnosis of Synucleinopathies. Mol. Neurodegener. 2021, 16, 83. [Google Scholar] [CrossRef] [PubMed]
- Nobili, A.; D’Amelio, M.; Viscomi, M.T. Nilotinib: From animal-based studies to clinical investigation in Alzheimer’s disease patients. Neural Regen. Res. 2023, 18, 803–804. [Google Scholar] [CrossRef]
- Xie, X.; Yuan, P.; Kou, L.; Chen, X.; Li, J.; Li, Y. Nilotinib in Parkinson’s disease: A systematic review and meta-analysis. Front. Aging Neurosci. 2022, 14, 996217. [Google Scholar] [CrossRef] [PubMed]
- La Barbera, L.; Vedele, F.; Nobili, A.; Krashia, P.; Spoleti, E.; Latagliata, E.C.; Cutuli, D.; Cauzzi, E.; Marino, R.; Viscomi, M.T.; et al. Nilotinib restores memory function by preventing dopaminergic neuron degeneration in a mouse model of Alzheimer’s Disease. Prog. Neurobiol. 2021, 202, 102031. [Google Scholar] [CrossRef]
- Adlimoghaddam, A.; Odero, G.G.; Glazner, G.; Turner, R.S.; Albensi, B.C. Nilotinib Improves Bioenergetic Profiling in Brain Astroglia in the 3xTg Mouse Model of Alzheimer’s Disease. Aging Dis. 2021, 12, 441–465. [Google Scholar] [CrossRef] [PubMed]
- Nishioka, H.; Tooi, N.; Isobe, T.; Nakatsuji, N.; Aiba, K. BMS-708163 and Nilotinib restore synaptic dysfunction in human embryonic stem cell-derived Alzheimer’s disease models. Sci. Rep. 2016, 6, 33427. [Google Scholar] [CrossRef]
- Leon, R.; Gutierrez, D.A.; Pinto, C.; Morales, C.; de la Fuente, C.; Riquelme, C.; Cortes, B.I.; Gonzalez-Martin, A.; Chamorro, D.; Espinosa, N.; et al. c-Abl tyrosine kinase down-regulation as target for memory improvement in Alzheimer’s disease. Front. Aging Neurosci. 2023, 15, 1180987. [Google Scholar] [CrossRef]
- Srivastava, A.; Johnson, M.; Renna, H.A.; Sheehan, K.M.; Ahmed, S.; Palaia, T.; Pinkhasov, A.; Gomolin, I.H.; De Leon, J.; Reiss, A.B. Therapeutic Potential of P110 Peptide: New Insights into Treatment of Alzheimer’s Disease. Life 2023, 13, 2156. [Google Scholar] [CrossRef]
- Vaz, M.; Silva, V.; Monteiro, C.; Silvestre, S. Role of Aducanumab in the Treatment of Alzheimer’s Disease: Challenges and Opportunities. Clin. Interv. Aging 2022, 17, 797–810. [Google Scholar] [CrossRef]
- Chowdhury, S.; Chowdhury, N.S. Novel anti-amyloid-beta (Abeta) monoclonal antibody lecanemab for Alzheimer’s disease: A systematic review. Int. J. Immunopathol. Pharmacol. 2023, 37, 3946320231209839. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Chi, Y.; Zhang, Q.; Ma, Y. Safety and efficacy of lecanemab for Alzheimer’s disease: A systematic review and meta-analysis of randomized clinical trials. Front. Aging Neurosci. 2023, 15, 1169499. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.P.; Clark, I.A.; Vissel, B. Questions concerning the role of amyloid-beta in the definition, aetiology and diagnosis of Alzheimer’s disease. Acta Neuropathol. 2018, 136, 663–689. [Google Scholar] [CrossRef] [PubMed]
- Galizzi, G.; Di Carlo, M. Mitochondrial DNA and Inflammation in Alzheimer’s Disease. Curr. Issues Mol. Biol. 2023, 45, 8586–8606. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Irwin, R.W.; Zhao, L.; Nilsen, J.; Hamilton, R.T.; Brinton, R.D. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 14670–14675. [Google Scholar] [CrossRef]
- Song, T.; Song, X.; Zhu, C.; Patrick, R.; Skurla, M.; Santangelo, I.; Green, M.; Harper, D.; Ren, B.; Forester, B.P.; et al. Mitochondrial dysfunction, oxidative stress, neuroinflammation, and metabolic alterations in the progression of Alzheimer’s disease: A meta-analysis of in vivo magnetic resonance spectroscopy studies. Ageing Res. Rev. 2021, 72, 101503. [Google Scholar] [CrossRef]
- Barros-Viegas, A.T.; Carmona, V.; Ferreiro, E.; Guedes, J.; Cardoso, A.M.; Cunha, P.; Pereira de Almeida, L.; Resende de Oliveira, C.; Pedro de Magalhaes, J.; Peca, J.; et al. miRNA-31 Improves Cognition and Abolishes Amyloid-beta Pathology by Targeting APP and BACE1 in an Animal Model of Alzheimer’s Disease. Mol. Ther. Nucleic Acids 2020, 19, 1219–1236. [Google Scholar] [CrossRef]
- Du, W.; Lei, C.; Dong, Y. MicroRNA-149 is downregulated in Alzheimer’s disease and inhibits beta-amyloid accumulation and ameliorates neuronal viability through targeting BACE1. Genet. Mol. Biol. 2021, 44, e20200064. [Google Scholar] [CrossRef]
- Postina, R.; Schroeder, A.; Dewachter, I.; Bohl, J.; Schmitt, U.; Kojro, E.; Prinzen, C.; Endres, K.; Hiemke, C.; Blessing, M.; et al. A disintegrin-metalloproteinase prevents amyloid plaque formation and hippocampal defects in an Alzheimer disease mouse model. J. Clin. Investig. 2004, 113, 1456–1464. [Google Scholar] [CrossRef]
- Bathina, S.; Das, U.N. Brain-derived neurotrophic factor and its clinical implications. Arch. Med. Sci. 2015, 11, 1164–1178. [Google Scholar] [CrossRef]
- Gao, L.; Zhang, Y.; Sterling, K.; Song, W. Brain-derived neurotrophic factor in Alzheimer’s disease and its pharmaceutical potential. Transl. Neurodegener. 2022, 11, 4. [Google Scholar] [CrossRef] [PubMed]
- Tampellini, D.; Capetillo-Zarate, E.; Dumont, M.; Huang, Z.; Yu, F.; Lin, M.T.; Gouras, G.K. Effects of synaptic modulation on beta-amyloid, synaptophysin, and memory performance in Alzheimer’s disease transgenic mice. J. Neurosci. 2010, 30, 14299–14304. [Google Scholar] [CrossRef]
- Harwell, C.S.; Coleman, M.P. Synaptophysin depletion and intraneuronal Abeta in organotypic hippocampal slice cultures from huAPP transgenic mice. Mol. Neurodegener. 2016, 11, 44. [Google Scholar] [CrossRef]
- Bhatia, S.; Rawal, R.; Sharma, P.; Singh, T.; Singh, M.; Singh, V. Mitochondrial Dysfunction in Alzheimer’s Disease: Opportunities for Drug Development. Curr. Neuropharmacol. 2022, 20, 675–692. [Google Scholar] [CrossRef]
- Perez Ortiz, J.M.; Swerdlow, R.H. Mitochondrial dysfunction in Alzheimer’s disease: Role in pathogenesis and novel therapeutic opportunities. Br. J. Pharmacol. 2019, 176, 3489–3507. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Su, B.; Fujioka, H.; Zhu, X. Dynamin-like protein 1 reduction underlies mitochondrial morphology and distribution abnormalities in fibroblasts from sporadic Alzheimer’s disease patients. Am. J. Pathol. 2008, 173, 470–482. [Google Scholar] [CrossRef] [PubMed]
- Trimmer, P.A.; Swerdlow, R.H.; Parks, J.K.; Keeney, P.; Bennett, J.P., Jr.; Miller, S.W.; Davis, R.E.; Parker, W.D., Jr. Abnormal mitochondrial morphology in sporadic Parkinson’s and Alzheimer’s disease cybrid cell lines. Exp. Neurol. 2000, 162, 37–50. [Google Scholar] [CrossRef]
- Calkins, M.J.; Reddy, P.H. Amyloid beta impairs mitochondrial anterograde transport and degenerates synapses in Alzheimer’s disease neurons. Biochim. Biophys. Acta 2011, 1812, 507–513. [Google Scholar] [CrossRef]
- Caspersen, C.; Wang, N.; Yao, J.; Sosunov, A.; Chen, X.; Lustbader, J.W.; Xu, H.W.; Stern, D.; McKhann, G.; Yan, S.D. Mitochondrial Abeta: A potential focal point for neuronal metabolic dysfunction in Alzheimer’s disease. FASEB J. 2005, 19, 2040–2041. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer (Tm) | Forward Sequence | Reverse Sequence |
|---|---|---|
| GAPDH (62 °C) | ACCATCATCCCTGCCTCTAC | CCTGTTGCTGTAGCCAAAT |
| APP (62 °C) | TTTGGCACTGCTCCTGCT | CCACAGAACATGGCAATC |
| BACE-1 (62 °C) | GCAGGGCTACTACGTGGAGA | CAGCACCCACTGCAAAGTTA |
| TFAM (63 °C) | AAGATTCCAAGAAGCTAAGGGTGA | CAGAGTCAGACAGATTTTTTCCAGTTT |
| SYNAPTOPHYSIN (65 °C) | CTGCAATGGGTCTTCGCCA | ACTCTCGGTCTTGTTGGC |
| ADAM10 (62 °C) | TCGAACCATCACCCTGCAACCT | GCCCACCAATGAGCCACAATCC |
| NRF-1 (63 °C) | GGCACTGTCTCACTTATCCAGGTT | CAGCCACGGCAGAATAATTCA |
| BDNF (64 °C) | AGCTATCCAGAGCATCTTCCA | ACCTGGTGGAACTTTATGAAACC |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Srivastava, A.; Renna, H.A.; Johnson, M.; Sheehan, K.; Ahmed, S.; Palaia, T.; Pinkhasov, A.; Gomolin, I.H.; Wisniewski, T.; De Leon, J.; et al. Nilotinib as a Prospective Treatment for Alzheimer’s Disease: Effect on Proteins Involved in Neurodegeneration and Neuronal Homeostasis. Life 2024, 14, 1241. https://doi.org/10.3390/life14101241
Srivastava A, Renna HA, Johnson M, Sheehan K, Ahmed S, Palaia T, Pinkhasov A, Gomolin IH, Wisniewski T, De Leon J, et al. Nilotinib as a Prospective Treatment for Alzheimer’s Disease: Effect on Proteins Involved in Neurodegeneration and Neuronal Homeostasis. Life. 2024; 14(10):1241. https://doi.org/10.3390/life14101241
Chicago/Turabian StyleSrivastava, Ankita, Heather A. Renna, Maryann Johnson, Katie Sheehan, Saba Ahmed, Thomas Palaia, Aaron Pinkhasov, Irving H. Gomolin, Thomas Wisniewski, Joshua De Leon, and et al. 2024. "Nilotinib as a Prospective Treatment for Alzheimer’s Disease: Effect on Proteins Involved in Neurodegeneration and Neuronal Homeostasis" Life 14, no. 10: 1241. https://doi.org/10.3390/life14101241