

Genetic Polymorphisms of Prokineticins and Prokineticin Receptors Associated with Human Disease


Abstract
1. Introduction
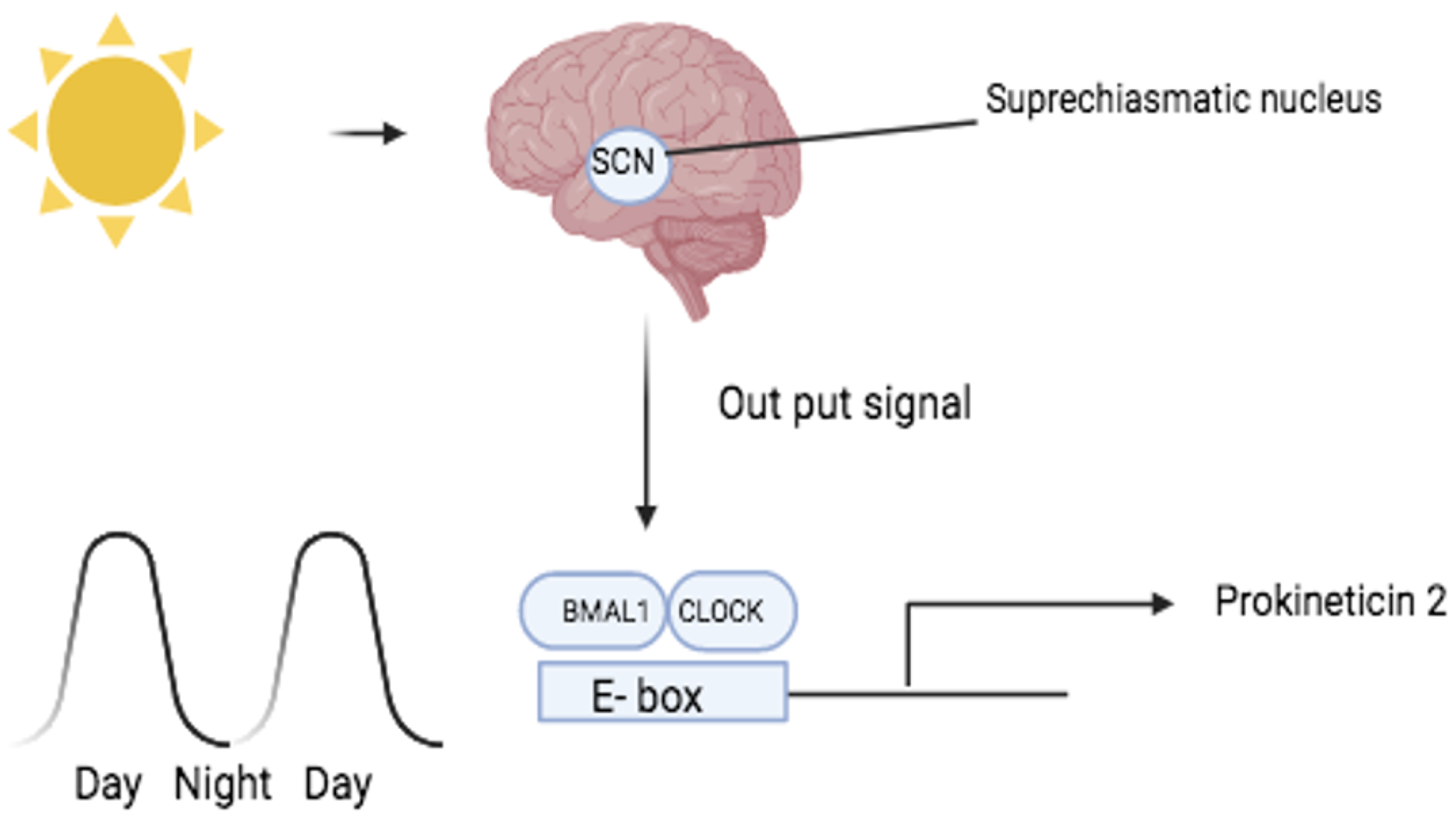
2. Disease Associated with Circadian Rhythms
3. Disease Associated with Psychiatric Disorders
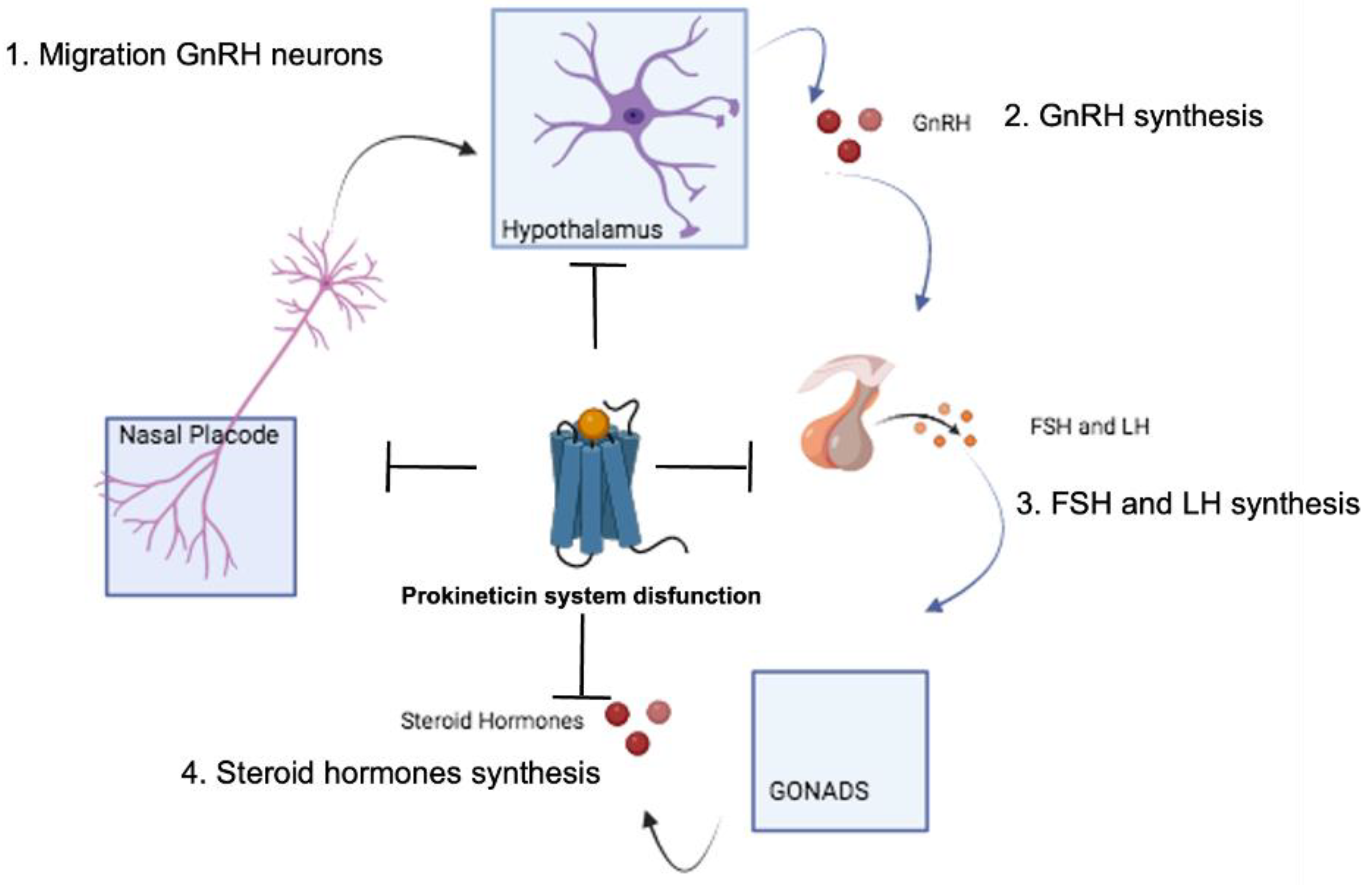
4. Diseases Associated with the Neuroendocrine System
4.1. Hypogonadotropic Hypogonadism and Kallmann Syndrome
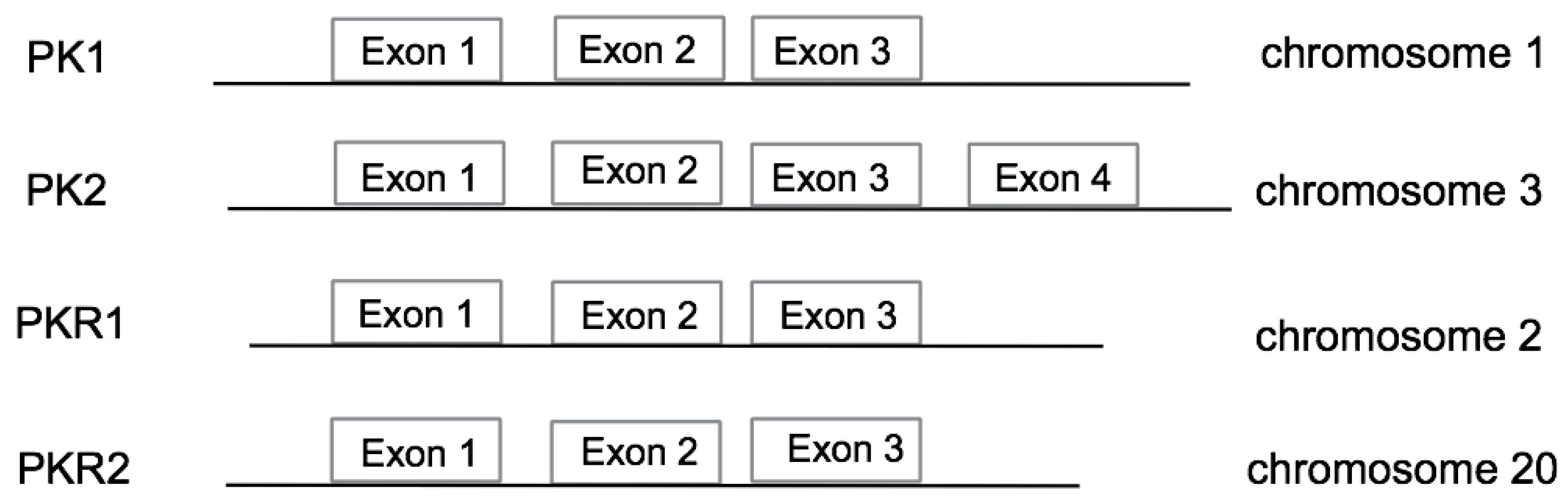
4.2. PK2 Mutations
4.3. PKR2 Mutations
4.4. Congenital Anosmia
4.5. Pituitary Dysplasia
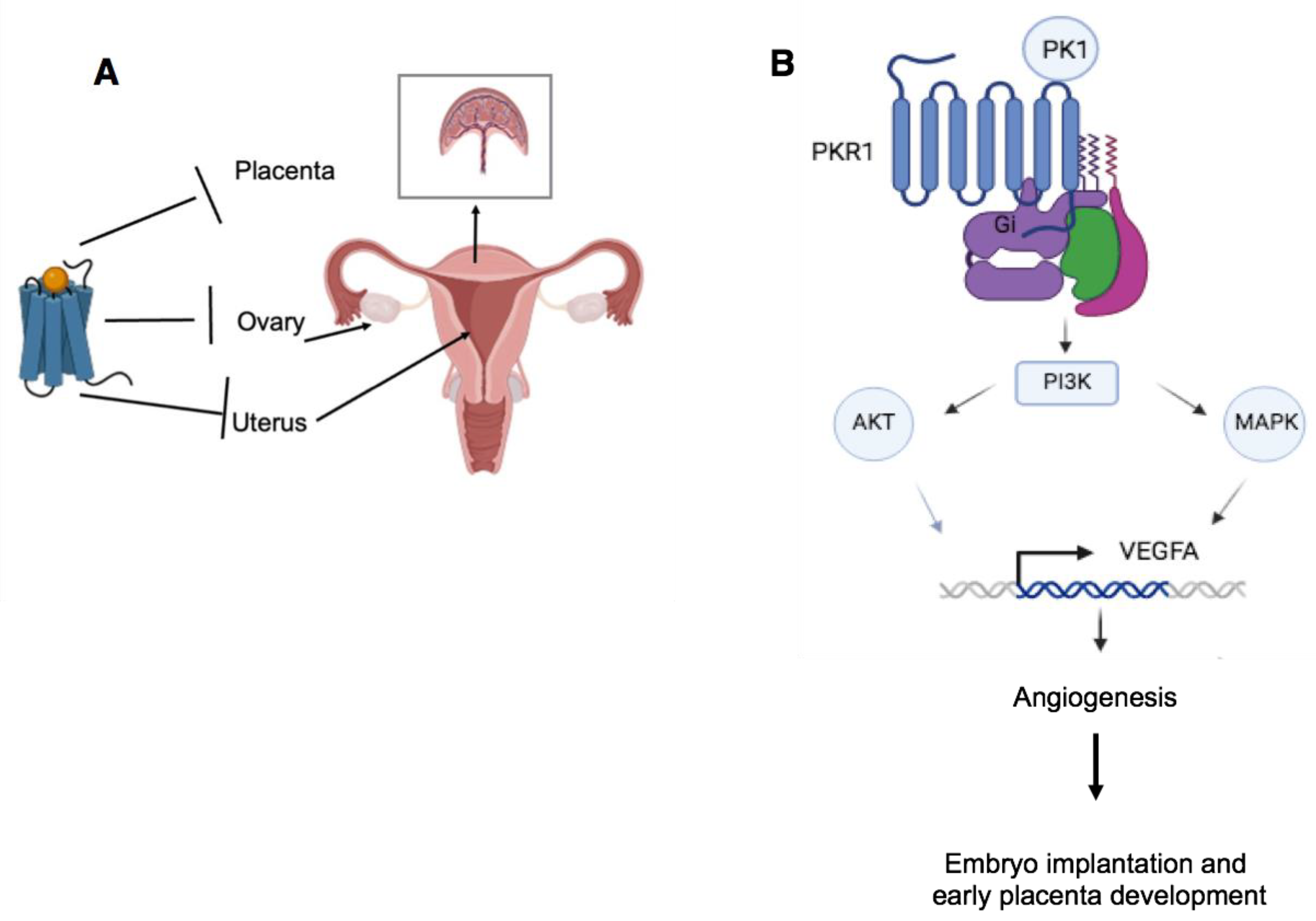
5. Disease of the Reproductive System
6. Disease of the Digestive System
7. Obesity
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lin, D.C.; Bullock, C.M.; Ehlert, F.J.; Chen, J.L.; Tian, H.; Zhou, Q.Y. Identification and molecular characterization of two closely related G protein-coupled receptors activated by prokineticins/endocrine gland vascular endothelial growth factor. J. Biol. Chem. 2002, 277, 19276–19280. [Google Scholar] [CrossRef] [PubMed]
- Masuda, Y.; Takatsu, Y.; Terao, Y.; Kumano, S.; Ishibashi, Y.; Suenaga, M.; Abe, M.; Fukusumi, S.; Watanabe, T.; Shintani, Y.; et al. Isolation and identification of EG-VEGF/prokineticins as cognate ligands for two orphan G-protein-coupled receptors. Biochem. Biophys. Res. Commun. 2002, 293, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Soga, T.; Matsumoto, S.; Oda, T.; Saito, T.; Hiyama, H.; Takasaki, J.; Kamohara, M.; Ohishi, T.; Matsushime, H.; Furuichi, K. Molecular cloning and characterization of prokineticin receptors. Biochim. Biophys. Acta 2002, 1579, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Vincenzi, M.; Kremić, A.; Jouve, A.; Lattanzi, R.; Miele, R.; Benharouga, M.; Alfaidy, N.; Migrenne-Li, S.; Kanthasamy, A.G.; Porciona, M.; et al. Therapeutic Potential of Targeting Prokineticin Receptors in Diseases. Pharmacol. Rev. 2023, 75, 1167–1199. [Google Scholar] [CrossRef]
- Lattanzi, R.; Miele, R. Prokineticin-Receptor Network: Mechanisms of Regulation. Life 2022, 12, 172. [Google Scholar] [CrossRef]
- Lattanzi, R.; Casella, I.; Fullone, M.R.; Maftei, D.; Miele, R. Mapping the interaction site for β-arrestin-2 in the prokineticin 2 receptor. Cell. Signal. 2024, 119, 111175. [Google Scholar] [CrossRef]
- Lattanzi, R.; Casella, I.; Fullone, M.R.; Maftei, D.; Miele, R. MRAP2 Inhibits β-Arrestin-2 Recruitment to the Prokineticin Receptor 2. Curr. Issues Mol. Biol. 2024, 46, 1607–1620. [Google Scholar] [CrossRef]
- Fullone, M.R.; Maftei, D.; Vincenzi, M.; Lattanzi, R.; Miele, R. Identification of Regions In-volved in the Physical Interaction between Melanocortin Receptor Accessory Protein 2 and Prokineticin Receptor 2. Biomolecules 2022, 12, 474. [Google Scholar] [CrossRef] [PubMed]
- Fullone, M.R.; Maftei, D.; Vincenzi, M.; Lattanzi, R.; Miele, R. Arginine 125 Is an Essential Residue for the Function of MRAP2. Int. J. Mol. Sci. 2022, 23, 9853. [Google Scholar] [CrossRef]
- Impellizzeri, D.; Maftei, D.; Severini, C.; Miele, R.; Balboni, G.; Siracusa, R.; Cordaro, M.; Di Paola, R.; Cuzzocrea, S.; Lattanzi, R. Blocking prokineticin receptors attenuates synovitis and joint destruction in collagen-induced arthritis. J. Mol. Med. 2023, 101, 558–569. [Google Scholar] [CrossRef]
- Lattanzi, R.; Severini, C.; Miele, R. Prokineticin 2 in cancer-related inflammation. Cancer Lett. 2022, 546, 215838. [Google Scholar] [CrossRef] [PubMed]
- Lattanzi, R.; Miele, R. Non-Peptide Agonists and Antagonists of the Prokineticin Receptors. Curr. Issues Mol. Biol. 2022, 44, 6323–6332. [Google Scholar] [CrossRef]
- Lattanzi, R.; Miele, R. Versatile Role of Prokineticins and Prokineticin Receptors in Neuroinflammation. Biomedicines 2021, 9, 1648. [Google Scholar] [CrossRef]
- Jilek, A.; Engel, E.; Beier, D.; Lepperdinger, G. Murine Bv8 gene maps near a synteny breakpoint of mouse chromosome 6 and human 3p21. Gene 2000, 256, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Kuei, C.; Sutton, S.; Wilson, S.; Yu, J.; Kamme, F.; Mazur, C.; Lovenberg, T.; Liu, C. Identification and pharmacological characterization of prokineticin 2 beta as a selective ligand for prokineticin receptor 1. Mol. Pharmacol. 2005, 67, 2070–2076. [Google Scholar] [CrossRef] [PubMed]
- Lattanzi, R.; Maftei, D.; Vincenzi, M.; Fullone, M.R.; Miele, R. Identification and Characterization of a New Splicing Variant of Prokineticin 2. Life 2022, 12, 248. [Google Scholar] [CrossRef]
- Lattanzi, R.; Maftei, D.; Negri, L.; Fusco, I.; Miele, R. PK2β ligand, a splice variant of prokineticin 2, is able to modulate and drive signaling through PKR1 receptor. Neuropeptides 2018, 71, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Parker, R.; Liu, M.; Eyre, H.J.; Copeland, N.G.; Gilbert, D.J.; Crawford, J.; Sutherland, G.R.; Jenkins, N.A.; Herzog, H. Y-receptor- like genes GPR72 and GPR73: Molecular cloning, genomic organisation and assignment to human chromosome 11q21.1 and 2p14 and mouse chromosome 9 and 6. Biochim. Biophys. Acta 2000, 1491, 369–375. [Google Scholar] [CrossRef]
- Lattanzi, R.; Maftei, D.; Fullone, M.R.; Miele, R. Identification and characterization of Prokineticin receptor 2 splicing TM4-7 variant and its modulation in an animal model of Alzheimer’s disease. Neuropeptides 2019, 73, 49–56. [Google Scholar] [CrossRef]
- Yang, L.-K.; Hou, Z.-S.; Tao, Y.-X. Biased signaling in naturally occurring mutations of G protein-coupled receptors associated with diverse human diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2021, 1867, 165973. [Google Scholar] [CrossRef]
- Barnard, A.R.; Nolan, P.M. When clocks go bad: Neurobehavioural consequences of disrupted circadian timing. PLoS Genet. 2008, 4, e1000040. [Google Scholar] [CrossRef] [PubMed]
- McClung, C.A. Circadian genes, rhythms and the biology of mood disorders. Pharmacol. Ther. 2007, 114, 222–232. [Google Scholar] [CrossRef]
- Li, J.D.; Hu, W.P.; Zhou, Q.Y. Disruption of the circadian output molecule prokineticin 2 results in anxiolytic and antidepressant-like effects in mice. Neuropsychopharmacology 2009, 34, 367–373. [Google Scholar] [CrossRef]
- Hu, W.P.; Li, J.D.; Zhang, C.; Boehmer, L.; Siegel, J.M.; Zhou, Q.Y. Altered circadian and homeostatic sleep regulation in prokineticin 2-deficient mice. Sleep 2007, 30, 247–256. [Google Scholar]
- Prosser, H.M.; Bradley, A.; Chesham, J.E.; Ebling, F.J.; Hastings, M.H.; Maywood, E.S. Prokineticin receptor 2 (Prokr2) is essential for the regulation of circadian behavior by the suprachiasmatic nuclei. Proc. Natl. Acad. Sci. USA 2007, 104, 648–653. [Google Scholar] [CrossRef] [PubMed]
- Detera-Wadleigh, S.D.; Badner, J.A.; Yoshikawa, T.; Sanders, A.R.; Goldin, L.R.; Turner, G.; Rollins, D.Y.; Moses, T.; Guroff, J.J.; Kazuba, D.; et al. Initial genome scan of the NIMH genetics initiative bipolar pedigrees: Chromosomes 4, 7, 9, 18, 19, 20, and 21q. Am. J. Med. Genet. 1997, 74, 254–262. [Google Scholar] [CrossRef]
- Fanous, A.H.; Neale, M.C.; Webb, B.T.; Straub, R.E.; O’Neill, F.A.; Walsh, D.; Riley, B.P.; Kendler, K.S. Novel linkage to chromosome 20p using latent classes of psychotic illness in 270 Irish high-density families. Biol. Psychiatry 2008, 64, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.; Berrettini, W.; Coryell, W.; Gershon, E.S.; Badner, J.A.; Kelsoe, J.R.; McInnis, M.G.; McMahon, F.J.; Murphy, D.L.; Nurnberger, J.I.; et al. Genome-wide parametric linkage analyses of 644 bipolar pedigrees suggest susceptibility loci at chromosomes 16 and 20. Psychiatr. Genet. 2008, 18, 191–198. [Google Scholar] [CrossRef]
- Kishi, T.; Kitajima, T.; Tsunoka, T.; Okumura, T.; Ikeda, M.; Okochi, T.; Kinoshita, Y.; Kawashima, K.; Yamanouchi, Y.; Ozaki, N.; et al. Possible Association of Prokineticin 2 Receptor Gene (PROKR2) with Mood Disorders in the Japanese Population. Neuromol. Med. 2009, 11, 114–122. [Google Scholar] [CrossRef]
- Currier, D.; Mann, M.J.; Oquendo, M.A.; Galfalvy, H.; Mann, J.J. Sex differences in the familial transmission of mood disorders. J. Affect. Disord. 2006, 95, 51–60. [Google Scholar] [CrossRef]
- Lehnkering, H.; Siegmund, R. Influence of chronotype, season, and sex of subject on sleep behavior of young adults. Chronobiol. Int. 2007, 24, 875–888. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.-H.; Cheng, M.Y. Prokineticin 2 and circadian clock output. FEBS J. 2005, 272, 5703–5709. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.Y.; Bittman, E.L.; Hattar, S.; Zhou, Q.Y. Regulation of prokineticin 2 expression by light and the circadian clock. BMC Neurosci. 2005, 6, 17. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.Y.; Bullock, C.M.; Li, C.; Lee, A.G.; Bermak, J.C.; Belluzzi, J.; Weaver, D.R.; Leslie, F.M.; Zhou, Q.Y. Prokineticin 2 transmits the behavioural circadian rhythm of the suprachi- asmatic nucleus. Nature 2006, 417, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Paulus, M.P.; Stewart, J.L. Neurobiology, clinical presentation, and treatment of methamphetamine use disorder: A review. JAMA Psychiatry 2020, 77, 959–966. [Google Scholar] [CrossRef]
- Wearne, T.A.; Cornish, J.L. A comparison of methamphetamine- induced psychosis and schizophrenia: A review of positive, negative, and cognitive symptomatology. Front. Psychiatry 2018, 9, 491. [Google Scholar] [CrossRef]
- Wise, R.A.; Robble, M.A. Dopamine and addiction. Annu. Rev. Psychol. 2020, 71, 79–106. [Google Scholar] [CrossRef]
- Jayanthi, S.; McCoy, M.T.; Chen, B.; Britt, J.P.; Kourrich, S.; Yau, H.J.; Ladenheim, B.; Krasnova, I.N.; Bonci, A.; Cadet, J.L. Methamphetamine downregulates striatal glutamate receptors via diverse epigenetic mechanisms. Biol. Psychiatry 2014, 76, 47–56. [Google Scholar] [CrossRef]
- Muller, C.P.; Carey, R.J.; Huston, J.P.; De Souza Silva, M.A. Serotonin and psychostimulant addiction: Focus on 5-HT1A-receptors. Prog. Neurobiol. 2007, 81, 133–178. [Google Scholar] [CrossRef]
- Morais, A.P.D.; Pita, I.R.; Fontes-Ribeiro, C.A.; Pereira, F.C. The neurobiological mechanisms of physical exercise in methamphetamine addiction. CNS Neurosci. Ther. 2018, 24, 85–97. [Google Scholar] [CrossRef]
- Chen, L.; Ru, Q.; Xiong, Q.; Zhou, M.; Yue, K.; Wu, Y. The role of Chinese herbal therapy in methamphetamine abuse and its induced psychiatric symptoms. Front. Pharmacol. 2021, 12, 679905. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Jeon, J.H. Recent advances in understanding Nrf2 agonism and its potential clinical application to metabolic and inflammatory diseases. Int. J. Mol. Sci. 2022, 23, 2846. [Google Scholar] [CrossRef] [PubMed]
- Guerina, A.A.; Nestlerc, E.J.; Berk, M.; Lawrencea, A.J.; Rosselle, S.L.; Kima, J.H. Genetics of methamphetamine use disorder: A systematic review and meta-analyses of gene association studies. Neurosci. Biobehav. Rev. 2021, 120, 48–74. [Google Scholar] [CrossRef]
- Jiang, Z.; Zhang, T.; Han, W.; Xiao, J.; Zhang, W.; Wang, X.; Liu, J.; Yang, Y.; Yang, C.; Guan, F.; et al. Identification of PROK2 gene polymorphisms as predictors of methamphetamine use disorder risk and indicators of craving scale in the Chinese Han population. Front. Pharmacol. 2023, 14, 1217382. [Google Scholar] [CrossRef]
- Wray, S. From nose to brain: Development of gonadotrophin-releasing hormone-1 neurones. J. Neuroendocrinol. 2010, 22, 743–753. [Google Scholar] [CrossRef]
- Wierman, M.E.; Kiseljak-Vassiliades, K.; Tobet, S. Gonadotropin-releasing hormone (GnRH) neuron migration: Initiation, maintenance and cessation as critical steps to ensure normal reproductive function. Front. Neuroendocrinol. 2011, 32, 43–52. [Google Scholar] [CrossRef] [PubMed]
- Pitteloud, N.; Durrani, S.; Raivio, T.; Sykiotis, G.P. Complex genetics in idiopathic hypogonadotropic hypogonadism. Front. Horm. Res. 2010, 39, 142–153. [Google Scholar]
- Abreu, A.P.; Kaiser, U.B.; Latronico, A.C. The role of prokineticins in the pathogenesis of hypogonadotropic hypogonadism. Neuroendocrinology 2010, 91, 283–290. [Google Scholar] [CrossRef]
- Sykiotis, G.P.; Plummer, L.; Hughes, V.A.; Au, M.; Durrani, S.; Nayak-Young, S.; Dwyer, A.A.; Quinton, R.; Hall, J.E.; Gusella, J.F.; et al. Oligogenic basis of isolated go-nadotropin-releasing hormone deficiency. Proc. Natl. Acad. Sci. USA 2010, 107, 15140–15144. [Google Scholar] [CrossRef]
- Sarfati, J.; Guiochon-Mantel, A.; Rondard, P.; Arnulf, I.; Garcia-Pinero, A.; Wolczynski, S.; Brailly-Tabard, S.; Bidet, M.; Arroyo, R.; Mathieu, M.; et al. A comparative phenotypic study of kallmann syndrome patients carrying monoallelic and biallelic mutations in the prokineticin 2 or prokineticin receptor 2 genes. J. Clin. Endocrinol. Metab. 2010, 95, 659–669. [Google Scholar] [CrossRef]
- Canto, P.; Munguia, P.; Soderlund, D.; Castro, J.J.; Mendez, J.P. Genetic analysis in patients with Kallmann syndrome: Coexistence of mutations in prokineticin receptor 2 and KAL1. J. Androl. 2009, 30, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Mkaouar, R.; Abdallah, L.C.B.; Naouali, C.; Lahbib, S.; Turki, Z.; Elouej, S.; Bouyacoub, Y.; Somai, M.; Mcelreavey, K.; Bashamboo, A.; et al. Oligogenic Inheritance Underlying Incomplete Penetrance of PROKR2 Mutations in Hypogonadotropic Hypogonadism. Front. Genet. 2021, 12, 665174. [Google Scholar] [CrossRef] [PubMed]
- Sugisawa, C.; Taniyama, M.; Sato, T.; Takahashi, Y.; Hasegawa, T.; Narumi, S. Biallelic PROKR2 variants and congenital hypogonadotropic hypogonadism: A case report and a literature review. Endocr. J. 2022, 69, 831–838. [Google Scholar] [CrossRef]
- Ng, K.L.; Li, J.D.; Cheng, M.Y.; Leslie, F.M.; Lee, A.G.; Zhou, Q.Y. Dependence of olfactory bulb neurogenesis on prokineticin 2 signaling. Science 2005, 308, 1923–1927. [Google Scholar] [CrossRef]
- Matsumoto, S.I.; Yamazaki, C.; Masumoto, K.H.; Nagano, M.; Naito, M.; Soga, T.; Hiyama, H.; Matsumoto, M.; Takasaki, J.; Shigeyoshi, Y.; et al. Abnormal development of the olfactory bulb and reproductive system in mice lacking prokineticin receptor PKR2. Proc. Natl. Acad. Sci. USA 2006, 103, 4140–4145. [Google Scholar] [CrossRef]
- Dodé, C.; Teixeira, L.; Levilliers, J.; Fouveaut, C.; Bouchard, P.; Kottler, M.-L.; Lespinasse, J.; Lienhardt-Roussie, A.; Mathieu, M.; Moerman, A.; et al. Kallmann syndrome: Mutations in the genes encoding prokineticin-2 and prokineticin receptor-2. PLoS Genet. 2006, 2, e175. [Google Scholar] [CrossRef]
- Pitteloud, N.; Zhang, C.; Pignatelli, D.; Li, J.D.; Raivio, T.; Cole, L.W.; Plummer, L.; Jacobson-Dickman, E.E.; Mellon, P.L.; Zhou, Q.Y.; et al. Loss-of-function mutation in the prokineticin 2 gene causes Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proc. Natl. Acad. Sci. USA 2007, 104, 17447–17452. [Google Scholar] [CrossRef] [PubMed]
- Cole, L.W.; Sidis, Y.; Zhang, C.; Quinton, R.; Plummer, L.; Pignatelli, D.; Hughes, V.A.; Dwyer, A.A.; Raivio, T.; Hayes, F.J.; et al. Mutations in prokineticin 2 and prokineticin receptor 2 genes in human gonadotrophin-releasing hormone deficiency: Molecular genetics and clinical spectrum. J. Clin. Endocrinol. Metab. 2008, 93, 3551–3559. [Google Scholar] [CrossRef]
- Zhou, S.; Li, P. The novel function of miR-3195 for mutant PROK2 (c.223-4C>A) degradation. Cell Biol. Int. 2021, 45, 404–410. [Google Scholar] [CrossRef]
- Monnier, C.; Dodé, C.; Fabre, L.; Teixeira, L.; Labesse, G.; Pin, J.P.; Hardelin, J.P.; Rondard, P. PROKR2 missense mutations associated with Kallmann syndrome impair receptor signalling activity. Hum. Mol. Genet. 2009, 18, 75–81. [Google Scholar] [CrossRef]
- Abreu, A.P.; Noel, S.D.; Xu, S.; Carroll, R.S.; Latronico, A.C.; Kaiser, U.B. Evidence of the importance of the rst intracellular loop of prokineticin receptor 2 in receptor function. Mol. Endocrinol. 2012, 26, 417–1427. [Google Scholar] [CrossRef] [PubMed]
- Libri, D.V.; Kleinau, G.; Vezzoli, V.; Busnelli, M.; Guizzardi, F.; Sinisi, A.A.; Pincelli, A.I.; Mancini, A.; Russo, G.; Beck-Peccoz, P.; et al. Germline prokineticin receptor 2 (PROKR2) variants associated with central hypogonadism cause differential modulation of distinct intracellular pathways. J. Clin. Endocrin. Metab. 2014, 99, E458–E463. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.N.; Ma, Y.T.; Liu, H.; Zhou, Q.Y.; Li, J.D. Functional rescue of Kallmann syndrome-associated prokineticinreceptor 2 (PKR2) mutants deficient in trafficking. J. Biol. Chem. 2014, 289, 15518–15526. [Google Scholar] [CrossRef]
- Song, Y.B.; Park, S.-Y.; Hwang, H.; Carrolla, R.S.; Hsuc, V.W.; Kaiser, U.B. Trafficking-defective mutant PROKR2 cycles between endoplasmic reticulum and Golgi to attenuate endoplasmic reticulum stress. Proc. Natl. Acad. Sci. USA 2022, 119, e2102248119. [Google Scholar] [CrossRef] [PubMed]
- Levit, A.; Yarnitzky, T.; Wiener, A.; Meidan, R.; Niv, M.Y. Modeling of human prokineticin recep-tors: Interactions with novel small-molecule binders and potential off-target drugs. PLoS ONE 2011, 6, e27990. [Google Scholar] [CrossRef]
- Gasser, A.; Brogi, S.; Urayama, K.; Nishi, T.; Kurose, H.; Ta, A.; Ribeiro, N.; Désaubry, L.; Nebigil, C.G. Discovery and cardioprotective effects of the rst non- peptide agonists of the G protein-coupled prokineticin receptor 1. PLoS ONE 2015, 10, e0121027. [Google Scholar] [CrossRef]
- Fullone, M.R.; Lattanzi, R.; Maftei, D.; Bonaccorsi, M.C.; Miele, R. Analysis of role of aromatic residues in extracellular loop 2 of Prokineticin receptor 2 in ligand binding probed with genetically encoded photo-crosslinkers. BBA-Biomembranes 2021, 1863, 183549. [Google Scholar] [CrossRef] [PubMed]
- Wootten, D.; Christopoulos, A.; Marti-Solano, M.; Babu, M.M.; Sexto, P.M. Mechanisms of signalling and biased agonism in G protein-coupled receptors. Nat. Rev. Mol. Cell Biol. 2018, 19, 638–653. [Google Scholar] [CrossRef]
- Sbai, O.; Monnier, C.; Dode, C.; Pin, J.P.; Hardelin, J.P.; Rondard, P. Biased signaling through G-protein-coupled PROKR2 receptors harboring missense mutations. FASEB J. 2014, 28, 3734–3744. [Google Scholar] [CrossRef]
- Cox, K.H.; Oliveira, L.M.B.; Plummer, L.; Corbin, B.; Gardella, T.; Balasubramanian, R.; Crowley, W.F. Modeling mutant/wild-type interactions to ascertain pathogenicity of PROKR2 missense variants in patients with isolated GnRH deficiency. Hum. Mol. Genet. 2018, 27, 338–350. [Google Scholar] [CrossRef]
- Zhao, Y.; Wu, J.; Jia, H.; Wang, X.; Zheng, R.; Jiang, F.; Chen, D.; Chen, Z.; Li, J. PROKR2mutations in idiopathic hypogonadotropic hypogonadism: Selective disruption of the binding to a G-protein leads to biased signaling. FASEB J. 2019, 33, 4538–4546. [Google Scholar] [CrossRef] [PubMed]
- Avbelj Stefanija, M.; Jeanpierre, M.; Sykiotis, G.P.; Young, J.; Quinton, R.; Abreu, A.P.; Plummer, L.; Au, M.G.; Dwyer, A.A.; Pitteloud, N.; et al. An ancient founder mutation in PROKR2 impairs human reproduction. Hum. Mol. Genet. 2012, 21, 4314–4324. [Google Scholar] [CrossRef] [PubMed]
- Lattanzi, R.; Maftei, D.; Fullone, M.R.; Miele, R. Trypanosoma cruzi trans-sialidase inducesSTAT3 and ERK activation by prokineticin receptor 2 binding. Cell Biochem. Funct. 2021, 39, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Chen, D.; Zhao, Y.; Men, M.; Chen, Z.; Jiang, F.; Zheng, R.; Stamou, M.I.; Plummer, L.; Balasubramanian, R.; et al. A functional spectrum of PROKR2 mutations identified in isolated hypogonadotropic hypogonadism. Hum. Mol. Genet. 2023, 32, 1722–1729. [Google Scholar] [CrossRef]
- Fukami, M.; Suzuki, E.; Izumi, Y.; Torii, T.; Narumi, S.; Igarashi, M.; Miyado, M.; Katsumi, M.; Fujisawa, Y.; Nakabayashi, K.; et al. Paradoxical gain-of-function mutant of the G-protein-coupled receptor PROKR2 promotes early puberty. J. Cell. Mol. Med. 2017, 21, 2623–2626. [Google Scholar] [CrossRef]
- Sposini, S.; Caltabiano, G.; Hanyaloglu, A.C.; Miele, R. Identification of transmembrane domains that regulate spatial arrangements and activity of prokineticin receptor 2 dimers. Mol. Cell. Endocrinol. 2015, 399, 362–372. [Google Scholar] [CrossRef]
- Marsango, S.; Bonaccorsi di Patti, M.C.; Barra, D.; Miele, R. Evidence that prokineticin receptor 2 exists as a dimer in vivo. Cell. Mol. Life Sci. 2011, 68, 2919–2929. [Google Scholar] [CrossRef]
- Aiello, F.; Cirillo, G.; Cassio, A.; Di Mase, R.; Tornese, G.; Umano, G.R.; Miraglia del Giudice, E.; Grandone, A. Molecular screening of PROKR2 gene in girls with idiopathic central precocious puberty. Ital. J. Pediatr. 2021, 47, 5. [Google Scholar] [CrossRef]
- Karstensen, H.G.; Tommerup, N. Isolated and syndromic forms of congenital anosmia. Clin. Genet. 2012, 81, 210–215. [Google Scholar] [CrossRef]
- Pfaffle, R.; Klammt, J. Pituitary transcription factors in the aetiology of combined pituitary hormone deficiency. Best Pract. Res. Clin. Endocrinol. Metab. 2011, 25, 43–60. [Google Scholar] [CrossRef]
- Reynaud, R.; Gueydan, M.; Saveanu, A.; Vallette-Kasic, S.; Enjalbert, A.; Brue, T.; Barlier, A. Genetic screening of combined pituitary hormone deficiency: Experience in 195 patients. J. Clin. Endocrinol. Metab. 2006, 91, 3329–3336. [Google Scholar] [CrossRef] [PubMed]
- Rottembourg, D.; Linglart, A.; Adamsbaum, C.; Lahlou, N.; Teinturier, C.; Bougneres, P.; Carel, J.C. Gonadotrophic status in adolescents with pituitary stalk interruption syndrome. Clin. Endocrinol. 2008, 69, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Reynaud, R.; Albarel, F.; Saveanu, A.; Kaffel, N.; Castinetti, F.; Lecomte, P.; Brauner, R.; Simonin, G.; Gaudart, J.; Carmona, E.; et al. Pituitary stalk interruption syndrome in 83 patients: Novel HESX1 mutation and severe hormonal prognosis in malformative forms. Eur. J. Endocrinol. 2011, 16, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Voutetakis, A. Pituitary stalk interruption syndrome. In Handbook of Clinical Neurology, Chapter 2; Elsevier: Amsterdam, The Netherlands, 2021; Volume 18, pp. 9–27. [Google Scholar]
- Reynaud, R.; Jayakody, S.A.; Monnier, C.; Saveanu, A.; Bouligand, J.; Guedj, A.M.; Simonin, G.; Lecomte, P.; Barlier, A.; Rondard, P.; et al. PROKR2 Variants in Multiple Hypopituitarism with Pituitary Stalk Interruption. J. Clin. Endocrinol. Metab. 2012, 97, E1068–E1073. [Google Scholar] [CrossRef]
- Asakura, Y.; Muroya, K.; Hanakawa, J.; Sato, T.; Aida, N.; Narumi, S.; Hasegawa, T.; Adachi, M. Combined pituitary hormone deficiency with unique pituitary dysplasia and morning glory syndrome related to a heterozygous PROKR2 mutation. Clin. Pediatr. Endocrinol. 2015, 24, 27–32. [Google Scholar] [CrossRef]
- Kardelen, A.D.; Najaflı, A.; Baş, F.; Karaman, B.; Toksoy, G.; Poyrazoğlu, S.; Avcı, S.; Altunoğlu, U.; Yavaş Abalı, Z.; Öztürk, A.P.; et al. PROKR2 Mutations in Patients with Short Stature Who Have Isolated Growth Hormone Deficiency and Multiple Pituitary Hormone Deficiency. J. Clin. Res. Pediatr. Endocrinol. 2023, 15, 338–347. [Google Scholar] [CrossRef]
- Martin, C.; Balasubramanian, R.; Dwyer, A.A.; Au, M.G.; Sidis, Y.; Kaiser, U.B.; Seminara, S.B.; Pitteloud, N.; Zhou, Q.Y.; Crowley, W.F., Jr. The role of the prokineticin 2 pathway in human reproduction: Evidence from the study of human and murine gene mutations. Endocr. Rev. 2010, 32, 225–246. [Google Scholar] [CrossRef]
- Maldonado-Perez, D.; Evans, I.; Denison, F.; Millar, R.P.; Jabbour, H.N. Potential roles of the prokineticins in reproduction. TRENDS Endocrinol. Metab. 2007, 18, 66–72. [Google Scholar] [CrossRef]
- Kisliouk, T.; Levy, N.; Hurwitz, A.; Meidan, R. Presence and regulation of endocrine gland vascular endothelial growth factor/prokineticin-1 and its receptors in ovarian cells. J. Clin. Endocrinol. Metab. 2003, 88, 3700–3707. [Google Scholar] [CrossRef]
- Battersby, S.; Critchley, H.O.; Morgan, K.; Millar, R.P.; Jabbour, H.N. Expression and regulation of the prokineticins (endocrine gland–derived vascular endothe- lial growth factor and Bv8) and their receptors in the human endometrium across the menstrual cycle. J. Clin. Endocrinol. Metab. 2004, 89, 2463–2469. [Google Scholar] [CrossRef]
- Fraser, H.M.; Bell, J.; Wilson, H.; Taylor, P.D.; Morgan, K.; Anderson, R.A.; Duncan, W.C. Localization and quantification of cyclic changes in the expression of endocrine gland vascular endothelial growth factor in the human corpus luteum. J. Clin. Endocrinol. Metab. 2005, 90, 427–434. [Google Scholar] [CrossRef] [PubMed]
- Brouillet, S.; Hoffmann, P.; Chauvet, S.; Salomon, A.; Chamboredon, S.; Sergent, F.; Benharouga, M.; Feige, J.J.; Alfaidy, N. Revisiting the role of hCG: New regulation of the angiogenic factor EG-VEGF and its receptors. Cell. Mol. Life Sci. 2012, 69, 1537–1550. [Google Scholar] [CrossRef] [PubMed]
- Caronia, L.M.; Martin, C.; Welt, C.K.; Sykiotis, G.P.; Quinton, R.; Thambundit, A.; Avbelj, M.; Dhruvakumar, S.; Plummer, L.; Hughes, V.A.; et al. A genetic basis for functional hypothalamic amenorrhea. N. Engl. J. Med. 2011, 364, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Goryszewska-Szczurek, E.; Baryla, M.; Kaczynski, P.; Waclawik, A. Prokineticin 1-prokineticin receptor 1 signaling in trophoblast promotes embryo implantation and placenta development. Sci. Rep. 2021, 11, 13715. [Google Scholar] [CrossRef] [PubMed]
- Goryszewska, E.; Kaczynski, P.; Balboni, G.; Waclawik, A. Prokineticin 1-prokineticin receptor 1 signaling promotes angiogenesis in the porcine endometrium during pregnancy. Biol. Reprod. 2020, 103, 654–668. [Google Scholar] [CrossRef]
- Hoffmann, P.; Saoudi, Y.; Benharouga, M.; Graham, C.H.; Schaal, J.P.; Mazouni, C.; Feige, J.J.; Alfaidy, N. Role of EG-VEGF in human placentation: Physiological and pathological implications. J. Cell. Mol. Med. 2009, 13, 2224–2235. [Google Scholar] [CrossRef]
- Alfaidy, N.; Hoffmann, P.; Boufettal, H.; Samouh, N.; Aboussaouira, T.; Benharouga, M.; Feige, J.J.; Brouillet, S. The multiple roles of EG-VEGF/PROK1 in normal and pathological placental angiogenesis. Biomed. Res. Int. 2014, 2014, 451906. [Google Scholar] [CrossRef]
- Brouillet, S.; Hoffmann, P.; Feige, J.J.; Alfaidy, N. EG-VEGF: A key endocrine factor in placental development. Trends Endocrinol. Metab. 2012, 23, 501–508. [Google Scholar] [CrossRef]
- Hoffmann, P.; Feige, J.J.; Alfaidy, N. Expression and oxygen regulation of endocrine gland-derived vascular endothelial growth factor/prokineticin-1and its receptors in human placenta during early pregnancy. Endocrinology 2006, 147, 1675–1684. [Google Scholar] [CrossRef]
- Guilini, C.; Urayama, K.; Turkeri, G.; Dedeoglu, D.B.; Kurose, H.; Messaddeq, N.; Nebigil, C.G. Divergent roles of prokineticin receptors in the endothelial cells: Angiogenesis and fenestration. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H844–H852. [Google Scholar] [CrossRef]
- Evans, J.; Catalano, R.D.; Morgan, K.; Critchley, H.O.; Millar, R.P.; Jabbour, H.N. Prokineticin 1 signaling and gene regulation in early human pregnancy. Endocrinology 2008, 149, 2877–2888. [Google Scholar] [CrossRef] [PubMed]
- Evans, J.; Catalano, R.D.; Brown, P.; Sherwin, R.; Critchley, H.O.; Fazleabas, A.T.; Jabbour, H.N. Prokineticin 1 mediates fetal-maternal dialogue regulating endometrial leukemia inhibitory factor. FASEB J. 2009, 23, 2165–2175. [Google Scholar] [CrossRef] [PubMed]
- Denison, F.C.; Battersby, S.; King, A.E.; Szuber, M.; Jabbour, H.N. Prokineticin-1: A novel mediator of the inflammatory response in third-trimester human placenta. Endocrinology 2008, 149, 3470–3477. [Google Scholar] [CrossRef]
- Cheng, X.-X.; Li, M.-Q.; Peng, T. Novel Insights into Prokineticin 1 Role in Pregnancy-related Diseases. Int. J. Med. Sci. 2024, 21, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.Z.; Gao, M.Z.; Yao, L.H.; Liang, A.J.; Zhao, X.M.; Sun, Z.G. Effect of high ovarian response on the expression of endocrine gland-derived vascular endothelial growth factor (EG-VEGF) in peri-implantation endometrium in IVF women. Int. J. Clin. Exp. Pathol. 2015, 8, 8902–8911. [Google Scholar]
- Gorowiec, M.R.; Catalano, R.D.; Norman, J.E.; Denison, F.C.; Jabbour, H.N. Prokineticin 1 induces inflammatory response in human myometrium a potential role in initiating term and preterm parturition. Am. J. Pathol. 2011, 179, 2709–2719. [Google Scholar] [CrossRef]
- Brouillet, S.; Murthi, P.; Hoffmann, P.; Salomon, A.; Sergent, F.; De Mazancourt, P.; Dakouane-Giudicelli, M.; Dieudonné, M.N.; Rozenberg, P.; Vaiman, D.; et al. EG-VEGF controls placental growth and survival in normal and pathological pregnancies: Case of fetal growth restriction (FGR). Cell. Mol. Life Sci. 2013, 70, 511–525. [Google Scholar] [CrossRef] [PubMed]
- Shaw, J.L.; Denison, F.C.; Evans, J.; Durno, K.; Williams, A.R.; Entrican, G.; Critchley, H.O.D.; Jabbour, H.N.; Horne, A.W. Evidence of prokineticin dysregulation in fallopian tube from women with ectopic pregnancy. Fertil. Steril. 2010, 94, 1601–1608.e1. [Google Scholar] [CrossRef]
- Ferrara, N.; Frantz, G.; LeCouter, J.; Dillard-Telm, L.; Pham, T.; Draksharapu, A.; Giordano, T.; Peale, F. Differential expression of the angiogenic factor genes vascular endothelial growth factor (VEGF) and endocrine gland-derived VEGF in normal and polycystic human ovaries. Am. J. Pathol. 2003, 162, 1881–1893. [Google Scholar] [CrossRef]
- Arias-Sosa, L.A.; Acosta, I.D.; Lucena-Quevedo, E.; Moreno-Ortiz, H.M.; Esteban-Pérez, C.; Maribel Forero-Castro, M. Genetic and epigenetic variations associated with idiopathic recurrent pregnancy loss. J. Assist. Reprod. Genet. 2018, 35, 355–366. [Google Scholar] [CrossRef]
- Su, M.T.; Lin, S.H.; Lee, I.W. Polymorphisms of endocrine gland-derived vascular endothelial growth factor gene and its receptor genes are associated with recurrent pregnancy loss. Hum. Reprod. 2010, 25, 2923–2930. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.L.; Zhang, Z.F.; Wang, J.; Miao, M.H.; Xu, J.H.; Shen, Y.P.; Chen, A.M.; Du, J.; Yuan, W.J. Zhejiang Association between polymorphisms of prokineticin receptor (PKR1 rs4627609 and PKR2 rs6053283) and recurrent pregnancy loss. J. Zhejiang Univ. Sci. B 2016, 17, 218–224. [Google Scholar] [CrossRef] [PubMed]
- Su, M.T.; Lin, S.H.; Chen, Y.C.; Wu, L.W.; Kuo, P.L. Prokineticin receptor variants (PKR1-I379V and PKR2-V331M) are protective genotypes in human early pregnancy. Reproduction 2013, 146, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Su, M.-T.; Huang, J.-Y.; Tsai, H.-L.; Chen, Y.-C.; Kuo, P.-L. A Common Variant of PROK1 (V67I) Acts as a Genetic Modifier in Early Human Pregnancy through Down-Regulation of Gene Expression. Int. J. Mol. Sci. 2016, 17, 162. [Google Scholar] [CrossRef]
- Su, M.T.; Lin, S.-H.; Chen, Y.-C.; Kuo, P.-L. Gene-gene interactions and gene polymorphisms of VEGFA and EG-VEGF gene systems in recurrent pregnancy loss. J. Assist. Reprod. Genet. 2014, 31, 699–705. [Google Scholar] [CrossRef]
- Alfaidy, N.; Brouillet, S.; Rajaraman, G.; Kalionis, B.; Hoffmann, P.; Barjat, Y.; Benharouga, M.; Murthi, P. The Emerging Role of the Prokineticins and Homeobox Genes in the Vascularization of the Placenta: Physiological and Pathological Aspects. Front. Physiol. 2020, 12, 591850. [Google Scholar] [CrossRef]
- Ngan, E.S.; Lee, K.Y.; Sit, F.Y.; Poon, H.C.; Chan, J.K.; Sham, M.H.; Lui, V.C.H.; Tam, P.K.H. Prokineticin-1 modulates proliferation and differentiation of enteric neural crest cells. Biochim. Biophys. Acta 2007, 1773, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Ngan, E.S.; Shum, C.K.; Poon, H.C.; Sham, M.H.; Garcia-Barcelo, M.M.; Lui, V.C.; Tam, P.K. Prokineticin-1 (Prok-1) works coordinately with glial cell line-derived neurotrophic factor (GDNF) to mediate proliferation and differentiation of enteric neural crest cells. Biochim. Biophys. Acta 2008, 1783, 467–478. [Google Scholar] [CrossRef]
- Ruiz-Ferrer, M.; Torroglosa, A.; Nunez-Torres, R.; de Agustin, J.C.; Antinolo, G.; Borrego, S. Expression of PROKR1 and PROKR2 in human enteric neural precursor cells and identification of sequence variants suggest a role in HSC. PLoS ONE 2011, 6, e23475. [Google Scholar] [CrossRef]
- Trang, K.; Struan, F.A. Grant Genetics and epigenetics in the obesity phenotyping scenario. Rev. Endocr. Metab. Disord. 2023, 10, 1–19. [Google Scholar]
- Berruien, N.N.A.; Smith, C.L. Emerging roles of melanocortin receptor accessory proteins (MRAP and MRAP2) in physiology and pathophysiology. Gene 2020, 757, 144949. [Google Scholar] [CrossRef] [PubMed]
- Baron, M.; Maillet, J.; Huyvaert, M.; Dechaume, A.; Boutry, R.; Loiselle, H.; Durand, E.; Toussaint, B.; Vaillant, E.; Philippe, J.; et al. Loss-of-function mutations in MRAP2 are pathogenic in hyperphagic obesity with hyperglycemia and hypertension. Nat. Med. 2019, 25, 1733–1738. [Google Scholar] [CrossRef] [PubMed]
- Chaly, A.L.; Srisai, D.; Gardner, E.E.; Sebag, J.A. The Melanocortin Receptor Accessory Protein 2 promotes food intake through inhibition of the Prokineticin Receptor-1. eLife 2016, 5, e12397. [Google Scholar] [CrossRef]
- Maftei, D.; Lattanzi, R.; Vincenzi, M.; Squillace, S.; Fullone, M.R.; Miele, R. The balance of concentration between Prokineticin 2β and Prokineticin 2 modulates the food intake by STAT3 signaling. BBA Adv. 2021, 1, 100028. [Google Scholar] [CrossRef]
- Han, Y.; Xia, G.; Harris, L.; Liu, P.; Guan, D. Reversal of Obesity by Enhancing Slow-wave Sleep via a Prokineticin Receptor Neural Circuit. bioRxiv 2024. bioRxiv: 591948. [Google Scholar]
- Gardiner, J.V.; Bataveljic, A.; Patel, N.A.; Bewick, G.A.; Roy, D.; Campbell, D.; Greenwood, H.C.; Murphy, K.G.; Hameed, S.; Jethwa, P.H.; et al. Prokineticin 2 is a hypothalamic neuropeptide that potently inhibits food intake. Diabetes 2010, 59, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Mortreux, M.; Foppen, E.; Denis, R.G.; Montaner, M.; Kassis, N.; Denom, J.; Vincent, M.; Fumeron, F.; Kujawski-Lafourcade, M.; AndreÃÅelli, F.; et al. New roles for prokineticin 2 in feeding behavior, insulin resistance and type 2 diabetes: Studies in mice and humans. Mol. Metab. 2019, 29, 182–196. [Google Scholar] [CrossRef]
- Lattanzi, R.; Fullone, M.R.; De Biase, A.; Maftei, D.; Vincenzi, M.; Miele, R. Biochemical Characterization of Prokineticin 2 binding to Prokineticin receptor 1 in Zebrafish. Neuropeptides 2024, 107, 102456, submitted. [Google Scholar] [CrossRef]
- Fullone, M.R.; Maftei, D.; Vincenzi, M.; Lattanzi, R.; Miele, R. MRAP2a binds and modulates activity and localization of Prokineticin receptor 1 In Zebrafish. Int. J. Mol. Sci. 2024, 25, 7816. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Polymorphism | Disease | References |
|---|---|---|---|
| PK1 | rs 62623571 | HSCR | Ruiz-Ferrer et al., 2011 |
| rs7514102 | RPL | Su et al., 2016 | |
| PK2 | rs1316780 rs10865660 rs3796224 rs1374913 | MDD, BP | Kishi et al., 2009 |
| rs75433452 | MUD | Jiang et al., 2023 | |
| PKR1 | rs4627609 rs347157 | RPL | Su et al., 2010 Cao et al., 2016 |
| PKR2 | rs17721321 rs6085086 rs3746684 rs3746682 rs4815787 | MDD, BP | Kishi et al., 2009 |
| rs6085086 rs3746682 rs4815787 | MUD | Guerina et al., 2021. | |
| rs3746682 | CPP | Aiello et al., 2021 | |
| rs6053283 rs5282731 | RPL | Su et al., 2010 Cao et al., 2016 | |
| Su et al., 2013; Su et al., 2014 | |||
| rs78861628 | HSCR | Rui-Ferrer et al., 2011 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lattanzi, R.; Miele, R. Genetic Polymorphisms of Prokineticins and Prokineticin Receptors Associated with Human Disease. Life 2024, 14, 1254. https://doi.org/10.3390/life14101254
Lattanzi R, Miele R. Genetic Polymorphisms of Prokineticins and Prokineticin Receptors Associated with Human Disease. Life. 2024; 14(10):1254. https://doi.org/10.3390/life14101254
Chicago/Turabian StyleLattanzi, Roberta, and Rossella Miele. 2024. "Genetic Polymorphisms of Prokineticins and Prokineticin Receptors Associated with Human Disease" Life 14, no. 10: 1254. https://doi.org/10.3390/life14101254
APA StyleLattanzi, R., & Miele, R. (2024). Genetic Polymorphisms of Prokineticins and Prokineticin Receptors Associated with Human Disease. Life, 14(10), 1254. https://doi.org/10.3390/life14101254