

Comparative Analysis of Gut Microbiota in Humans Living with and Without Companion Animals

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Participants
2.2. Sample Collection and DNA Extraction
2.3. 16S rRNA Gene Sequencing
2.4. Bioinformatic Analysis and Statistical Analysis
3. Results
3.1. Clinical Data of Participants
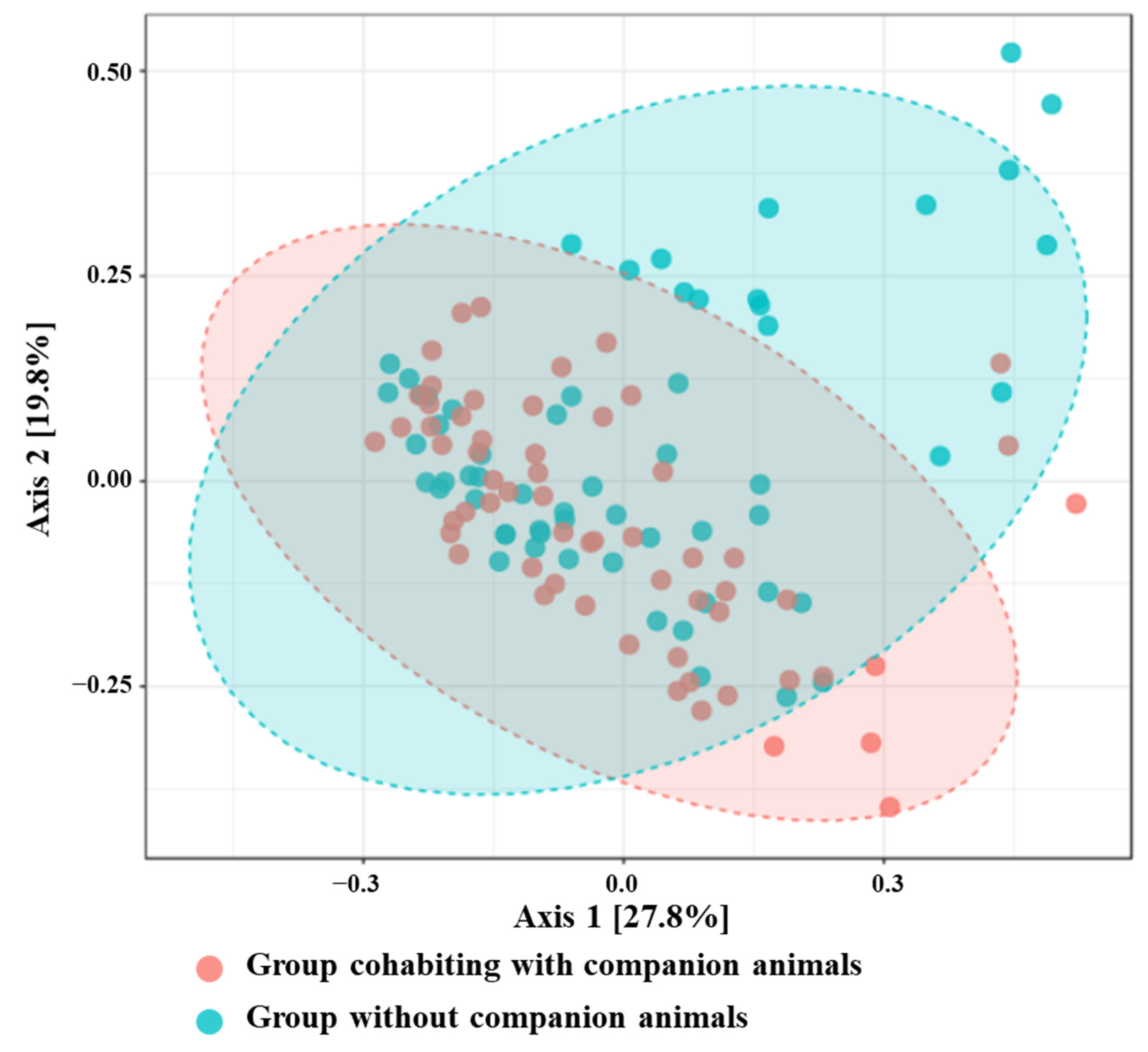
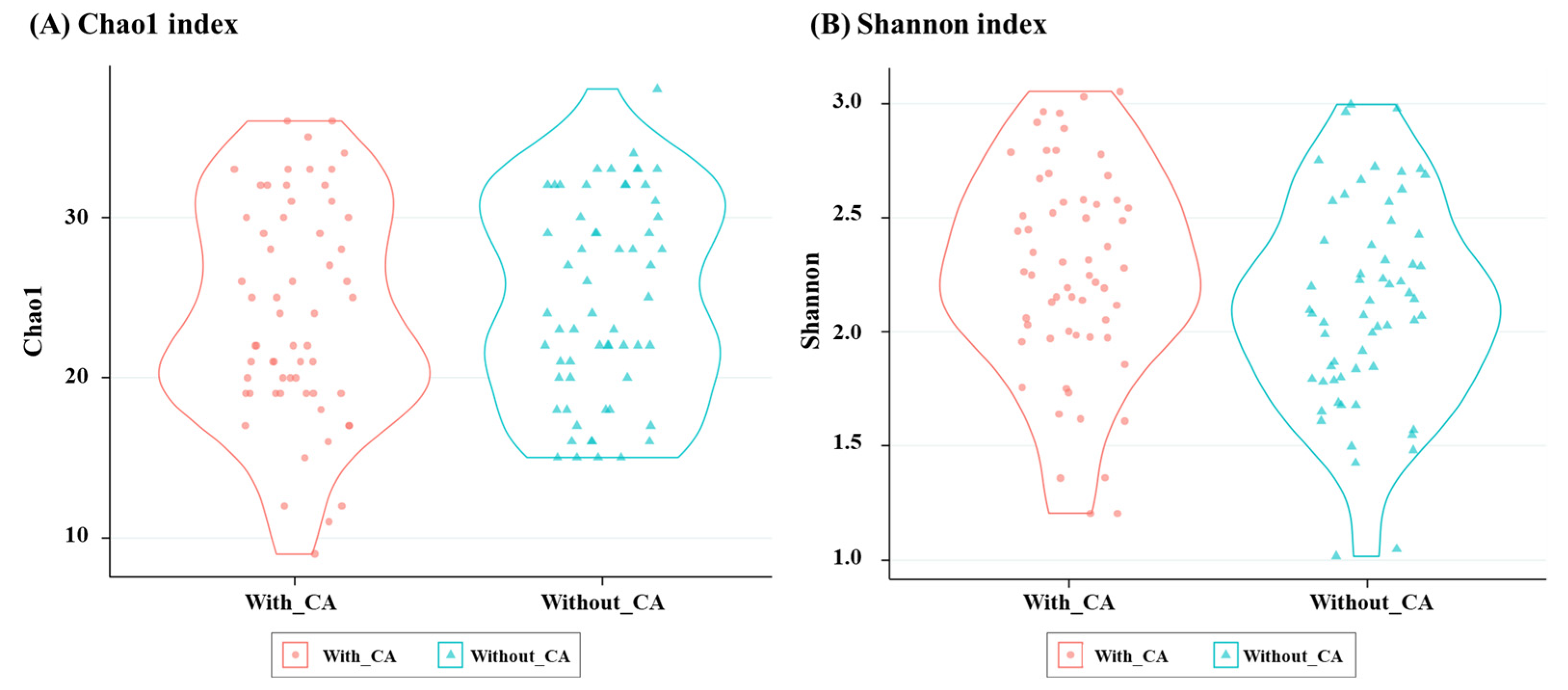
3.2. Difference in the Gut Microbiota: Alpha Diversity and Beta Diversity
3.3. Sharing/Transfer of the Gut Microbiota in the Same Environment
3.4. Overall Composition of the Intestinal Microbiota
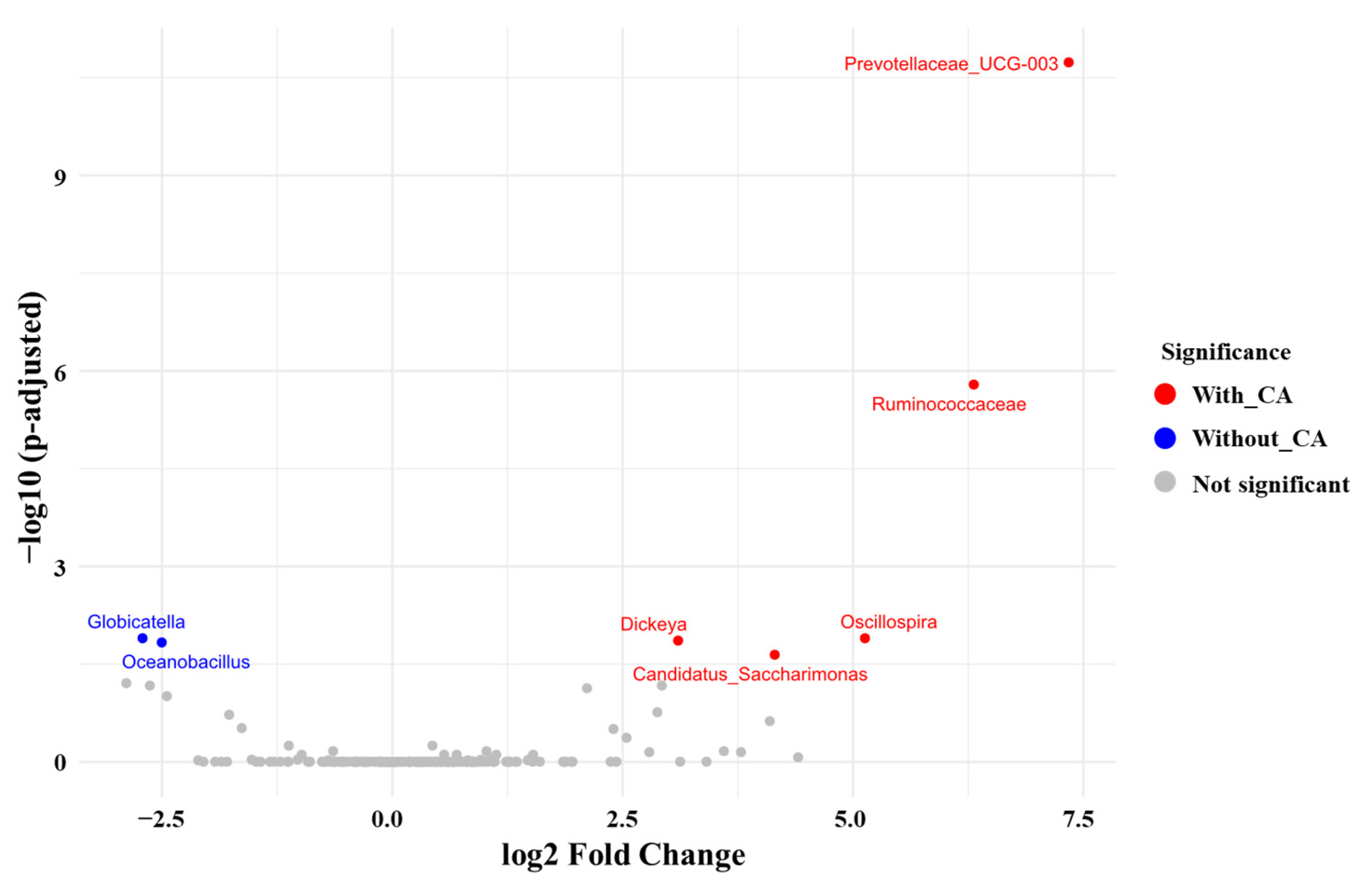
3.5. Differences in the Gut Microbiota
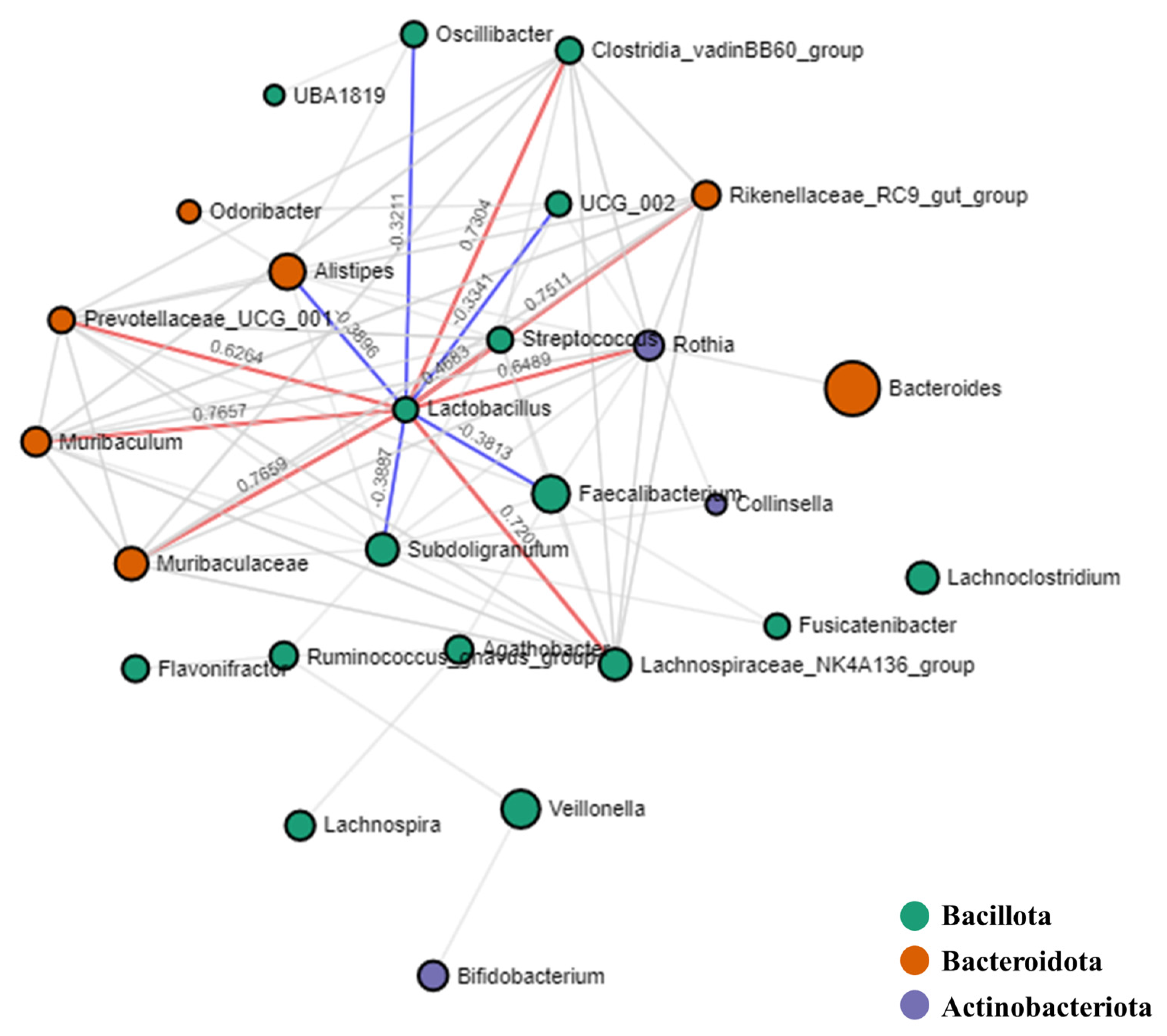
3.6. Microbial Interactions: Correlation Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Alessandri:, G.; Milani, C.; Mancabelli, L.; Mangifesta, M.; Lugli, G.A.; Viappiani, A.; Duranti, S.; Turroni, F.; Ossiprandi, M.C.; van Sinderen, D.; et al. Metagenomic Dissection of the Canine Gut Microbiota: Insights into Taxonomic, Metabolic and Nutritional Features. Environ. Microbiol. 2019, 21, 1331–1343. [Google Scholar] [CrossRef] [PubMed]
- Kates, A.E.; Jarrett, O.; Skarlupka, J.H.; Sethi, A.; Duster, M.; Watson, L.; Suen, G.; Poulsen, K.; Safdar, N. Household Pet Ownership and the Microbial Diversity of the Human Gut Microbiota. Front. Cell. Infect. Microbiol. 2020, 10, 73. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yi, H.; Liu, S.; Zhang, Y.; Su, Y.; Liu, X.; Bi, S.; Lai, H.; Zeng, Z.; Li, G. Probiotics Improve Eating Disorders in Mandarin Fish (Siniperca chuatsi) Induced by a Pellet Feed Diet via Stimulating Immunity and Regulating Gut Microbiota. Microorganisms 2021, 9, 1288. [Google Scholar] [CrossRef]
- Illescas, O.; Rodríguez-Sosa, M.; Gariboldi, M. Mediterranean Diet to Prevent the Development of Colon Diseases: A Meta-Analysis of Gut Microbiota Studies. Nutrients 2021, 13, 2234. [Google Scholar] [CrossRef]
- Odamaki, T.; Kato, K.; Sugahara, H.; Hashikura, N.; Takahashi, S.; Xiao, J.; Abe, F.; Osawa, R. Age-Related Changes in Gut Microbiota Composition from Newborn to Centenarian: A Cross-Sectional Study. BMC Microbiol. 2016, 16, 90. [Google Scholar] [CrossRef]
- Redfern, A.; Suchodolski, J.; Jergens, A. Role of the Gastrointestinal Microbiota in Small Animal Health and Disease. Vet. Rec. 2017, 181, 370. [Google Scholar] [CrossRef]
- Do, K.-H.; Ko, S.-H.; Kim, K.B.; Seo, K.; Lee, W.-K. Comparative Study of Intestinal Microbiome in Patients with Ulcerative Colitis and Healthy Controls in Korea. Microorganisms 2023, 11, 2750. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, M.; Raes, J.; Pelletier, E.; Paslier, D.L.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.-M.; et al. Enterotypes of the Human Gut Microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef]
- Ley, R.E.; Bäckhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity Alters Gut Microbial Ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef]
- Caugant, D.A.; Levin, B.R.; Selander, R.K. Distribution of Multilocus Genotypes of Escherichia coli within and between Host Families. J. Hyg. 1984, 92, 377–384. [Google Scholar] [CrossRef]
- Zoetendal, E.G.; Akkermans, A.D.L.; Vliet, W.M.A.; de Visser, J.A.G.M.; de Vos, W.M. The Host Genotype Affects the Bacterial Community in the Human Gastronintestinal Tract. Microb. Ecol. Health Dis. 2009, 13, 129–134. [Google Scholar] [CrossRef]
- Stewart, J.A.; Chadwick, V.S.; Murray, A. Investigations into the Influence of Host Genetics on the Predominant Eubacteria in the Faecal Microflora of Children. J. Méd. Microbiol. 2005, 54, 1239–1242. [Google Scholar] [CrossRef] [PubMed]
- Rajilić-Stojanović, M.; Smidt, H.; Vos, W.M.D. Diversity of the Human Gastrointestinal Tract Microbiota Revisited. Environ. Microbiol. 2007, 9, 2125–2136. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A Core Gut Microbiome in Obese and Lean Twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human Gut Microbiome Viewed across Age and Geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef]
- Hwang, I.-C.; Vasquez, R.; Song, J.H.; Engstrand, L.; Valeriano, V.D.; Kang, D.-K. Alterations in the Gut Microbiome and Its Metabolites Are Associated with the Immune Response to Mucosal Immunization with Lactiplantibacillus plantarum-Displaying Recombinant SARS-CoV-2 Spike Epitopes in Mice. Front. Cell. Infect. Microbiol. 2023, 13, 1242681. [Google Scholar] [CrossRef]
- Song, S.J.; Lauber, C.; Costello, E.K.; Lozupone, C.A.; Humphrey, G.; Berg-Lyons, D.; Caporaso, J.G.; Knights, D.; Clemente, J.C.; Nakielny, S.; et al. Cohabiting Family Members Share Microbiota with One Another and with Their Dogs. eLife 2013, 2, e00458. [Google Scholar] [CrossRef]
- Coelho, L.P.; Kultima, J.R.; Costea, P.I.; Fournier, C.; Pan, Y.; Czarnecki-Maulden, G.; Hayward, M.R.; Forslund, S.K.; Schmidt, T.S.B.; Descombes, P.; et al. Similarity of the Dog and Human Gut Microbiomes in Gene Content and Response to Diet. Microbiome 2018, 6, 72. [Google Scholar] [CrossRef]
- Hooda, S.; Minamoto, Y.; Suchodolski, J.S.; Swanson, K.S. Current State of Knowledge: The Canine Gastrointestinal Microbiome. Anim. Health Res. Rev. 2012, 13, 78–88. [Google Scholar] [CrossRef]
- Deng, P.; Swanson, K.S. Gut Microbiota of Humans, Dogs and Cats: Current Knowledge and Future Opportunities and Challenges. Br. J. Nutr. 2015, 113, S6–S17. [Google Scholar] [CrossRef]
- Moon, C.D.; Young, W.; Maclean, P.H.; Cookson, A.L.; Bermingham, E.N. Metagenomic Insights into the Roles of Proteobacteria in the Gastrointestinal Microbiomes of Healthy Dogs and Cats. Microbiologyopen 2018, 7, e00677. [Google Scholar] [CrossRef] [PubMed]
- Grigor’eva, I.N. Gallstone Disease, Obesity and the Firmicutes/Bacteroidetes Ratio as a Possible Biomarker of Gut Dysbiosis. J. Pers. Med. 2020, 11, 13. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhang, Q.; Zhao, Y.; Zou, Y.; Chen, M.; Zhou, S.; Wang, Z. The Relationship of Megamonas species with Nonalcoholic Fatty Liver Disease in Children and Adolescents Revealed by Metagenomics of Gut Microbiota. Sci. Rep. 2022, 12, 22001. [Google Scholar] [CrossRef] [PubMed]
- Stull, J.W.; Brophy, J.; Weese, J.S. Reducing the Risk of Pet-Associated Zoonotic Infections. Can. Méd. Assoc. J. 2015, 187, 736–743. [Google Scholar] [CrossRef] [PubMed]
- Azad, M.B.; Konya, T.; Maughan, H.; Guttman, D.S.; Field, C.J.; Sears, M.R.; Becker, A.B.; Scott, J.A.; Kozyrskyj, A.L.; Investigators, C.S. Infant Gut Microbiota and the Hygiene Hypothesis of Allergic Disease: Impact of Household Pets and Siblings on Microbiota Composition and Diversity. Allergy Asthma Clin. Immunol. 2013, 9, 15. [Google Scholar] [CrossRef]
- Tun, H.M.; Konya, T.; Takaro, T.K.; Brook, J.R.; Chari, R.; Field, C.J.; Guttman, D.S.; Becker, A.B.; Mandhane, P.J.; Turvey, S.E.; et al. Exposure to Household Furry Pets Influences the Gut Microbiota of Infants at 3–4 Months Following Various Birth Scenarios. Microbiome 2017, 5, 40. [Google Scholar] [CrossRef]
- Liu, S.; Wang, Z.; Zhu, R.; Wang, F.; Cheng, Y.; Liu, Y. Three Differential Expression Analysis Methods for RNA Sequencing: Limma, EdgeR, DESeq2. J. Vis. Exp. 2021, 175, e62528. [Google Scholar] [CrossRef]
- Friedman, J.; Alm, E.J. Inferring Correlation Networks from Genomic Survey Data. PLoS Comput. Biol. 2012, 8, e1002687. [Google Scholar] [CrossRef]
- Kurtz, Z.D.; Müller, C.L.; Miraldi, E.R.; Littman, D.R.; Blaser, M.J.; Bonneau, R.A. Sparse and Compositionally Robust Inference of Microbial Ecological Networks. PLoS Comput. Biol. 2015, 11, e1004226. [Google Scholar] [CrossRef]
- Yun, S.; Seo, Y.; Lee, Y.; Lee, D.T. Gut Microbiome Related to Metabolic Diseases after Moderate-to-Vigorous Intensity Exercise. J. Exerc. Sci. Fit. 2024, 22, 375–382. [Google Scholar] [CrossRef]
- Yeo, S.; Park, H.; Kim, H.; Ryu, C.B.; Huh, C.S. Selenobaculum gbiensis Gen. Nov. Sp. Nov., a New Bacterium Isolated from the Gut Microbiota of a Patient with Crohn’s Disease. Sci. Rep. 2023, 13, 14835. [Google Scholar] [CrossRef] [PubMed]
- Park, H.-E.; Kim, Y.J.; Kim, M.; Kim, H.; Do, K.-H.; Kim, J.K.; Ham, J.-S.; Lee, W.-K. Effects of Queso Blanco Cheese Containing Bifidobacterium Longum KACC 91563 on Fecal Microbiota, Metabolite and Serum Cytokine in Healthy Beagle Dogs. Anaerobe 2020, 64, 102234. [Google Scholar] [CrossRef] [PubMed]
- El-Bahnasawy, M.M.; Khalil, H.H.M.; Morsy, T.A. Babesiosis in an Egyptian Boy Aquired from Pet Dog, and a General Review. J. Egypt. Soc. Parasitol. 2011, 41, 99–108. [Google Scholar] [PubMed]
- Hetem, D.J.; Pekelharing, M.; Thijsen, S.F.T. Probable Transmission of Yersinia enterocolitica from a Pet Dog with Diarrhoea to a 1-Year-Old Infant. BMJ Case Rep. 2013, 2013, bcr2013200046. [Google Scholar] [CrossRef]
- Maraki, S.; Kastanis, G.; Stafylaki, D.; Masunt, S.; Kapsetakis, P.; Scoulica, E. Pasteurella multocida Wound Infection Transmitted by a Pet Dog. Germs 2018, 8, 214–217. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Participants Cohabiting with Companion Animals | Participants Without Companion Animals | ||||
|---|---|---|---|---|---|---|
| Adult (n = 40) | Children (n = 20) | Sub-Total (n = 60) | Adult (n = 40) | Children (n = 20) | Sub-Total (n = 60) | |
| Age | 41.10 ± 5.60 | 9.55 ± 2.91 | 30.58 ± 15.76 | 42.63 ± 3.99 | 9.90 ± 2.05 | 31.72 ± 15.93 |
| Male/Female | 20/20 | 11/9 | 31/29 | 20/20 | 9/11 | 29/31 |
| Total Bilirubin (mg/dL) | 0.5 ± 0.3 | 0.3 ± 0.1 | 0.4 ± 0.3 | 0.5 ± 0.3 | 0.3 ± 0.1 | 0.4 ± 0.3 |
| ALP (1) (U/L) | 69.5 ± 21.2 | 282.2 ± 73.0 | 140.4 ± 110.6 | 69.6 ± 17.4 | 275.1 ± 65.7 | 138.1 ± 105.5 |
| AST (2) (U/L) | 21.8 ± 9.0 | 27.8 ± 5.2 | 23.8 ± 8.4 | 22.1 ± 15.6 | 24.6 ± 5.5 | 22.9 ± 13.1 |
| ALT (3) (U/L) | 25.4 ± 27.3 | 18.5 ± 21.0 | 23.1 ± 25.4 | 22.6 ± 23.5 | 14.3 ± 6.0 | 19.8 ± 19.8 |
| γ- GTP (4) (U/L) | 31.0 ± 22.6 | 14.3 ± 8.2 | 25.4 ± 20.5 | 28.0 ± 21.0 | 13.0 ± 4.0 | 23.0 ± 18.6 |
| Glucose (mg/dL) | 100.6 ± 22.6 | 90.1 ± 11.6 | 97.1 ± 20.2 | 94.9 ± 17.7 | 86.0 ± 11.4 | 92.0 ± 16.4 |
| Total Cholesterol (mg/dL) | 201.0 ± 41.2 | 168.6 ± 28.3 | 190.2 ± 40.3 | 186.8 ± 35.3 | 163.9 ± 28.6 | 179.1 ± 34.8 |
| Free Fatty Acid (μ Eq/L) | 392.0 ± 221.5 | 386.9 ± 189.0 | 390.3 ± 209.6 | 369.9 ± 253.2 | 407.1 ± 229.3 | 382.3 ± 244.2 |
| HDL (5) Cholesterol (mg/dL) | 56.7 ± 13.5 | 56.8 ± 9.5 | 56.8 ± 12.2 | 58.9 ± 16.6 | 54.6 ± 11.2 | 60.8 ± 15.2 |
| LDL (6) Cholesterol (mg/dL) | 119.3 ± 37.5 | 94.8 ± 23.4 | 111.1 ± 35.3 | 107.5 ± 31.5 | 89.4 ± 26.6 | 101.5 ± 30.9 |
| Triglyceride (mg/dL) | 210.7 ± 167.2 | 133.5 ± 93.0 | 185.0 ± 150.4 | 193.4 ± 152.4 | 95.0 ± 45.1 | 160.6 ± 134.9 |
| Insulin (μ IU/mL) | 30.1 ± 33.4 | 39.5 ± 78.0 | 33.2 ± 52.1 | 24.3 ± 22.1 | 18.2 ± 18.7 | 22.3 ± 21.1 |
| White Blood Cells (×103/μL) ‡ | 7.4 ± 1.8 | 8.1 ± 1.6 | 7.6 ± 1.8 ‡ | 6.6 ± 1.6 | 7.5 ± 1.2 | 6.9 ± 1.5 ‡ |
| Red Blood Cells (×106/μL) | 4.6 ± 0.6 | 4.5 ± 0.4 | 4.6 ± 0.5 | 4.5 ± 0.5 | 4.5 ± 0.3 | 4.5 ± 0.4 |
| Hemoglobin (g/dL) † | 14.1 ± 1.6 † | 13.0 ± 1.1 | 13.8 ± 1.6 | 13.5 ± 1.6 † | 12.9 ± 0.7 | 13.3 ± 1.4 |
| Hematocrit (%) | 42.8 ± 4.4 | 39.0 ± 3.2 | 41.6 ± 4.4 | 41.5 ± 4.3 | 39.0 ± 3.0 | 40.7 ± 4.1 |
| Platelet (×103/μL) ‡ | 295. ± 62.1 | 345.9 ± 57.2 | 311.5 ± 64.6 ‡ | 265.1 ± 70.5 | 322.3 ± 61.8 | 284.2 ± 72.4 ‡ |
| MCV (7) (fL) | 92.8 ± 4.7 | 86.0 ± 3.7 | 90.6 ± 5.5 | 92.1 ± 5.4 | 86.1 ± 3.3 | 90.1 ± 5.6 |
| MCH (8) (pg) | 30.6 ± 1.6 | 28.6 ± 1.3 | 29.9 ± 1.8 | 29.8 ± 2.4 | 28.4 ± 1.0 | 29.3 ± 2.1 |
| MCHC (9) (g/dL) ‡ | 32.9 ± 1.0 | 33.3 ± 0.9 | 33.1 ± 1.0 ‡ | 32.4 ± 1.3 | 33.0 ± 1.0 | 32.6 ± 1.3 ‡ |
| Segmented Neutrophil (%) | 58.0 ± 6.8 | 49.8 ± 10.5 | 55.4 ± 9.0 | 55.3 ± 8.4 | 47.8 ± 8.4 | 52.8 ± 9.0 |
| Lymphocyte (%) | 31.6 ± 6.0 | 38.3 ± 8.7 | 33.7 ± 7.6 | 34.0 ± 7.9 | 42.1 ± 8.2 | 36.7 ± 8.8 |
| Monocyte (%) | 7.5 ± 1.7 | 6.9 ± 1.4 | 7.3 ± 1.6 | 7.2 ± 1.6 | 6.6 ± 1.3 | 7.0 ± 1.5 |
| Eosinophil (%) | 2.3 ± 1.3 | 4.3 ± 3.3 | 2.9 ± 2.3 | 2.9 ± 2.1 | 3.1 ± 2.4 | 2.9 ± 2.2 |
| Basophil (%) | 0.7 ± 0.3 | 0.7 ± 0.3 | 0.7 ± 0.3 | 0.7 ± 0.3 | 0.5 ± 0.2 | 0.6 ± 0.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Do, K.-H.; Park, J.; Kim, N.; Ryu, D.; Kim, M.-G.; Ahn, H.; Kim, H.; Hwang, J.-G.; Park, M.-K.; Seo, K.-W.; et al. Comparative Analysis of Gut Microbiota in Humans Living with and Without Companion Animals. Life 2024, 14, 1621. https://doi.org/10.3390/life14121621
Do K-H, Park J, Kim N, Ryu D, Kim M-G, Ahn H, Kim H, Hwang J-G, Park M-K, Seo K-W, et al. Comparative Analysis of Gut Microbiota in Humans Living with and Without Companion Animals. Life. 2024; 14(12):1621. https://doi.org/10.3390/life14121621
Chicago/Turabian StyleDo, Kyung-Hyo, Jiwon Park, Nahee Kim, Dahye Ryu, Min-Gyu Kim, Hyunjung Ahn, Hakhyun Kim, Jun-Gi Hwang, Min-Kyu Park, Kwang-Won Seo, and et al. 2024. "Comparative Analysis of Gut Microbiota in Humans Living with and Without Companion Animals" Life 14, no. 12: 1621. https://doi.org/10.3390/life14121621
APA StyleDo, K.-H., Park, J., Kim, N., Ryu, D., Kim, M.-G., Ahn, H., Kim, H., Hwang, J.-G., Park, M.-K., Seo, K.-W., & Lee, W.-K. (2024). Comparative Analysis of Gut Microbiota in Humans Living with and Without Companion Animals. Life, 14(12), 1621. https://doi.org/10.3390/life14121621