
Characterization of the Complete Mitochondrial Genome of Wintersweet (Chimonanthus praecox) and Comparative Analysis within Magnoliids
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials, Library Preparation, and Genome Sequence
2.2. Mitochondrial Genome Assembly and Annotation
2.3. Repetitive Sequence Analysis
2.4. Codon Usage Analysis
2.5. Synonymous and Nonsynonymous Substitution Ratio
2.6. Phylogenetic Analysis
3. Results and Discussion
3.1. Mitogenome Structure, Organization, and Composition
3.2. Genomic Features of the C. praecox Mitogenome
3.3. Codon Usage Analysis
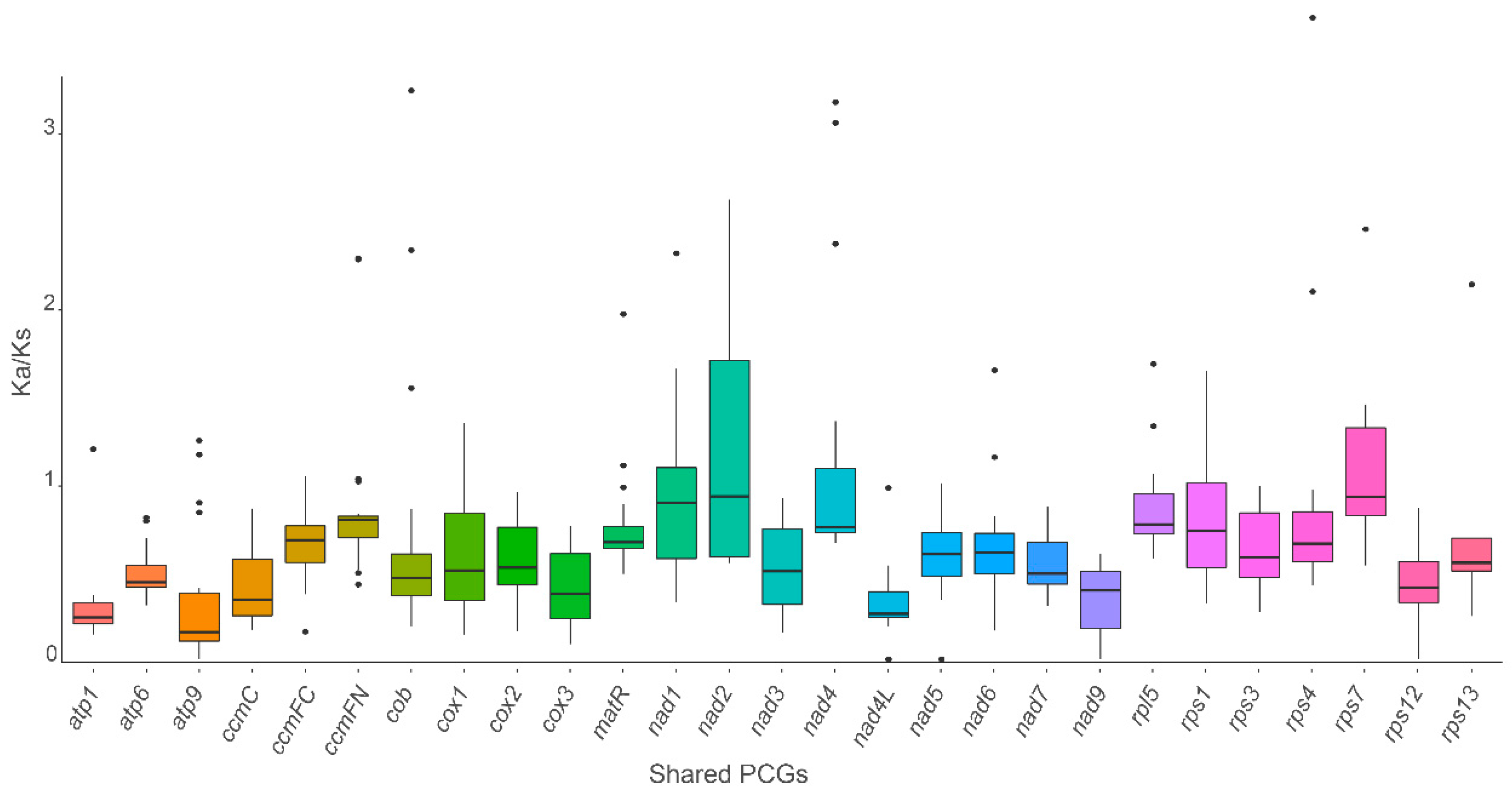
3.4. Synonymous and Nonsynonymous Substitution Ratio
3.5. Repetitive Sequence Analysis
3.6. Phylogenetic Analysis of C. praecox within Magnoliids
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nicely, K.A. A monographic study of the Calycanthaceae. Castanea 1965, 30, 38–81. [Google Scholar]
- Zhao, Y.; Zhang, Y.; Wang, Z.Z. Chemical Composition and Biological Activities of Essential Oil from Flower of Chimonanthus praecox (L.) Link. Lishizhen Med. Mater. Med. Res. 2010, 21, 622–625. [Google Scholar]
- Ueyama, Y.; Hashimoto, S.; Nii, H.; Furukawa, K. The volatile constituents of the flower concrete of Chimonanthus praecox Link. from China. Flavour Fragr. J. 1990, 5, 85–88. [Google Scholar] [CrossRef]
- Zhang, J.W.; Gao, J.M.; Xu, T.; Zhang, X.C.; Ma, Y.T.; Jarussophon, S.; Konishi, Y. Antifungal activity of alkaloids from the seeds of Chimonanthus praecox. Chem. Biodivers. 2009, 6, 838–845. [Google Scholar] [CrossRef] [PubMed]
- Lou, H.-Y.; Zhang, Y.; Ma, X.-P.; Jiang, S.; Wang, X.-P.; Yi, P.; Liang, G.-Y.; Wu, H.-M.; Feng, J.; Jin, F.-Y.; et al. Novel sesquiterpenoids isolated from Chimonanthus praecox and their antibacterial activities. Chin. J. Nat. Med. 2018, 16, 621–627. [Google Scholar] [CrossRef] [PubMed]
- Soltis, P.S.; Soltis, D.E. The origin and diversification of angiosperms. Am. J. Bot. 2004, 91, 1614–1626. [Google Scholar] [CrossRef] [PubMed]
- The Angiosperm Phylogeny Group. An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG III. Bot. J. Linn. Soc. 2009, 161, 105–121. [Google Scholar] [CrossRef]
- Palmer, J.D.; Soltis, D.E.; Chase, M.W. The plant tree of life: An overview and some points of view. Am. J. Bot. 2004, 91, 1437–1445. [Google Scholar] [CrossRef] [PubMed]
- Endress, P.K. The evolution of floral biology in basal angiosperms. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2010, 365, 411–421. [Google Scholar] [CrossRef]
- Qin, L.; Hu, Y.; Wang, J.; Wang, X.; Zhao, R.; Shan, H.; Li, K.; Xu, P.; Wu, H.; Yan, X.; et al. Insights into angiosperm evolution, floral development and chemical biosynthesis from the Aristolochia fimbriata genome. Nat. Plants 2021, 7, 1239–1253. [Google Scholar] [CrossRef]
- Jansen, R.K.; Cai, Z.; Raubeson, L.A.; Daniell, H.; dePamphilis, C.W.; Leebens-Mack, J.; Müller, K.F.; Guisinger-Bellian, M.; Haberle, R.C.; Hansen, A.K.; et al. Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc. Natl. Acad. Sci. USA 2007, 104, 19369–19374. [Google Scholar] [CrossRef]
- Chaw, S.-M.; Liu, Y.-C.; Wu, Y.-W.; Wang, H.-Y.; Lin, C.-Y.I.; Wu, C.-S.; Ke, H.-M.; Chang, L.-Y.; Hsu, C.-Y.; Yang, H.-T.; et al. Stout camphor tree genome fills gaps in understanding of flowering plant genome evolution. Nat. Plants 2019, 5, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Hao, Z.; Guang, X.; Zhao, C.; Wang, P.; Xue, L.; Zhu, Q.; Yang, L.; Sheng, Y.; Zhou, Y.; et al. Liriodendron genome sheds light on angiosperm phylogeny and species-pair differentiation. Nat. Plants 2019, 5, 18–25. [Google Scholar] [CrossRef]
- Rendon-Anaya, M.; Ibarra-Laclette, E.; Mendez-Bravo, A.; Lan, T.; Zheng, C.; Carretero-Paulet, L.; Perez-Torres, C.A.; Chacon-Lopez, A.; Hernandez-Guzman, G.; Chang, T.H.; et al. The avocado genome informs deep angiosperm phylogeny, highlights introgressive hybridization, and reveals pathogen-influenced gene space adaptation. Proc. Natl. Acad. Sci. USA 2019, 116, 17081–17089. [Google Scholar] [CrossRef]
- Hu, L.; Xu, Z.; Wang, M.; Fan, R.; Yuan, D.; Wu, B.; Wu, H.; Qin, X.; Yan, L.; Tan, L.; et al. The chromosome-scale reference genome of black pepper provides insight into piperine biosynthesis. Nat. Commun. 2019, 10, 4702. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-P.; Sun, W.-H.; Xiong, Y.-F.; Jiang, Y.-T.; Liu, X.-D.; Liao, X.-Y.; Zhang, D.-Y.; Jiang, S.-Z.; Li, Y.; Liu, B. The Phoebe genome sheds light on the evolution of magnoliids. Hortic. Res. 2020, 7, 146. [Google Scholar] [CrossRef]
- Lv, Q.; Qiu, J.; Liu, J.; Li, Z.; Zhang, W.; Wang, Q.; Fang, J.; Pan, J.; Chen, Z.; Cheng, W.; et al. The Chimonanthus salicifolius genome provides insight into magnoliid evolution and flavonoid biosynthesis. Plant J. Cell Mol. Biol. 2020, 103, 1910–1923. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.-H.; Xiang, S.; Zhang, Q.-G.; Xiao, L.; Zhang, D.; Zhang, P.; Chen, D.-Q.; Hao, Y.; Liu, D.-K.; Ding, L. The camphor tree genome enhances the understanding of magnoliid evolution. J. Genet. Genom. Yi Chuan Xue Bao 2022, 49, 249–253. [Google Scholar] [CrossRef]
- Sun, M.; Soltis, D.E.; Soltis, P.S.; Zhu, X.; Burleigh, J.G.; Chen, Z. Deep phylogenetic incongruence in the angiosperm clade Rosidae. Mol. Phylogenetics Evol. 2015, 83, 156–166. [Google Scholar] [CrossRef]
- Soltis, D.E.; Soltis, P.S. Nuclear genomes of two magnoliids. Nat. Plants 2019, 5, 6–7. [Google Scholar] [CrossRef]
- Guo, X.; Fang, D.; Sahu, S.K.; Yang, S.; Guang, X.; Folk, R.; Smith, S.A.; Chanderbali, A.S.; Chen, S.; Liu, M.; et al. Chloranthus genome provides insights into the early diversification of angiosperms. Nat. Commun. 2021, 12, 6930. [Google Scholar] [CrossRef]
- Yang, L.; Su, D.; Chang, X.; Foster, C.S.; Sun, L.; Huang, C.-H.; Zhou, X.; Zeng, L.; Ma, H.; Zhong, B. Phylogenomic insights into deep phylogeny of angiosperms based on broad nuclear gene sampling. Plant Commun. 2020, 1, 100027. [Google Scholar] [CrossRef]
- Shen, Z.; Ding, X.; Cheng, J.; Wu, F.; Yin, H.; Wang, M. Phylogenetic studies of magnoliids: Advances and perspectives. Front. Plant Sci. 2023, 13, 1100302. [Google Scholar] [CrossRef]
- Dong, S.; Chen, L.; Liu, Y.; Wang, Y.; Zhang, S.; Yang, L.; Lang, X.; Zhang, S. The draft mitochondrial genome of Magnolia biondii and mitochondrial phylogenomics of angiosperms. PLoS ONE 2020, 15, e0231020. [Google Scholar] [CrossRef]
- Zhao, Y.; Ren, Y.; Xu, Y.; Yan, M.; Huo, Y.; Zhao, X.; Yuan, Z. The complete chloroplast genome sequence of Chimonanthus praecox cv. concolor. Mitochondrial DNA Part B 2019, 4, 3236–3237. [Google Scholar] [CrossRef] [PubMed]
- Zhou, G.-C.; Wang, J.-Y.; Li, W.; Zhang, M.; Meng, G.-Q.; Wang, H.-Y.; Chen, X.; Wu, Y.-H.; Wu, P.; Wang, Y.-L. Complete chloroplast genome sequence of Chimonanthus praecox link (Calycanthaceae): An endemic plant species in China. Mitochondrial DNA Part B 2020, 5, 3469–3471. [Google Scholar] [CrossRef]
- Shang, J.; Tian, J.; Cheng, H.; Yan, Q.; Li, L.; Jamal, A.; Xu, Z.; Xiang, L.; Saski, C.A.; Jin, S. The chromosome-level wintersweet (Chimonanthus praecox) genome provides insights into floral scent biosynthesis and flowering in winter. Genome Biol. 2020, 21, 200. [Google Scholar] [CrossRef] [PubMed]
- Ogihara, Y.; Yamazaki, Y.; Murai, K.; Kanno, A.; Terachi, T.; Shiina, T.; Miyashita, N.; Nasuda, S.; Nakamura, C.; Mori, N. Structural dynamics of cereal mitochondrial genomes as revealed by complete nucleotide sequencing of the wheat mitochondrial genome. Nucleic Acids Res. 2005, 33, 6235–6250. [Google Scholar] [CrossRef]
- Unseld, M.; Marienfeld, J.R.; Brandt, P.; Brennicke, A. The mitochondrial genome of Arabidopsis thaliana contains 57 genes in 366,924 nucleotides. Nat. Genet. 1997, 15, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Alverson, A.J.; Wei, X.; Rice, D.W.; Stern, D.B.; Barry, K.; Palmer, J.D. Insights into the evolution of mitochondrial genome size from complete sequences of Citrullus lanatus and Cucurbita pepo (Cucurbitaceae). Mol. Biol. Evol. 2010, 27, 1436–1448. [Google Scholar] [CrossRef]
- Notsu, Y.; Masood, S.; Nishikawa, T.; Kubo, N.; Akiduki, G.; Nakazono, M.; Hirai, A.; Kadowaki, K. The complete sequence of the rice (Oryza sativa L.) mitochondrial genome: Frequent DNA sequence acquisition and loss during the evolution of flowering plants. Mol. Genet. Genom. 2002, 268, 434–445. [Google Scholar] [CrossRef]
- Alverson, A.J.; Rice, D.W.; Dickinson, S.; Barry, K.; Palmer, J.D. Origins and recombination of the bacterial-sized multichromosomal mitochondrial genome of cucumber. Plant Cell 2011, 23, 2499–2513. [Google Scholar] [CrossRef]
- Mower, J.P. Variation in protein gene and intron content among land plant mitogenomes. Mitochondrion 2020, 53, 203–213. [Google Scholar] [CrossRef]
- Feng, Y.; Wang, Y.; Lu, H.; Li, J.; Akhter, D.; Liu, F.; Zhao, T.; Shen, X.; Li, X.; Whelan, J.; et al. Assembly and phylogenomic analysis of cotton mitochondrial genomes provide insights into the history of cotton evolution. Crop J. 2023, 11, 1782–1792. [Google Scholar] [CrossRef]
- Wang, J.; Kan, S.; Liao, X.; Zhou, J.; Tembrock, L.R.; Daniell, H.; Jin, S.; Wu, Z. Plant organellar genomes: Much done, much more to do. Trends Plant Sci. 2024; in press. [Google Scholar] [CrossRef] [PubMed]
- Arseneau, J.-R.; Steeves, R.; Laflamme, M. Modified low-salt CTAB extraction of high-quality DNA from contaminant-rich tissues. Mol. Ecol. Resour. 2017, 17, 686–693. [Google Scholar] [CrossRef]
- Patel, R.K.; Jain, M. NGS QC Toolkit: A toolkit for quality control of next generation sequencing data. PLoS ONE 2012, 7, e30619. [Google Scholar] [CrossRef] [PubMed]
- Prjibelski, A.; Antipov, D.; Meleshko, D.; Lapidus, A.; Korobeynikov, A. Using SPAdes De Novo Assembler. Curr. Protoc. Bioinform. 2020, 70, e102. [Google Scholar] [CrossRef]
- Wick, R.R.; Schultz, M.B.; Zobel, J.; Holt, K.E. Bandage: Interactive visualization of de novo genome assemblies. Bioinformatics 2015, 31, 3350–3352. [Google Scholar] [CrossRef]
- Lohse, M.; Drechsel, O.; Kahlau, S.; Bock, R. OrganellarGenomeDRAW—A suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 2013, 41, W575–W581. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Sloan, D.B.; Taylor, D.R. Testing for selection on synonymous sites in plant mitochondrial DNA: The role of codon bias and RNA editing. J. Mol. Evol. 2010, 70, 479–491. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Soltis, P.S.; Soltis, D.E.; Chase, M.W. Angiosperm phylogeny inferred from multiple genes as a tool for comparative biology. Nature 1999, 402, 402–404. [Google Scholar] [CrossRef]
- Zeng, L.; Zhang, Q.; Sun, R.; Kong, H.; Zhang, N.; Ma, H. Resolution of deep angiosperm phylogeny using conserved nuclear genes and estimates of early divergence times. Nat. Commun. 2014, 5, 4956. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Feng, Y.; Zhai, W.; Chen, M.; Wu, G. The complete mitochondrial genome of Schisandra sphenanthera (Schisandraceae). Mitochondrial DNA Part B 2018, 3, 1246–1247. [Google Scholar] [CrossRef]
- Parvathy, S.T.; Udayasuriyan, V.; Bhadana, V. Codon usage bias. Mol. Biol. Rep. 2022, 49, 539–565. [Google Scholar] [CrossRef] [PubMed]
- Richardson, A.O.; Rice, D.W.; Young, G.J.; Alverson, A.J.; Palmer, J.D. The “fossilized” mitochondrial genome of Liriodendron tulipifera: Ancestral gene content and order, ancestral editing sites, and extraordinarily low mutation rate. BMC Biol. 2013, 11, 29. [Google Scholar] [CrossRef]
- Gui, S.; Wu, Z.; Zhang, H.; Zheng, Y.; Zhu, Z.; Liang, D.; Ding, Y. The mitochondrial genome map of Nelumbo nucifera reveals ancient evolutionary features. Sci. Rep. 2016, 6, 30158. [Google Scholar] [CrossRef]
- Li, S.-N.; Yang, Y.-Y.; Xu, L.; Xing, Y.-P.; Zhao, R.; Ao, W.-L.; Zhang, T.-T.; Zhang, D.-C.; Song, Y.-Y.; Bao, G.-H.; et al. The complete mitochondrial genome of Aconitum kusnezoffii Rchb. (Ranales, Ranunculaceae). Mitochondrial DNA B Resour. 2021, 6, 779–781. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Chen, X.; Long, L.; Jost, M.; Zhao, R.; Liu, L.; Mower, J.P.; de Pamphilis, C.W.; Wanke, S.; Jiao, Y. De novo Assembly and Comparative Analyses of Mitochondrial Genomes in Piperales. Genome Biol. Evol. 2023, 15, evad041. [Google Scholar] [CrossRef]
- Bi, C.; Lu, N.; Xu, Y.; He, C.; Lu, Z. Characterization and Analysis of the Mitochondrial Genome of Common Bean (Phaseolus vulgaris) by Comparative Genomic Approaches. Int. J. Mol. Sci. 2020, 21, 3778. [Google Scholar] [CrossRef]
- Shidhi, P.R.; Biju, V.C.; Anu, S.; Vipin, C.L.; Deelip, K.R.; Achuthsankar, S.N. Genome Characterization, Comparison and Phylogenetic Analysis of Complete Mitochondrial Genome of Evolvulus alsinoides Reveals Highly Rearranged Gene Order in Solanales. Life 2021, 11, 769. [Google Scholar] [CrossRef]
- Gualberto, J.M.; Mileshina, D.; Wallet, C.; Niazi, A.K.; Weber-Lotfi, F.; Dietrich, A. The plant mitochondrial genome: Dynamics and maintenance. Biochimie 2014, 100, 107–120. [Google Scholar] [CrossRef]
- Čechová, J.; Lýsek, J.; Bartas, M.; Brázda, V. Complex analyses of inverted repeats in mitochondrial genomes revealed their importance and variability. Bioinformatics 2018, 34, 1081–1085. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Group of Genes | Gene Names |
|---|---|
| Complex I (NADH dehydrogenase) | nad1 a, nad2 a, nad3, nad4 ad, nad4L, nad5 a, nad6, nad7 ad, nad9 |
| Complex II (succinate dehydrogenase) | sdh3, sdh4 |
| Complex III (ubiquinol cytochrome c reductase) | cob |
| Complex IV (cytochrome c oxidase) | cox1, cox2 ac, cox3 |
| Complex V (ATP synthase) | atp1 b(2), atp4, atp6, atp8, atp9 |
| Cytochrome c biogenesis | ccmB, ccmC, ccmFc a, ccmFn |
| Ribosomal proteins (SSU) | rps1, rps2, rps3 a, rps4, rps7, rps10 a, rps11, rps12, rps13, rps14, rps19 b(2) |
| Ribosomal proteins (LSU) | rpl5, rpl10, rpl16 |
| Maturases | matR |
| Transport membrane protein | mttB |
| Ribosomal RNAs | rrn5, rrnL b(2), rrnS |
| Transfer RNAs | trnA, trnC, trnD b(2), trnE b(2), trnF b(2), trnG, trnH, trnK b(2), trnL b(3), trnM b(5), trnN, trnP b(2), trnQ b(2), trnR, trnS b(2), trnW, trnY b(2) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, X.; Feng, Y.; Zhang, J. Characterization of the Complete Mitochondrial Genome of Wintersweet (Chimonanthus praecox) and Comparative Analysis within Magnoliids. Life 2024, 14, 182. https://doi.org/10.3390/life14020182
Yu X, Feng Y, Zhang J. Characterization of the Complete Mitochondrial Genome of Wintersweet (Chimonanthus praecox) and Comparative Analysis within Magnoliids. Life. 2024; 14(2):182. https://doi.org/10.3390/life14020182
Chicago/Turabian StyleYu, Xianxian, Yanlei Feng, and Jie Zhang. 2024. "Characterization of the Complete Mitochondrial Genome of Wintersweet (Chimonanthus praecox) and Comparative Analysis within Magnoliids" Life 14, no. 2: 182. https://doi.org/10.3390/life14020182