
Unveiling the Mystery of Adult-Onset Still’s Disease: A Compelling Case Report

 ,
,  ,
, 
 ,
, 
Abstract
1. Introduction
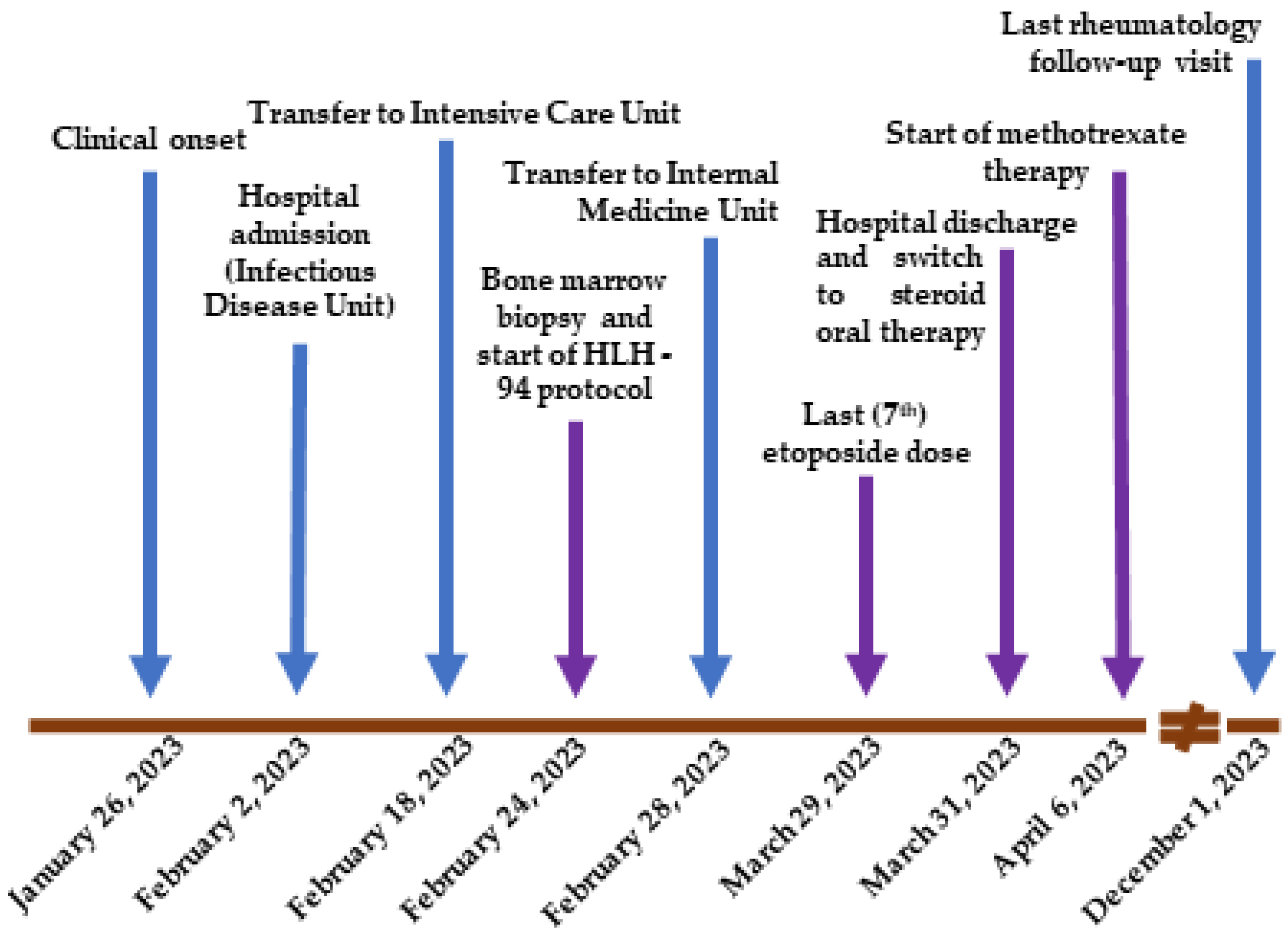
2. Case Presentation
3. Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Efthimiou, P.; Kontzias, A.; Hur, P.; Rodha, K.; Ramakrishna, G.S.; Nakasato, P. Adult-Onset Still’s Disease in Focus: Clinical Manifestations, Diagnosis, Treatment, and Unmet Needs in the Era of Targeted Therapies. Semin. Arthritis Rheum. 2021, 51, 858–874. [Google Scholar] [CrossRef] [PubMed]
- Magadur-Joly, G.; Billaud, E.; Barrier, J.H.; Pennec, Y.L.; Masson, C.; Renou, P.; Prost, A. Epidemiology of Adult Still’s Disease: Estimate of the Incidence by a Retrospective Study in West France. Ann. Rheum. Dis. 1995, 54, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, M.; Ohta, A.; Tsunematsu, T.; Kasukawa, R.; Mizushima, Y.; Kashiwagi, H.; Kashiwazaki, S.; Tanimoto, K.; Matsumoto, Y.; Ota, T.; et al. Preliminary Criteria for Classification of Adult Still’s Disease. J. Rheumatol. 1992, 19, 424–430. [Google Scholar] [PubMed]
- Fautrel, B.; Zing, E.; Golmard, J.-L.; Le Moel, G.; Bissery, A.; Rioux, C.; Rozenberg, S.; Piette, J.-C.; Bourgeois, P. Proposal for a New Set of Classification Criteria for Adult-Onset Still Disease. Medicine 2002, 81, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Arlet, J.B.; Huong, D.L.T.; Marinho, A.; Amoura, Z.; Wechsler, B.; Papo, T.; Piette, J.C. Reactive Haemophagocytic Syndrome in Adult-Onset Still’s Disease: A Report of Six Patients and a Review of the Literature. Ann. Rheum. Dis. 2006, 65, 1596–1601. [Google Scholar] [CrossRef]
- Bae, C.B.; Jung, J.Y.; Kim, H.A.; Suh, C.H. Reactive Hemophagocytic Syndrome in Adult-Onset Still Disease: Clinical Features, Predictive Factors, and Prognosis in 21 Patients. Medicine 2015, 94, e451. [Google Scholar] [CrossRef]
- Efthimiou, P.; Kadavath, S.; Mehta, B. Life-Threatening Complications of Adult-Onset Still’s Disease. Clin. Rheumatol. 2014, 33, 305–314. [Google Scholar] [CrossRef]
- Bergsten, E.; Horne, A.C.; Aricó, M.; Astigarraga, I.; Egeler, R.M.; Filipovich, A.H.; Ishii, E.; Janka, G.; Ladisch, S.; Lehmberg, K.; et al. Confirmed Efficacy of Etoposide and Dexamethasone in HLH Treatment: Long-Term Results of the Cooperative HLH-2004 Study. Blood 2017, 130, 2728–2738. [Google Scholar] [CrossRef]
- Ramos-Casals, M.; Brito-Zerón, P.; López-Guillermo, A.; Khamashta, M.A.; Bosch, X. Adult Haemophagocytic Syndrome. Lancet 2014, 383, 1503–1516. [Google Scholar] [CrossRef]
- Jordan, M.B.; Allen, C.E.; Weitzman, S.; Filipovich, A.H.; McClain, K.L. How I Treat Hemophagocytic Lymphohistiocytosis. Blood 2011, 118, 4041–4052. [Google Scholar] [CrossRef]
- Filipovich, A.H. Hemophagocytic Lymphohistiocytosis (HLH) and Related Disorders. Hematol. Am. Soc. Hematol. Educ. Program 2009, 1, 127–131. [Google Scholar] [CrossRef]
- Shakoory, B.; Geerlinks, A.; Wilejto, M.; Kernan, K.; Hines, M.; Romano, M.; Piskin, D.; Ravelli, A.; Sinha, R.; Aletaha, D.; et al. The 2022 EULAR/ACR Points to Consider at the Early Stages of Diagnosis and Management of Suspected Haemophagocytic Lymphohistiocytosis/Macrophage Activation Syndrome (HLH/MAS). Arthritis Rheumatol. 2023, 75, 1714–1732. [Google Scholar] [CrossRef]
- Trottestam, H.; Horne, A.; Aricò, M.; Egeler, R.M.; Filipovich, A.H.; Gadner, H.; Imashuku, S.; Ladisch, S.; Webb, D.; Janka, G.; et al. Chemoimmunotherapy for Hemophagocytic Lymphohistiocytosis: Long-Term Results of the HLH-94 Treatment Protocol. Blood 2011, 118, 4577–4584. [Google Scholar] [CrossRef] [PubMed]
- Henter, J.-I.; Horne, A.; Aricó, M.; Egeler, R.M.; Filipovich, A.H.; Imashuku, S.; Ladisch, S.; McClain, K.; Webb, D.; Winiarski, J.; et al. HLH-2004: Diagnostic and Therapeutic Guidelines for Hemophagocytic Lymphohistiocytosis. Pediatr. Blood Cancer 2007, 48, 124–131. [Google Scholar] [CrossRef] [PubMed]
- La Rosée, P.; Horne, A.C.; Hines, M.; Greenwood, T.V.B.; Machowicz, R.; Berliner, N.; Birndt, S.; Gil-Herrera, J.; Girschikofsky, M.; Jordan, M.B.; et al. Recommendations for the Management of Hemophagocytic Lymphohistiocytosis in Adults. Blood 2019, 133, 2465–2477. [Google Scholar] [CrossRef]
- Oen, K.; Duffy, C.N.M.; Tse, S.M.L.; Ramsey, S.; Ellsworth, J.; Chédeville, G.; Chetaille, A.L.; Saint-Cyr, C.; Cabral, D.A.; Spiegel, L.R.; et al. Early Outcomes and Improvement of Patients with Juvenile Idiopathic Arthritis Enrolled in a Canadian Multicenter Inception Cohort. Arthritis Care Res. 2010, 62, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Pouchot, J.; Sampalis, J.S.; Beaudet, F.; Carette, S.; Décary, F.; Salusinsky-Sternbach, M.; Hill, R.O.; Gutkowski, A.; Harth, M.; Myhal, D.; et al. Adult Still’s Disease: Manifestations, Disease Course, and Outcome in 62 Patients. Medicine 1991, 70, 118–136. [Google Scholar] [CrossRef] [PubMed]
- Kalyoncu, U.; Solmaz, D.; Emmungil, H.; Yazici, A.; Kasifoglu, T.; Kimyon, G.; Balkarli, A.; Bes, C.; Ozmen, M.; Alibaz-Oner, F.; et al. Response Rate of Initial Conventional Treatments, Disease Course, and Related Factors of Patients with Adult-Onset Still’s Disease: Data from a Large Multicenter Cohort. J. Autoimmun. 2016, 69, 59–63. [Google Scholar] [CrossRef] [PubMed]
- Ruscitti, P.; Sota, J.; Vitale, A.; Lopalco, G.; Iannone, F.; Morrone, M.; Giardini, H.A.M.; D’Agostin, M.A.; de Brito Antonelli, I.P.; Almaghlouth, I.; et al. The Administration of Methotrexate in Patients with Still’s Disease, “Real-Life” Findings from AIDA Network Still Disease Registry. Semin. Arthritis Rheum. 2023, 62, 152244. [Google Scholar] [CrossRef]
- Gerfaud-Valentin, M.; Maucort-Boulch, D.; Hot, A.; Iwaz, J.; Ninet, J.; Durieu, I.; Broussolle, C.; Sève, P. Adult-Onset Still Disease: Manifestations, Treatment, Outcome, and Prognostic Factors in 57 Patients. Medicine 2014, 93, 91–99. [Google Scholar] [CrossRef]
- Yamamoto, T. Cutaneous Manifestations Associated with Adult-Onset Still’s Disease: Important Diagnostic Values. Rheumatol. Int. 2012, 32, 2233–2237. [Google Scholar] [CrossRef]
- Ichida, H.; Kawaguchi, Y.; Sugiura, T.; Takagi, K.; Katsumata, Y.; Gono, T.; Ota, Y.; Kataoka, S.; Kawasumi, H.; Yamanaka, H. Clinical Manifestations of Adult-Onset Still’s Disease Presenting with Erosive Arthritis: Association with Low Levels of Ferritin and Interleukin-18. Arthritis Care Res. 2014, 66, 642–646. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.H.Y.; Weisman, M.H. Severe Sore Throat as a Presenting Symptom of Adult Onset Still’s Disease: A Case Series and Review of the Literature. J. Rheumatol. 1997, 24, 592–597. [Google Scholar] [PubMed]
- Gerfaud-Valentin, M.; Jamilloux, Y.; Iwaz, J.; Sève, P. Adult-Onset Still’s Disease. Autoimmun. Rev. 2014, 13, 708–722. [Google Scholar] [CrossRef] [PubMed]
- Cheema, G.S.; Quismorio, F.P. Pulmonary Involvement in Adult-Onset Still’s Disease. Curr. Opin. Pulm. Med. 1999, 5, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Drouot, M.H.; Hachulla, E.; Houvenagel, E.; Hatron, P.Y.; Flipo, R.M.; Goullard, L.; Ducloux, G.; Devulder, B. Cardiac Complications in Adult Onset Still Disease: From Pericarditis to Tamponade as Manifestations. Rev. Med. Interne 1994, 15, 740–743. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.W.; Suh, Y.J.; Song, H.J.; Choi, J.H.; Park, H.S.; Cho, S.R.; Suh, C.H. Pure Red Cell Aplasia and Adult-Onset Still’s Disease. Clin. Rheumatol. 2004, 23, 368–370. [Google Scholar] [CrossRef] [PubMed]
- Fautrel, B. Adult-Onset Still Disease. Best. Pr. Res. Clin. Rheumatol. 2008, 22, 773–792. [Google Scholar] [CrossRef] [PubMed]
- Kontzias, A.; Efthimiou, P. Adult-Onset Still’s Disease: Pathogenesis, Clinical Manifestations and Therapeutic Advances. Drugs 2008, 68, 319–337. [Google Scholar] [CrossRef]
- Khobragade, A.K.; Chogle, A.R.; Ram, R.P.; Mascarenhas, J.; Kothari, S.; Kawadkar, S.; Deshpande, S.S.; Nair, D.; Makhija, J. Reversible Posterior Leukoencephalopathy Syndrome in a Case of Adult Onset Still’s Disease with Concurrent Thrombotic Thrombocytopenic Purpura: Response to High Dose Immunoglobulin Infusions. J. Assoc. Physicians India 2012, 60, 59–62. [Google Scholar]
- Saito, K.; Temmoku, J.; Sumichika, Y.; Yoshida, S.; Takano, E.; Watanabe, S.; Matsumoto, H.; Fujita, Y.; Matsuoka, N.; Asano, T.; et al. Adult-Onset Still’s Disease with Acute Kidney Injury Requiring Hemodialysis: A Case Report and Literature Review. Intern. Med. 2023, 62, 2901–2906. [Google Scholar] [CrossRef] [PubMed]
- Bagnari, V.; Colina, M.; Ciancio, G.; Govoni, M.; Trotta, F. Adult-Onset Still’s Disease. Rheumatol. Int. 2010, 30, 855–862. [Google Scholar] [CrossRef] [PubMed]
- Giacomelli, R.; Ruscitti, P.; Shoenfeld, Y. A Comprehensive Review on Adult Onset Still’s Disease. J. Autoimmun. 2018, 93, 24–36. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Li, Z.; Zhang, L.; Lian, H.; Ma, H.; Wang, D.; Zhao, X.; Zhang, Q.; Wang, T.; Zhang, R. Clinical Features and Outcomes of Patients with Hemophagocytic Lymphohistiocytosis at Onset of Systemic Autoinflammatory Disorder and Compare with Epstein–Barr Virus (EBV)-Related Hemophagocytic Lymphohistiocytosis. Medicine 2020, 99, e18503. [Google Scholar] [CrossRef] [PubMed]
- Zafran, M.; Wassef, N. Complex Presentation of Adult-Onset Still’s Disease. BMJ Case Rep. 2019, 12, e228210. [Google Scholar] [CrossRef]
- Tsuboi, H.; Segawa, S.; Yagishita, M.; Toko, H.; Honda, F.; Kitada, A.; Miki, H.; Ohyama, A.; Hagiwara, S.; Kondo, Y.; et al. Activation Mechanisms of Monocytes/Macrophages in Adult-Onset Still Disease. Front. Immunol. 2022, 13, 953730. [Google Scholar] [CrossRef]
- Li, X.; Qu, B.; Nie, Y.; Zhu, G.; Li, W.; Mu, F. Clinical Features of Macrophage Activation Syndrome in the Adult Northern Chinese Population. Lupus 2014, 23, 785–792. [Google Scholar] [CrossRef]
- Macovei, L.A.; Burlui, A.; Bratoiu, I.; Rezus, C.; Cardoneanu, A.; Richter, P.; Szalontay, A.; Rezus, E. Adult-Onset Still’s Disease—A Complex Disease, a Challenging Treatment. Int. J. Mol. Sci. 2022, 23, 12810. [Google Scholar] [CrossRef]
- Lee, W.-S.; Yoo, W.-H. Rituximab for Refractory Adult-Onset Still’s Disease with Thrombotic Microangiopathy. Rheumatology 2014, 53, 1717–1718. [Google Scholar] [CrossRef]
- Mehta, P.; Cron, R.Q.; Hartwell, J.; Manson, J.J.; Tattersall, R.S. Silencing the Cytokine Storm: The Use of Intravenous Anakinra in Haemophagocytic Lymphohistiocytosis or Macrophage Activation Syndrome. Lancet Rheumatol. 2020, 2, e358–e367. [Google Scholar] [CrossRef]
- Fardet, L.; Galicier, L.; Lambotte, O.; Marzac, C.; Aumont, C.; Chahwan, D.; Coppo, P.; Hejblum, G. Development and Validation of the Hscore, a Score for the Diagnosis of Reactive Hemophagocytic Syndrome. Arthritis Rheumatol. 2014, 66, 2613–2620. [Google Scholar] [CrossRef]
- Bailly, C. Etoposide: A Rider on the Cytokine Storm. Cytokine 2023, 168, 156234. [Google Scholar] [CrossRef]
- Cron, R.Q.; Goyal, G.; Chatham, W.W. Cytokine Storm Syndrome. Annu. Rev. Med. 2023, 74, 321–337. [Google Scholar] [CrossRef]
- Nam, S.H.; Ahn, S.M.; Oh, J.S.; Hong, S.; Lee, C.-K.; Yoo, B.; Kim, Y.-G. Macrophage Activation Syndrome in Rheumatic Disease: Clinical Characteristics and Prognosis of 20 Adult Patients. PLoS ONE 2022, 17, e0267715. [Google Scholar] [CrossRef]
- Carter, S.J.; Tattersall, R.S.; Ramanan, A.V. Macrophage Activation Syndrome in Adults: Recent Advances in Pathophysiology, Diagnosis and Treatment. Rheumatology 2019, 58, 5–17. [Google Scholar] [CrossRef]
- Sung, L.; King, S.M.; Carcao, M.; Trebo, M.; Weitzman, S.S. Adverse Outcomes in Primary Hemophagocytic Lymphohistiocytosis. J. Pediatr. Hematol. Oncol. 2002, 24, 550–554. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.A.; Tyrrell, P.; Valani, R.; Benseler, S.; Abdelhaleem, M.; Weitzman, S. Experience With Hemophagocytic Lymphohistiocytosis/Macrophage Activation Syndrome at a Single Institution. J. Pediatr. Hematol. Oncol. 2009, 31, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Li, T.; Ye, S.; Lv, L.; Chen, S.; Wang, X.; Bao, C.; Fu, Q. Short-Term, Low-Dose Etoposide in Refractory Adult-Onset Still’s Disease-Associated Macrophage Activation Syndrome. Clin. Rheumatol. 2022, 41, 2817–2823. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Admission | VP-16 1st Dose | VP-16 2nd Dose | VP-16 3rd Dose | Vp-16 4th Dose | VP-16 5th Dose | Vp-16 6th Dose | VP-16 7th Dose | Discharge | f/up 2 mos | f/up 8 mos | Local Laboratory NR | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WBCs | 10.84 | 12.97 | 3.05 | 2.99 | 2.36 | 6.65 | 3.91 | 9.16 | 4.55 | 6.64 | 7.76 | 4.50–11.00 × 109/L |
| Neutrophils | 9.42 | 10.99 | 2.59 | 2.42 | 1.54 | 3.18 | 1.61 | 5.31 | 2.24 | 4.03 | 4.85 | 1.80–7.70 × 109/L |
| Lymphocytes | 1.00 | 0.67 | 0.39 | 0.51 | 0.81 | 2.53 | 2.02 | 2.96 | 1.83 | 1.93 | 2.20 | 1.00–4.50 × 109/L |
| Hb | 9.2 | 7.9 | 8.9 | 8.7 | 9 | 8.5 | 8.2 | 8.1 | 8.5 | 12.2 | 11.0 | 11.5–15.5 g/dL |
| PLTs | 296 | 30 | 36 | 46 | 56 | 76 | 238 | 176 | 152 | 197 | 191 | 130–400 × 109/L |
| INR | 1.38 | 1.75 | 1.64 | 1.50 | 1.47 | 1.19 | 1.09 | 1.05 | 1.02 | 1.01 | 1.01 | 0.8–1.2 Units |
| Fibrinogen | 112 | 96 | 98 | 85 | 83 | 107 | 66 | 137 | 209 | / | / | 200–393 mg/dL |
| Glycemia | 158 | 183 | 162 | 88 | 76 | 70 | 106 | 73 | 79 | 80 | 78 | 74–100 mg/dL |
| Creatinine | 0.72 | 0.55 | 0.37 | 0.22 | 0.40 | 0.49 | 0.47 | 0.42 | 0.44 | 0.65 | 0.65 | 0.5–121 mg/dL |
| Na | 136 | 137 | 137 | 135 | 136 | 137 | 136 | 139 | 141 | 143 | 144 | 134–146 mmol/L |
| K | 3.5 | 4.4 | 4.0 | 4.2 | 4.1 | 4.1 | 4.0 | 4.0 | 4.0 | 3.8 | 4.0 | 3.4–4.5 mmol/L |
| Triglycerides | 309 | / | / | 226 | 64 | / | / | / | / | / | / | <150 mg/dL |
| CRP | 11.38 | 2.10 | 1.04 | 0.32 | 0.22 | / | / | 0.12 | / | 0.03 | 0.02 | 0–0.50 mg/dL |
| PCT | 0.3 | 0.2 | 0.3 | <0.05 | / | / | / | / | / | / | <0.05 | <0.5 µg/L |
| Ferritin | 1059 | 1608 | 1338 | 553 | 574 | 628 | 531 | 580 | 555 | 19 | 38 | 13–150 µg/L |
| LDH | 970 | 1727 | 735 | 562 | 530 | 461 | 475 | 568 | / | 443 | 27 | 208–450 U/L |
| Total bilirubin | 0.55 | 1.63 | 1.58 | 1.24 | 1.36 | 1.43 | 1.00 | 0.66 | 1.05 | 0.70 | 0.81 | 0.30–1.20 mg/dL |
| AST | 49 | 590 | 100 | 39 | 27 | 21 | 27 | 30 | 28 | 24 | 19 | 0–40 U/L |
| ALT | 42 | 620 | 349 | 188 | 127 | 55 | 56 | 62 | 61 | 15 | 17 | 0–40 U/L |
| GGT | 74 | 199 | 288 | 239 | 177 | 93 | 79 | 85 | 85 | 26 | 34 | 0–50 U/L |
| ALP | 187 | 139 | 104 | 87 | 79 | 66 | 61 | 58 | 66 | 31 | 70 | 46–116 U/L |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sola, D.; Smirne, C.; Bruggi, F.; Bottino Sbaratta, C.; Tamen Njata, A.C.; Valente, G.; Pavanelli, M.C.; Vitetta, R.; Bellan, M.; De Paoli, L.; et al. Unveiling the Mystery of Adult-Onset Still’s Disease: A Compelling Case Report. Life 2024, 14, 195. https://doi.org/10.3390/life14020195
Sola D, Smirne C, Bruggi F, Bottino Sbaratta C, Tamen Njata AC, Valente G, Pavanelli MC, Vitetta R, Bellan M, De Paoli L, et al. Unveiling the Mystery of Adult-Onset Still’s Disease: A Compelling Case Report. Life. 2024; 14(2):195. https://doi.org/10.3390/life14020195
Chicago/Turabian StyleSola, Daniele, Carlo Smirne, Francesco Bruggi, Chiara Bottino Sbaratta, Aubin Cardin Tamen Njata, Guido Valente, Maria Cristina Pavanelli, Rosetta Vitetta, Mattia Bellan, Lorenzo De Paoli, and et al. 2024. "Unveiling the Mystery of Adult-Onset Still’s Disease: A Compelling Case Report" Life 14, no. 2: 195. https://doi.org/10.3390/life14020195
APA StyleSola, D., Smirne, C., Bruggi, F., Bottino Sbaratta, C., Tamen Njata, A. C., Valente, G., Pavanelli, M. C., Vitetta, R., Bellan, M., De Paoli, L., & Pirisi, M. (2024). Unveiling the Mystery of Adult-Onset Still’s Disease: A Compelling Case Report. Life, 14(2), 195. https://doi.org/10.3390/life14020195