

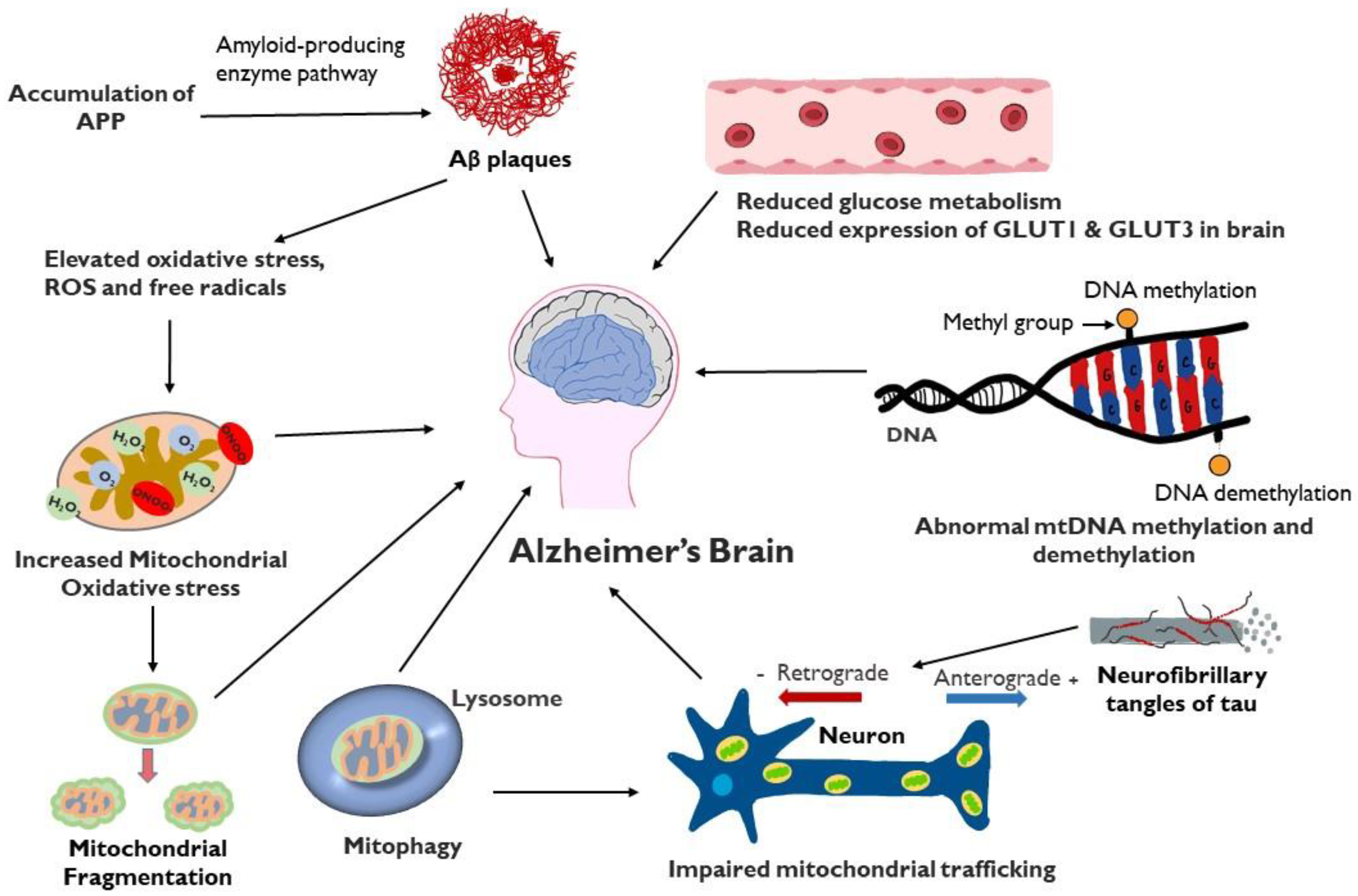
Mitochondria in Alzheimer’s Disease Pathogenesis

 ,
,  , ,
, ,  and
and
Abstract
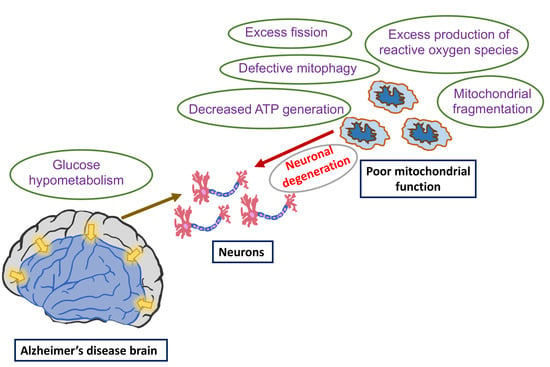
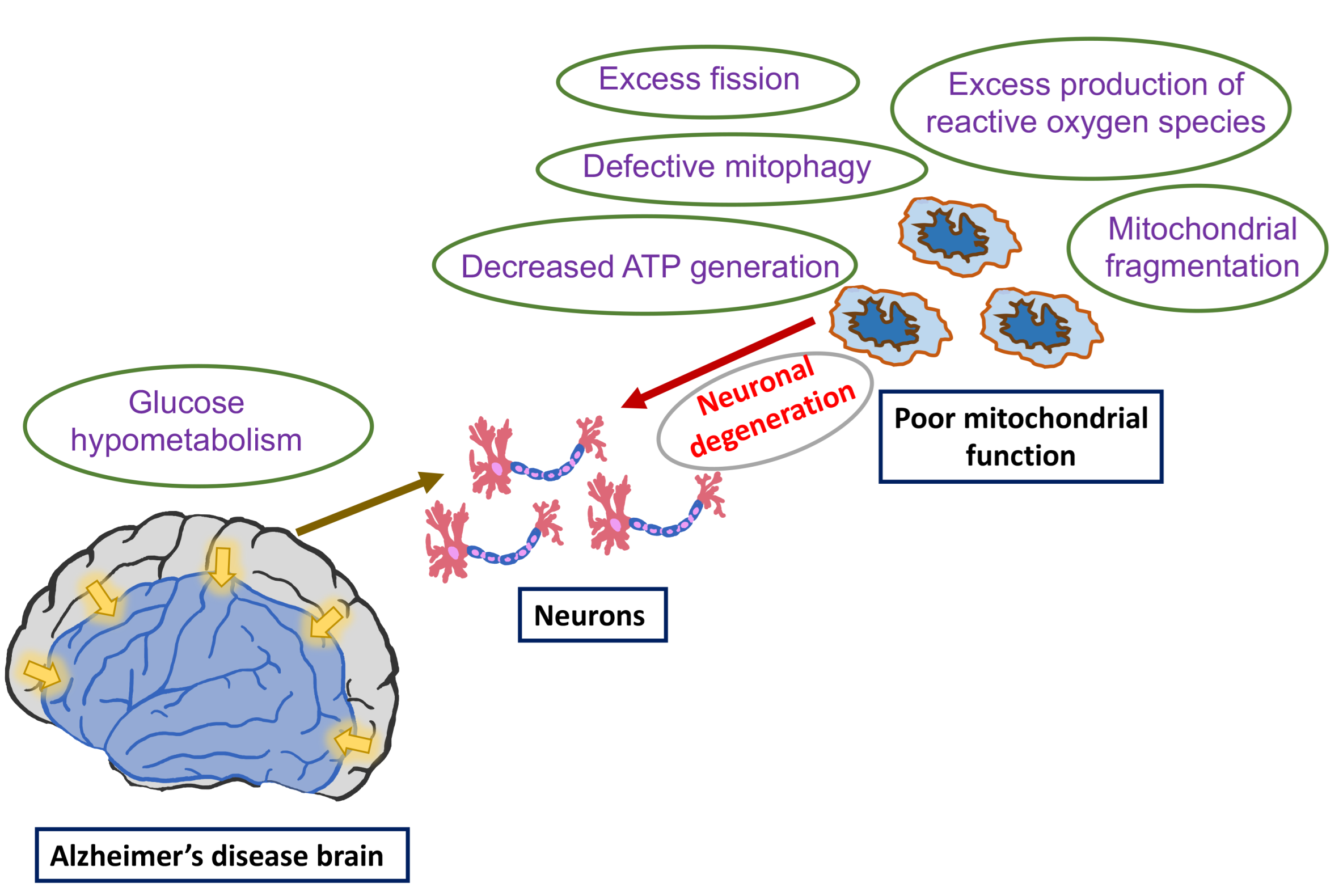
1. Introduction
2. Mitochondrial ATP Production and Oxidative Stress in Neurons
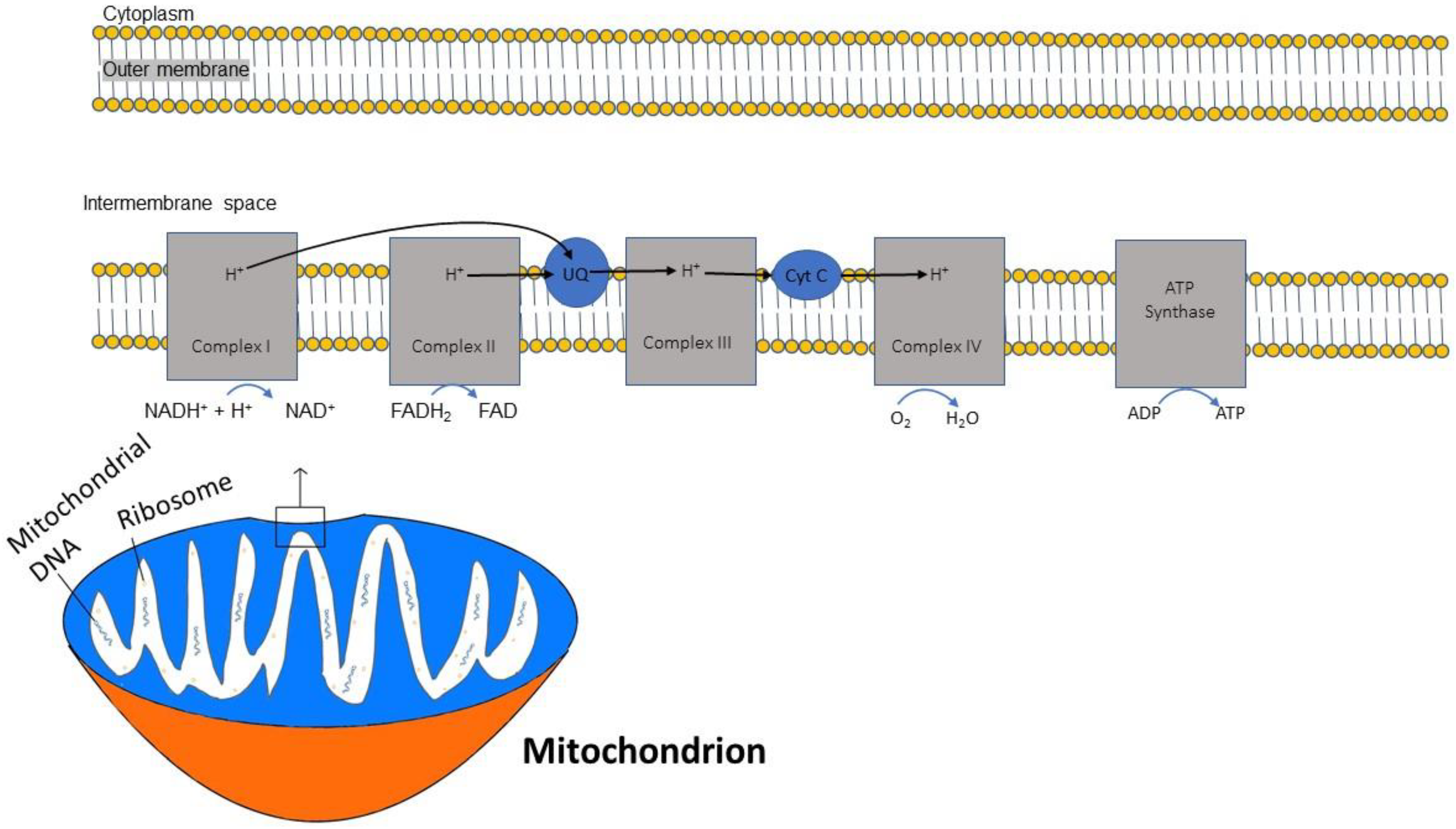
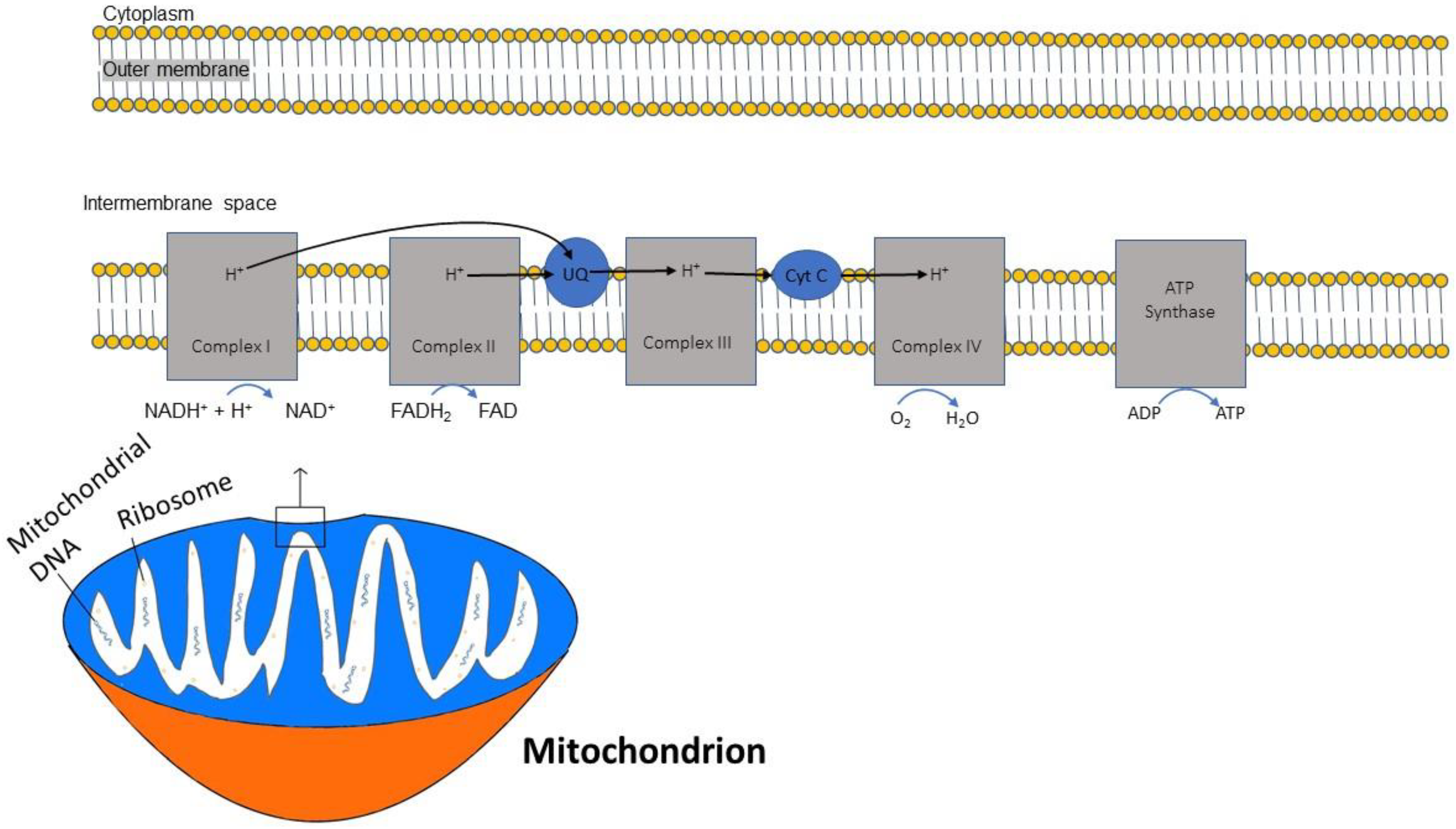
2.1. Structural Characteristics
2.2. Energy Production by Mitochondria and Mitochondrial Oxidative Stress
2.3. ATP and Oxidative Phosphorylation
2.4. Nicotinamide Adenine Dinucleotide (NAD+) and Complex I
3. Mitochondrial Trafficking
4. Mitophagy, Mitochondrial Dynamics and AD
4.1. Mitophagy
4.2. Aβ and Tau in Mitophagy and Mitochondrial Movement
4.3. Mitochondrial Fission and Fusion
4.4. Effects of Amyloid and Tau on Fission and Fusion
5. Mitochondrial DNA Methylation
6. Glucose Metabolism Reduced in AD
7. Apolipoprotein (Apo)E Gene Impact on Mitochondria and Bioenergetics
8. Mitochondria in the Treatment of AD
9. Limitations of the Hypothesis That AD Is Driven by Mitochondrial Dysfunction
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- d’Errico, P.; Meyer-Luehmann, M. Mechanisms of Pathogenic Tau and Aβ Protein Spreading in Alzheimer’s Disease. Front. Aging Neurosci. 2020, 12, 265. [Google Scholar] [CrossRef] [PubMed]
- Volloch, V.; Rits-Volloch, S. Effect of Lecanemab in Early Alzheimer’s Disease: Mechanistic Interpretation in the Amyloid Cascade Hypothesis 2.0 Perspective. J. Alzheimers Dis. 2023, 93, 1277–1284. [Google Scholar] [CrossRef]
- Varadharajan, A.; Davis, A.D.; Ghosh, A.; Jagtap, T.; Xavier, A.; Menon, A.J.; Roy, D.; Gandhi, S.; Gregor, T. Guidelines for pharmacotherapy in Alzheimer’s disease—A primer on FDA-approved drugs. J. Neurosci. Rural Pract. 2023, 14, 566–573. [Google Scholar] [CrossRef]
- Birnbaum, J.H.; Wanner, D.; Gietl, A.F.; Saake, A.; Kündig, T.M.; Hock, C.; Nitsch, R.M.; Tackenberg, C. Oxidative stress and altered mitochondrial protein expression in the absence of amyloid-β and tau pathology in iPSC-derived neurons from sporadic Alzheimer’s disease patients. Stem Cell. Res. 2018, 27, 121–130. [Google Scholar] [CrossRef]
- Tapias, V.; González-Andrés, P.; Peña, L.F.; Barbero, A.; Núñez, L.; Villalobos, C. Therapeutic Potential of Heterocyclic Compounds Targeting Mitochondrial Calcium Homeostasis and Signaling in Alzheimer’s Disease and Parkinson’s Disease. Antioxidants 2023, 12, 1282. [Google Scholar] [CrossRef] [PubMed]
- Dentoni, G.; Castro-Aldrete, L.; Naia, L.; Ankarcrona, M. The Potential of Small Molecules to Modulate the Mitochondria-Endoplasmic Reticulum Interplay in Alzheimer’s Disease. Front. Cell Dev. Biol. 2022, 10, 920228. [Google Scholar] [CrossRef]
- Terada, T.; Obi, T.; Bunai, T.; Matsudaira, T.; Yoshikawa, E.; Ando, I.; Futatsubashi, M.; Tsukada, H.; Ouchi, Y. In vivo mitochondrial and glycolytic impairments in patients with Alzheimer disease. Neurology 2020, 94, e1592–e1604. [Google Scholar] [CrossRef] [PubMed]
- Bonda, D.J.; Wang, X.; Perry, G.; Smith, M.A.; Zhu, X. Mitochondrial dynamics in Alzheimer’s disease: Opportunities for future treatment strategies. Drugs Aging 2010, 27, 181–192. [Google Scholar] [CrossRef]
- Mei, T.; Li, Y.; Orduña Dolado, A.; Li, Z.; Andersson, R.; Berliocchi, L.; Rasmussen, L.J. Pooled analysis of frontal lobe transcriptomic data identifies key mitophagy gene changes in Alzheimer’s disease brain. Front. Aging Neurosci. 2023, 15, 1101216. [Google Scholar] [CrossRef]
- Granzotto, A.; Sensi, S.L. Once upon a time, the Amyloid Cascade Hypothesis. Ageing Res. Rev. 2024, 93, 102161. [Google Scholar] [CrossRef]
- Goldberg, T.E.; Lee, S.; Devanand, D.P.; Schneider, L.S. Comparison of relative change with effect size metrics in Alzheimer’s disease clinical trials. J. Neurol. Neurosurg. Psychiatry 2023, 95, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Herst, P.M.; Rowe, M.R.; Carson, G.M.; Berridge, M.V. Functional Mitochondria in Health and Disease. Front. Endocrinol. 2017, 8, 296. [Google Scholar] [CrossRef]
- Rangaraju, V.; Lewis, T.L., Jr.; Hirabayashi, Y.; Bergami, M.; Motori, E.; Cartoni, R.; Kwon, S.K.; Courchet, J. Pleiotropic Mitochondria: The Influence of Mitochondria on Neuronal Development and Disease. J. Neurosci. 2019, 39, 8200–8208. [Google Scholar] [CrossRef] [PubMed]
- Atlante, A.; Valenti, D. Mitochondria Have Made a Long Evolutionary Path from Ancient Bacteria Immigrants within Eukaryotic Cells to Essential Cellular Hosts and Key Players in Human Health and Disease. Curr. Issues Mol. Biol. 2023, 45, 4451–4479. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.; Richard, T.H.; Gohil, V.M. Mitochondrial phospholipid metabolism in health and disease. J. Cell Sci. 2023, 136, jcs260857. [Google Scholar] [CrossRef]
- Camandola, S.; Mattson, M.P. Brain metabolism in health, aging, and neurodegeneration. EMBO J. 2017, 36, 1474–1492. [Google Scholar] [CrossRef]
- Nicholls, D.G.; Budd, S.L. Mitochondria and neuronal survival. Physiol. Rev. 2000, 80, 315–360. [Google Scholar] [CrossRef]
- Harris, J.J.; Jolivet, R.; Attwell, D. Synaptic energy use and supply. Neuron 2012, 75, 762–777. [Google Scholar] [CrossRef]
- Friedman, J.R.; Nunnari, J. Mitochondrial form and function. Nature 2014, 505, 335–343. [Google Scholar] [CrossRef]
- Schon, E.A.; DiMauro, S.; Hirano, M. Human mitochondrial DNA: Roles of inherited and somatic mutations. Nat. Rev. Genet. 2012, 13, 878–890. [Google Scholar] [CrossRef] [PubMed]
- Caruana, N.J.; Stroud, D.A. The road to the structure of the mitochondrial respiratory chain supercomplex. Biochem. Soc. Trans. 2020, 48, 621–629. [Google Scholar] [CrossRef] [PubMed]
- Xie, N.; Zhang, L.; Gao, W.; Huang, C.; Huber, P.E.; Zhou, X.; Li, C.; Shen, G.; Zou, B. NAD+ metabolism: Pathophysiologic mechanisms and therapeutic potential. Signal Transduct. Target. Ther. 2020, 5, 227. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling. Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial electron transport chain: Oxidative phosphorylation, oxidant production, and methods of measurement. Redox Bio. 2020, 37, 101674. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Tirichen, H.; Yaigoub, H.; Xu, W.; Wu, C.; Li, R.; Li, Y. Mitochondrial Reactive Oxygen Species and Their Contribution in Chronic Kidney Disease Progression Through Oxidative Stress. Front. Physiol. 2021, 12, 627837. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Raptova, R.; Alomar, S.Y.; Alwasel, S.H.; Nepovimova, E.; Kuca, K.; Valko, M. Reactive oxygen species, toxicity, oxidative stress, and antioxidants: Chronic diseases and aging. Arch. Toxicol. 2023, 97, 2499–2574. [Google Scholar] [CrossRef]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003–2014. [Google Scholar] [CrossRef]
- Bauer, T.M.; Murphy, E. Role of Mitochondrial Calcium and the Permeability Transition Pore in Regulating Cell Death. Circ. Res. 2020, 126, 280–293. [Google Scholar] [CrossRef]
- Ahmad, W.; Ijaz, B.; Shabbiri, K.; Ahmed, F.; Rehman, S. Oxidative toxicity in diabetes and Alzheimer’s disease: Mechanisms behind ROS/ RNS generation. J. Biomed. Sci. 2017, 24, 76. [Google Scholar] [CrossRef]
- Khotina, V.A.; Vinokurov, A.Y.; Bagheri Ekta, M.; Sukhorukov, V.N.; Orekhov, A.N. Creation of Mitochondrial Disease Models Using Mitochondrial DNA Editing. Biomedicines 2023, 11, 532. [Google Scholar] [CrossRef]
- Han, Y.; Liu, D.; Cheng, Y.; Ji, Q.; Liu, M.; Zhang, B.; Zhou, S. Maintenance of mitochondrial homeostasis for Alzheimer’s disease: Strategies and challenges. Redox Biol. 2023, 63, 102734. [Google Scholar] [CrossRef] [PubMed]
- Tarafdar, A.; Pula, G. The Role of NADPH Oxidases and Oxidative Stress in Neurodegenerative Disorders. Int. J. Mol. Sci. 2018, 19, 3824. [Google Scholar] [CrossRef]
- Biffi, A.; Sabuncu, M.R.; Desikan, R.S.; Schmansky, N.; Salat, D.H.; Rosand, J.; Anderson, C.D.; Alzheimer’s disease Neuroimaging Initiative (ADNI). Genetic variation of oxidative phosphorylation genes in stroke and Alzheimer’s disease. Neurobiol. Aging 2014, 35, 1956.e1–1956.e8. [Google Scholar] [CrossRef]
- Venkataraman, A.V.; Mansur, A.; Rizzo, G.; Bishop, C.; Lewis, Y.; Kocagoncu, E.; Lingford-Hughes, A.; Huiban, M.; Passchier, J.; Rowe, J.B.; et al. Widespread cell stress and mitochondrial dysfunction occur in patients with early Alzheimer’s disease. Sci. Transl. Med. 2022, 14, eabk1051. [Google Scholar] [CrossRef]
- Zhang, C.; Rissman, R.A.; Feng, J. Characterization of ATP alternations in an Alzheimer’s disease transgenic mouse model. J. Alzheimers Dis. 2015, 44, 375–378. [Google Scholar] [CrossRef] [PubMed]
- Armand-Ugon, M.; Ansoleaga, B.; Berjaoui, S.; Ferrer, I. Reduced Mitochondrial Activity is Early and Steady in the Entorhinal Cortex but it is Mainly Unmodified in the Frontal Cortex in Alzheimer’s Disease. Curr. Alzheimer Res. 2017, 14, 1327–1334. [Google Scholar] [CrossRef] [PubMed]
- Finney, C.A.; Delerue, F.; Gold, W.A.; Brown, D.A.; Shvetcov, A. Artificial intelligence-driven meta-analysis of brain gene expression identifies novel gene candidates and a role for mitochondria in Alzheimer’s disease. Comput. Struct. Biotechnol. J. 2022, 21, 388–400. [Google Scholar] [CrossRef]
- Yaku, K.; Okabe, K.; Nakagawa, T. NAD metabolism: Implications in aging and longevity. Ageing Res. Rev. 2018, 47, 1–17. [Google Scholar] [CrossRef]
- Lautrup, S.; Sinclair, D.A.; Mattson, M.P.; Fang, E.F. NAD+ in Brain Aging and Neurodegenerative Disorders. Cell Metab. 2019, 30, 630–655. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhou, P.; Zhao, Z.; Li, J.; Fan, Z.; Li, X.; Cui, Z.; Fu, A. Improvement Effect of Mitotherapy on the Cognitive Ability of Alzheimer’s Disease through NAD+/SIRT1-Mediated Autophagy. Antioxidants 2023, 12, 2006. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias, A.J.; Perrone, R.; Grozio, A.; Verdin, E. NAD+ metabolism and its roles in cellular processes during ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 119–141. [Google Scholar] [CrossRef] [PubMed]
- Martire, S.; Mosca, L.; d’Erme, M. PARP-1 involvement in neurodegeneration: A focus on Alzheimer’s and Parkinson’s diseases. Mech. Ageing Dev. 2015, 146–148, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.; Feng, Y.; Zheng, J.; Ge, X.; Zhang, Y.; Wu, D.; Zhao, J.; Zhai, Q. Nmnat2 delays axon degeneration in superior cervical ganglia dependent on its NAD synthesis activity. Neurochem. Int. 2010, 56, 101–106. [Google Scholar] [CrossRef]
- Cheng, X.S.; Shi, F.X.; Zhao, K.P.; Lin, W.; Li, X.Y.; Zhang, J.; Bu, Y.Y.; Zhu, R.; Li, X.H.; Duan, D.X.; et al. Nmnat2 attenuates amyloidogenesis and up-regulates ADAM10 in AMPK activity-dependent manner. Aging 2021, 13, 23620–23636. [Google Scholar] [CrossRef]
- Angeletti, C.; Amici, A.; Gilley, J.; Loreto, A.; Trapanotto, A.G.; Antoniou, C.; Merlini, E.; Coleman, M.P.; Orsomando, G. SARM1 is a multi-functional NAD(P)ase with prominent base exchange activity, all regulated bymultiple physiologically relevant NAD metabolites. iScience 2022, 25, 103812. [Google Scholar] [CrossRef]
- Figley, M.D.; Gu, W.; Nanson, J.D.; Shi, Y.; Sasaki, Y.; Cunnea, K.; Malde, A.K.; Jia, X.; Luo, Z.; Saikot, F.K.; et al. SARM1 is a metabolic sensor activated by an increased NMN/NAD+ ratio to trigger axon degeneration. Neuron 2021, 109, 1118–1136.e11. [Google Scholar] [CrossRef]
- Yang, S.; Park, J.H.; Lu, H.C. Axonal energy metabolism, and the effects in aging and neurodegenerative diseases. Mol. Neurodegener. 2023, 18, 49. [Google Scholar] [CrossRef]
- Miao, X.; Wu, Q.; Du, S.; Xiang, L.; Zhou, S.; Zhu, J.; Chen, Z.; Wang, H.; Pan, X.; Fan, Y.; et al. SARM1 Promotes Neurodegeneration and Memory Impairment in Mouse Models of Alzheimer’s Disease. Aging Dis. 2023; Advance online publication. [Google Scholar] [CrossRef]
- Campbell, J.M. Supplementation with NAD+ and Its Precursors to Prevent Cognitive Decline across Disease Contexts. Nutrients 2022, 14, 3231. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Gao, Y.; Zeng, M.; Wang, Y.; Wei, T.F.; Lu, Y.B.; Zhang, W.P. Nicotinamide ribose ameliorates cognitive impairment of aged and Alzheimer’s disease model mice. Metab. Brain Dis. 2019, 34, 353–366. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Wei, Y.; Lautrup, S.; Yang, B.; Wang, Y.; Cordonnier, S.; Mattson, M.P.; Croteau, D.L.; Bohr, V.A. NAD+ supplementation reduces neuroinflammation and cell senescence in a transgenic mouse model of Alzheimer’s disease via cGAS-STING. Proc. Natl. Acad. Sci. USA 2021, 118, e2011226118. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Lautrup, S.; Cordonnier, S.; Wang, Y.; Croteau, D.L.; Zavala, E.; Zhang, Y.; Moritoh, K.; O’Connell, J.F.; Baptiste, B.A.; et al. NAD+ supplementation normalizes key Alzheimer’s features and DNA damage responses in a new AD mouse model with introduced DNA repair deficiency. Proc. Natl. Acad. Sci. USA 2018, 115, E1876–E1885. [Google Scholar] [CrossRef] [PubMed]
- Guedes-Dias, P.; Holzbaur, E.L.F. Axonal transport: Driving synaptic function. Science 2019, 366, eaaw9997. [Google Scholar] [CrossRef]
- Berth, S.H.; Lloyd, T.E. Disruption of axonal transport in neurodegeneration. J. Clin. Investg. 2023, 133, e168554. [Google Scholar] [CrossRef] [PubMed]
- Niescier, R.F.; Kwak, S.K.; Joo, S.H.; Chang, K.T.; Min, K.T. Dynamics of Mitochondrial Transport in Axons. Front. Cell. Neurosci. 2016, 10, 123. [Google Scholar] [CrossRef]
- Hung, C. Importance of retrograde axonal transport in mitochondrial health and distribution. Cell Death Discov. 2021, 7, 106. [Google Scholar] [CrossRef]
- Mandal, A.; Wong, H.C.; Pinter, K.; Mosqueda, N.; Beirl, A.; Lomash, R.M.; Won, S.; Kindt, K.S.; Drerup, C.M. Retrograde Mitochondrial Transport Is Essential for Organelle Distribution and Health in Zebrafish Neurons. J. Neurosci. 2021, 41, 1371–1392. [Google Scholar] [CrossRef]
- Cai, Q.; Gerwin, C.; Sheng, Z.H. Syntabulin-mediated anterograde transport of mitochondria along neuronal processes. J. Cell Biol. 2005, 170, 959–969. [Google Scholar] [CrossRef]
- Li, A.; Gao, M.; Liu, B.; Qin, Y.; Chen, L.; Liu, H.; Wu, H.; Gong, G. Mitochondrial autophagy: Molecular mechanisms and implications for cardiovascular disease. Cell Death Dis. 2022, 13, 444. [Google Scholar] [CrossRef]
- Pickles, S.; Vigié, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef]
- Babbar, M.; Basu, S.; Yang, B.; Croteau, D.L.; Bohr, V.A. Mitophagy and DNA damage signaling in human aging. Mech. Ageing Dev. 2020, 186, 111207. [Google Scholar] [CrossRef] [PubMed]
- Martín-Maestro, P.; Gargini, R.; Perry, G.; Avila, J.; García-Escudero, V. PARK2 enhancement is able to compensate mitophagy alterations found in sporadic Alzheimer’s disease. Hum. Mol. Genet. 2016, 25, 792–806. [Google Scholar] [CrossRef]
- Kim, I.; Rodriguez-Enriquez, S.; Lemasters, J.J. Selective degradation of mitochondria by mitophagy. Arch. Biochem. Biophys. 2007, 462, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Varte, V.; Munkelwitz, J.W.; Rincon-Limas, D.E. Insights from Drosophila on Aβ- and tau-induced mitochondrial dysfunction: Mechanisms and tools. Front. Neurosci. 2023, 17, 1184080. [Google Scholar] [CrossRef]
- Wang, Z.T.; Lu, M.H.; Zhang, Y.; Ji, W.L.; Lei, L.; Wang, W.; Fang, L.P.; Wang, L.W.; Yu, F.; Wang, J.; et al. Disrupted-in-schizophrenia-1 protects synaptic plasticity in a transgenic mouse model of Alzheimer’s disease as a mitophagy receptor. Aging Cell 2019, 18, e12860. [Google Scholar] [CrossRef]
- Kerr, J.S.; Adriaanse, B.A.; Greig, N.H.; Mattson, M.P.; Cader, M.Z.; Bohr, V.A.; Fang, E.F. Mitophagy and Alzheimer’s Disease: Cellular and Molecular Mechanisms. Trends Neurosci. 2017, 40, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Cenini, G.; Lloret, A.; Cascella, R. Oxidative stress in neurodegenerative diseases: From a mitochondrial point of view. Oxid. Med. Cell. Longev. 2019, 2, 607–618. [Google Scholar] [CrossRef]
- Du, F.; Yu, Q.; Kanaan, N.M.; Yan, S.S. Mitochondrial oxidative stress contributes to the pathological aggregation and accumulation of tau oligomers in Alzheimer’s disease. Hum. Mol. Genet. 2022, 31, 2498–2507. [Google Scholar] [CrossRef]
- Audano, M.; Schneider, A.; Mitro, N. Mitochondria, lysosomes, and dysfunction: Their meaning in neurodegeneration. J. Neurochem. 2018, 147, 291–309. [Google Scholar] [CrossRef]
- Cardoso, S.; Carvalho, C.; Correia, S.C.; Seiça, R.M.; Moreira, P.I. Alzheimer’s Disease: From Mitochondrial Perturbations to Mitochondrial Medicine. Brain Pathol. 2016, 26, 632–647. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Oliver, D.M. Amyloid Beta and Phosphorylated Tau-Induced Defective Autophagy and Mitophagy in Alzheimer’s Disease. Cells 2019, 8, 488. [Google Scholar] [CrossRef] [PubMed]
- Averchuk, A.S.; Ryazanova, M.V.; Baranich, T.I.; Stavrovskaya, A.V.; Rozanova, N.A.; Novikova, S.V.; Salmina, A.B. The Neurotoxic Effect of β-Amyloid Is Accompanied by Changes in the Mitochondrial Dynamics and Autophagy in Neurons and Brain Endothelial Cells in the Experimental Model of Alzheimer’s Disease. Bull. Exp. Biol. Med. 2023, 175, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Hansson Petersen, C.A.; Alikhani, N.; Behbahani, H.; Wiehager, B.; Pavlov, P.F.; Alafuzoff, I.; Leinonen, V.; Ito, A.; Winblad, B.; Glaser, E.; et al. The amyloid beta-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. USA 2008, 105, 13145–13150. [Google Scholar] [CrossRef] [PubMed]
- Dou, Y.; Tan, Y. Presequence protease reverses mitochondria-specific amyloid-β-induced mitophagy to protect mitochondria. FASEB J. 2023, 37, e22890. [Google Scholar] [CrossRef] [PubMed]
- Alikhani, N.; Guo, L.; Yan, S.; Du, H.; Pinho, C.M.; Chen, J.X.; Glaser, E.; Yan, S.S. Decreased Proteolytic Activity of the Mitochondrial Amyloid-β Degrading Enzyme, PreP Peptidasome, in Alzheimer’s Disease Brain Mitochondria. J. Alzheimers Dis. 2011, 27, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Limorenko, G.; Lashuel, H.A. Revisiting the grammar of Tau aggregation and pathology formation: How new insights from brain pathology are shaping how we study and target tauopathies. Chem. Soc. Rev. 2022, 51, 513–565. [Google Scholar] [CrossRef]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 5–21. [Google Scholar] [CrossRef]
- Saavedra, J.; Nascimento, M.; Liz, M.A.; Cardoso, I. Key brain cell interactions and contributions to the pathogenesis of Alzheimer’s disease. Front. Cell Dev. Biol. 2022, 10, 1036123. [Google Scholar] [CrossRef]
- Reddy, P.H. Abnormal tau, mitochondrial dysfunction, impaired axonal transport of mitochondria, and synaptic deprivation in Alzheimer’s disease. Brain Res. 2011, 1415, 136–148. [Google Scholar] [CrossRef]
- Mietelska-Porowska, A.; Wasik, U.; Goras, M.; Filipek, A.; Niewiadomska, G. Tau protein modifications and interactions: Their role in function and dysfunction. Int. J. Mol. Sci. 2014, 15, 4671–4713. [Google Scholar] [CrossRef]
- Eckert, A.; Schulz, K.L.; Rhein, V.; Götz, J. Convergence of amyloid-beta and tau pathologies on mitochondria in vivo. Mol. Neurobiol. 2010, 41, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Cario, A.; Berger, C.L. Tau, microtubule dynamics, and axonal transport: New paradigms for neurodegenerative disease. Bioessays 2023, 45, e2200138. [Google Scholar] [CrossRef] [PubMed]
- Medeiros, R.; Baglietto-Vargas, D.; LaFerla, F.M. The role of tau in Alzheimer’s disease and related disorders. Neurosci. Ther. 2011, 17, 514–524. [Google Scholar] [CrossRef] [PubMed]
- Combs, B.; Mueller, R.L.; Morfini, G.; Brady, S.T.; Kanaan, N.M. Tau and Axonal Transport Misregulation in Tauopathies. Adv. Exp. Med. Biol. 2019, 1184, 81–95. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ye, M.; Ma, L. The emerging role of autophagy and mitophagy in tauopathies: From pathogenesis to translational implications in Alzheimer’s disease. Front. Aging Neurosci. 2022, 14, 1022821. [Google Scholar] [CrossRef] [PubMed]
- Morton, H.; Kshirsagar, S.; Orlov, E.; Bunquin, L.E.; Sawant, N.; Boleng, L.; George, M.; Basu, T.; Ramasubramanian, B.; Pradeepkiran, J.A.; et al. Defective mitophagy and synaptic degeneration in Alzheimer’s disease: Focus on aging, mitochondria and synapse. Free Radic. Biol. Med. 2021, 172, 652–667. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Li, X.C.; Wang, Z.H.; Luo, Y.; Zhang, X.; Liu, X.P.; Feng, Q.; Wang, Q.; Yue, Z.; Chen, Z.; et al. Tau accumulation impairs mitophagy via increasing mitochondrial membrane potential and reducing mitochondrial Parkin. Oncotarget 2016, 7, 17356–17368. [Google Scholar] [CrossRef] [PubMed]
- Lechado-Terradas, A.; Schepers, S.; Zittlau, K.I.; Sharma, K.; Ok, O.; Fitzgerald, J.C.; Geimer, S.; Westermann, B.; Macek, B.; Kahle, P.J. Parkin-dependent mitophagy occurs via proteasome-dependent steps sequentially targeting separate mitochondrial sub-compartments for autophagy. Autophagy Rep. 2022, 1, 576–602. [Google Scholar] [CrossRef]
- Cummins, N.; Tweedie, A.; Zuryn, S.; Bertran-Gonzalez, J.; Götz, J. Disease-associated tau impairs mitophagy by inhibiting Parkin translocation to mitochondria. EMBO J. 2019, 38, e99360. [Google Scholar] [CrossRef]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Das, R.; Chakrabarti, O. Mitochondrial hyperfusion: A friend or a foe. Biochem. Soc. Trans. 2020, 48, 631–644. [Google Scholar] [CrossRef]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta 2014, 8, 1240–1247. [Google Scholar] [CrossRef]
- Manczak, M.; Calkins, M.J.; Reddy, P.H. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer’s disease: Implications for neuronal damage. Hum. Mol. Genet. 2011, 20, 2495–2509. [Google Scholar] [CrossRef] [PubMed]
- Dhapola, R.; Sarma, P.; Medhi, B.; Prakash, A.; Reddy, D.H. Recent Advances in Molecular Pathways and Therapeutic Implications Targeting Mitochondrial Dysfunction for Alzheimer’s Disease. Mol. Neurobiol. 2022, 59, 535–555. [Google Scholar] [CrossRef]
- Kim, D.I.; Lee, K.H.; Gabr, A.A.; Choi, G.E.; Kim, J.S.; Ko, S.H.; Han, H.J. Abeta-induced Drp1 phosphorylation through Akt activation promotes excessive mitochondrial fission leading to neuronal apoptosis. Biochim. Biophys. Acta 2016, 1863, 2820–2834. [Google Scholar] [CrossRef] [PubMed]
- Abtahi, S.L.; Masoudi, R.; Haddadi, M. The distinctive role of tau and amyloid beta in mitochondrial dysfunction through alteration in Mfn2 and Drp1 mRNA Levels: A comparative study in Drosophila melanogaster. Gene 2020, 754, 144854. [Google Scholar] [CrossRef]
- Silva, D.F.; Selfridge, J.E.; Lu, J.; E, L.; Roy, N.; Hutfles, L.; Burns, J.M.; Michaelis, E.K.; Yan, S.; Cardoso, S.M.; et al. Bioenergetic flux, mitochondrial mass and mitochondrial morphology dynamics in AD and MCI cybrid cell lines. Hum. Mol. Genet. 2013, 22, 3931–3946. [Google Scholar] [CrossRef]
- Nakamura, T.; Lipton, S.A. Redox regulation of mitochondrial fission, protein misfolding, synaptic damage, and neuronal cell death: Potential implications for Alzheimer’s and Parkinson’s diseases. Apoptosis 2010, 15, 1354–1363. [Google Scholar] [CrossRef]
- Harland, M.; Torres, S.; Liu, J.; Wang, X. Neuronal mitochondria modulation of LPS-induced neuroinflammation. J. Neurosci. 2020, 40, 1756–1765. [Google Scholar] [CrossRef] [PubMed]
- Batista, A.F.; Rody, T.; Forny-Germano, L.; Cerdeiro, S.; Bellio, M.; Ferreira, S.T.; Munoz, D.P.; De Felice, F.G. Interleukin-1β mediates alterations in mitochondrial fusion/fission proteins and memory impairment induced by amyloid-β oligomers. J. Neuroinflammation 2021, 18, 54. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, M.; Reddy, P.H. Reduced VDAC1, Maintained Mitochondrial Dynamics and Enhanced Mitochondrial Biogenesis in a Transgenic Tau Mouse Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 8561. [Google Scholar] [CrossRef] [PubMed]
- Li, X.C.; Hu, Y.; Wang, Z.H.; Luo, Y.; Zhang, Y.; Liu, X.P.; Feng, Q.; Wang, Q.; Ye, K.; Liu, G.P.; et al. Human wild-type full-length tau accumulation disrupts mitochondrial dynamics and the functions via increasing mitofusins. Sci. Rep. 2016, 6, 24756. [Google Scholar] [CrossRef] [PubMed]
- Bera, A.; Lavanya, G.; Reshmi, R.; Dev, K.; Kumar, R. Mechanistic and therapeutic role of Drp1 in the pathogenesis of Alzheimer’s disease. Eur. J. Neurosci. 2022, 9, 5516–5531. [Google Scholar] [CrossRef]
- Lauretti, E.; Dincer, O.; Praticò, D. Glycogen synthase kinase-3 signaling in Alzheimer’s disease. Biochim. Biophys. Acta Mol. Cell. Res. 2020, 1867, 118664. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.; Atluri, V.; Kaushik, A.; Yndart, A.; Nair, M. Alzheimer’s disease: Pathogenesis, diagnostics, and therapeutics. Int. J. Nanomed. 2019, 14, 5541–5554. [Google Scholar] [CrossRef] [PubMed]
- Amaral, A.C.; Perez-Nievas, B.G.; Siao Tick Chong, M.; Gonzalez-Martinez, A.; Argente-Escrig, H.; Rubio-Guerra, S.; Commins, C.; Muftu, S.; Eftekharzadeh, B.; Hudry, E.; et al. Isoform-selective decrease of glycogen synthase kinase-3-beta (GSK-3β) reduces synaptic tau phosphorylation, transcellular spreading, and aggregation. iScience 2021, 24, 102058. [Google Scholar] [CrossRef]
- Chou, C.H.; Lin, C.C.; Yang, M.C.; Wei, C.C.; Liao, H.D.; Lin, R.C.; Tu, W.Y.; Kao, T.C.; Hsu, C.M.; Cheng, J.T.; et al. GSK3beta-mediated Drp1 phosphorylation induced elongated mitochondrial morphology against oxidative stress. PLoS ONE 2012, 7, e49112. [Google Scholar] [CrossRef]
- Kandimalla, R.; Reddy, P.H. Multiple faces of dynamin-related protein 1 and its role in Alzheimer’s disease pathogenesis. Biochim. Biophys. Acta 2016, 4, 814–828. [Google Scholar] [CrossRef]
- Yan, J.; Liu, X.H.; Han, M.Z.; Wang, Y.M.; Sun, X.L.; Yu, N.; Li, T.; Su, B.; Chen, Z.Y. Blockage of GSK3β-mediated Drp1 phosphorylation provides neuroprotection in neuronal and mouse models of Alzheimer’s disease. Neurobiol. Aging 2015, 36, 211–227. [Google Scholar] [CrossRef]
- Ly, P.T.; Wu, Y.; Zou, H.; Wang, R.; Zhou, W.; Kinoshita, A.; Zhang, M.; Yang, Y.; Cai, F.; Woodgett, J.; et al. Inhibition of GSK3β-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J. Clin. Investg. 2013, 123, 224–235. [Google Scholar] [CrossRef]
- Du, F.; Yu, Q.; Yan, S.; Hu, G.; Lue, L.F.; Walker, D.G.; Wu, L.; Yan, S.F.; Tieu, K.; Yan, S.S. PINK1 signalling rescues amyloid pathology and mitochondrial dysfunction in Alzheimer’s disease. Brain 2017, 140, 3233–3251. [Google Scholar] [CrossRef]
- Du, F.; Yu, Q.; Yan, S.S. PINK1 Activation Attenuates Impaired Neuronal-Like Differentiation and Synaptogenesis and Mitochondrial Dysfunction in Alzheimer’s Disease Trans-Mitochondrial Cybrid Cells. J. Alzheimers Dis. 2021, 81, 1749–1761. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Tan, J.; Wang, R.; Wan, H.; He, Y.; Yan, X.; Guo, J.; Gao, Q.; Li, J.; Shang, S.; et al. PINK1 phosphorylates Drp1S616 to regulate mitophagy-independent mitochondrial dynamics. EMBO Rep. 2020, 21, e48686. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Song, P.; Du, L.; Tian, W.; Yue, W.; Liu, M.; Li, D.; Wang, B.; Zhu, Y.; Cao, C.; et al. Parkin ubiquitinates Drp1 for proteasome-dependent degradation: Implication of dysregulated mitochondrial dynamics in Parkinson disease. J. Biol. Chem. 2011, 286, 11649–11658. [Google Scholar] [CrossRef] [PubMed]
- Coppedè, F.; Stoccoro, A. Mitoepigenetics and Neurodegenerative Diseases. Front. Endocrinol. 2019, 10, 86. [Google Scholar] [CrossRef] [PubMed]
- Coppedè, F. Mitochondrial DNA methylation and mitochondria-related epigenetics in neurodegeneration. Neural Regen. Res. 2024, 19, 405–406. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Xu, L.; Han, M.; Liu, X.; Li, F.; Zhou, X.; Wang, Y.; Bi, J. Altered mitochondrial DNA methylation and mitochondrial DNA copy number in an app/ps1 transgenic mouse model of Alzheimer disease. Biochem. Biophys. Res. Commun. 2019, 520, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Filograna, R.; Mennuni, M.; Alsina, D.; Larsson, N.G. Mitochondrial DNA copy number in human disease: The more the better? FEBS Lett. 2021, 595, 976–1002. [Google Scholar] [CrossRef] [PubMed]
- Castellani, C.A.; Longchamps, R.J.; Sun, J.; Guallar, E.; Arking, D.E. Thinking outside the nucleus: Mitochondrial DNA copy number in health and disease. Mitochondrion 2020, 53, 214–223. [Google Scholar] [CrossRef]
- Xu, Y.; Cheng, L.; Sun, J.; Li, F.; Liu, X.; Wei, Y.; Han, M.; Zhu, Z.; Bi, J.; Lai, C.; et al. Hypermethylation of Mitochondrial Cytochrome b and Cytochrome c Oxidase II Genes with Decreased Mitochondrial DNA Copy Numbers in the APP/PS1 Transgenic Mouse Model of Alzheimer’s Disease. Neurochem. Res. 2021, 46, 564–572. [Google Scholar] [CrossRef]
- Petrosillo, G.; De Benedictis, V.; Ruggiero, F.M.; Paradies, G. Decline in cytochrome c oxidase activity in rat-brain mitochondria with aging. Role of peroxidized cardiolipin and beneficial effect of melatonin. J. Bioenerg. Biomembr. 2013, 45, 431–440. [Google Scholar] [CrossRef]
- Kadenbach, B. Complex IV–The regulatory center of mitochondrial oxidative phosphorylation. Mitochondrion 2021, 58, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Zhang, X.; Wan, Z.; Tian, F.; Ling, J.; Tan, J.; Peng, X. Characterization of Mitochondrial DNA Methylation of Alzheimer’s Disease in Plasma Cell-Free DNA. Diagnostics 2023, 13, 2351. [Google Scholar] [CrossRef] [PubMed]
- Klein, H.U.; Trumpff, C.; Yang, H.S.; Lee, A.J.; Picard, M.; Bennett, D.A.; De Jager, P.L. Characterization of mitochondrial DNA quantity and quality in the human aged and Alzheimer’s disease brain. Mol. Neurodegener. 2021, 16, 75. [Google Scholar] [CrossRef]
- Costantini, L.C.; Barr, L.J.; Vogel, J.L.; Henderson, S.T. Hypometabolism as a therapeutic target in Alzheimer’s disease. BMC Neurosci. 2008, 9, S16. [Google Scholar] [CrossRef]
- Mergenthaler, P.; Lindauer, U.; Dienel, G.A.; Meisel, A. Sugar for the brain: The role of glucose in physiological and pathological brain function. Trends Neurosci. 2013, 36, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Kim, S.H.; Bishayee, K. Dysfunctional Glucose Metabolism in Alzheimer’s Disease Onset and Potential Pharmacological Interventions. Int. J. Mol. Sci. 2022, 23, 9540. [Google Scholar] [CrossRef]
- Patel, V.; Mill, J.; Okonkwo, O.C.; Salamat, S.; Li, L.; Raife, T. Global Energy Metabolism Deficit in Alzheimer Disease Brain. J. Prev. Alzheimers Dis. 2024, 11, 171–178. [Google Scholar] [CrossRef]
- Mosconi, L. Brain glucose metabolism in the early and specific diagnosis of Alzheimer’s disease FDG-PET studies in MCI and AD. Eur. J. Nucl. Med. Mol. Imaging 2005, 32, 486–510. [Google Scholar] [CrossRef]
- Myoraku, A.; Klein, G.; Landau, S.; Tosun, D.; Alzheimer’s Disease Neuroimaging, I. Regional uptakes from early frame amyloid PET and (18)F-FDG PET scans are comparable independent of disease state. Eur. J. Hybrid. Imaging 2022, 6, 2. [Google Scholar] [CrossRef] [PubMed]
- Mosconi, L.; Rinne, J.O.; Tsui, W.H.; Murray, J.; Li, Y.; Glodzik, L.; McHugh, P.; Williams, S.; Cummings, M.; Pirraglia, E.; et al. Amyloid and metabolic positron emission tomography imaging of cognitively normal adults with Alzheimer’s parents. Neurobiol. Aging 2013, 34, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Blazhenets, G.; Ma, Y.; Sörensen, A.; Schiller, F.; Rücker, G.; Eidelberg, D.; Frings, L.; Meyer, P.T.; Alzheimer Disease Neuroimaging Initiative. Predictive value of 18F-florbetapir and 18F-FDG PET for conversion from mild cognitive impairment to Alzheimer dementia. J. Nucl. Med. 2020, 61, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Mosconi, L.; Mistur, R.; Switalski, R.; Tsui, W.H.; Glodzik, L.; Li, Y.; Pirraglia, E.; De Santi, S.; Reisberg, B.; Wisniewski, T.; et al. FDG-PET changes in brain glucose metabolism from normal cognition to pathologically verified Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 811–822. [Google Scholar] [CrossRef] [PubMed]
- Ou, Y.N.; Xu, W.; Li, J.Q.; Guo, Y.; Cui, M.; Chen, K.L.; Huang, Y.Y.; Dong, Q.; Tan, L.; Yu, J.T.; et al. FDG-PET as an independent biomarker for Alzheimer’s biological diagnosis: A longitudinal study. Alzheimers Res. Ther. 2019, 11, 57. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Shen, Z.; Wang, Q.; Zhang, B.; Zhuang, Z.; Lin, J.; Shen, Y.; Chen, Y.; Dai, Z.; Wu, R. Reduced Cerebral Glucose Uptake in an Alzheimer’s Rat Model With Glucose-Weighted Chemical Exchange Saturation Transfer Imaging. Front. Aging Neurosci. 2021, 13, 618690. [Google Scholar] [CrossRef]
- Zhou, Y.; Dougherty, J.H., Jr.; Hubner, K.F.; Bai, B.; Cannon, R.L.; Hutson, R.K. Abnormal connectivity in the posterior cingulate and hippocampus in early Alzheimer’s disease and mild cognitive impairment. Alzheimers Dement. 2008, 4, 265–270. [Google Scholar] [CrossRef]
- Ferrari, B.L.; Neto, G.C.C.; Nucci, M.P.; Mamani, J.B.; Lacerda, S.S.; Felicio, A.C.; Amaro, E., Jr.; Gamarra, L.F. The accuracy of hippocampal volumetry and glucose metabolism for the diagnosis of patients with suspected Alzheimer’s disease, using automatic quantitative clinical tools. Medicine 2019, 98, e17824. [Google Scholar] [CrossRef]
- Weise, C.M.; Chen, K.; Chen, Y.; Kuang, X.; Savage, C.R.; Reiman, E.M.; Alzheimer’s Disease Neuroimaging Initiative. Left lateralized cerebral glucose metabolism declines in amyloid-β positive persons with mild cognitive impairment. Neuroimage Clin. 2018, 20, 286–296. [Google Scholar] [CrossRef]
- Kyrtata, N.; Emsley, H.C.A.; Sparasci, O.; Parkes, L.M.; Dickie, B.R. A Systematic Review of Glucose Transport Alterations in Alzheimer’s Disease. Front. Neurosci. 2021, 15, 626636. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, Y.T.K.; Ha, H.T.T.; Nguyen, T.H.; Nguyen, L.N. The role of SLC transporters for brain health and disease. Cell. Mol. Life Sci. 2022, 79, 20. [Google Scholar] [CrossRef] [PubMed]
- Patching, S.G. Glucose transporters at the blood–brain barrier: Function, regulation and gateways for drug delivery. Mol. Neurobiol. 2017, 54, 1046–1077. [Google Scholar] [CrossRef] [PubMed]
- Szablewski, L. Brain Glucose Transporters: Role in Pathogenesis and Potential Targets for the Treatment of Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 8142. [Google Scholar] [CrossRef] [PubMed]
- Ashok, B.S.; Ajith, T.A.; Sivanesan, S. Hypoxia-inducible factors as neuroprotective agent in Alzheimer’s disease. Clin. Exp. Pharmacol. Physiol. 2017, 44, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.X. Decreased glucose transporters correlate to abnormal hyperphosphorylation of tau in Alzheimer disease. FEBS Lett. 2008, 582, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Karelina, K.; Obrietan, K. CREB: A multifaceted regulator of neuronal plasticity and protection. J. Neurochem. 2011, 116, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Pugazhenthi, S.; Wang, M.; Pham, S.; Sze, C.I.; Eckman, C.B. Downregulation of CREB expression in Alzheimer’s brain and in Aβ-treated rat hippocampal neurons. Mol. Neurodegener. 2011, 6, 60. [Google Scholar] [CrossRef] [PubMed]
- Jin, N.; Qian, W.; Yin, X.; Zhang, L.; Iqbal, K.; Grundke-Iqbal, I.; Gong, C.X.; Liu, F. CREB regulates the expression of neuronal glucose transporter 3: A possible mechanism related to impaired brain glucose uptake in Alzheimer’s disease. Nucleic Acids Res. 2013, 41, 3240–3256. [Google Scholar] [CrossRef]
- Huang, C.W.; Rust, N.C.; Wu, H.F.; Yin, A.; Zeltner, N.; Yin, H.; Hart, G.W. Low glucose induced Alzheimer’s disease-like biochemical changes in human induced pluripotent stem cell-derived neurons is due to dysregulated O-GlcNAcylation. Alzheimers Dement. 2023, 19, 4872–4885. [Google Scholar] [CrossRef]
- Hart, G.W. Nutrient regulation of signaling and transcription. J. Biol. Chem. 2019, 294, 2211–2231. [Google Scholar] [CrossRef]
- Dos Santos, J.P.A.; Vizuete, A.; Hansen, F.; Biasibetti, R.; Gonçalves, C.A. Early and persistent O-GlcNAc protein modification in the streptozotocin model of Alzheimer’s disease. J. Alzheimers Dis. 2018, 61, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Förster, S.; Welleford, A.S.; Triplett, J.C.; Sultana, R.; Schmitz, B.; Butterfield, D.A. Increased O-GlcNAc levels correlate with decreased O-GlcNAcase levels in Alzheimer disease brain. Biochim. Biophys. Acta 2014, 1842, 1333–1339. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.W.; Rust, N.C.; Wu, H.F.; Hart, G.W. Altered O-GlcNAcylation and mitochondrial dysfunction, a molecular link between brain glucose dysregulation and Sporadic Alzheimer’s disease. Neural Regen. Res. 2023, 18, 779–783. [Google Scholar] [CrossRef] [PubMed]
- Pinho, T.S.; Correia, S.C.; Perry, G.; Ambrósio, A.F.; Moreira, P.I. Diminished O-GlcNAcylation in Alzheimer’s disease is strongly correlated with mitochondrial anomalies. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2048–2059. [Google Scholar] [CrossRef]
- Permanne, B.; Sand, A.; Ousson, S.; Nény, M.; Hantson, J.; Schubert, R.; Wiessner, C.; Quattropani, A.; Beher, D. O-GlcNAcase inhibitor ASN90 is a multimodal drug candidate for tau and α-synuclein proteinopathies. ACS Chem. Neurosci. 2022, 13, 1296–1314. [Google Scholar] [CrossRef]
- Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Hart, G.W.; Gong, C.X. O-GlcNAcylation regulates phosphorylation of tau: A mechanism involved in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 10804–10809. [Google Scholar] [CrossRef]
- Tan, E.P.; McGreal, S.R.; Graw, S.; Tessman, R.; Koppel, S.J.; Dhakal, P.; Zhang, Z.; Machacek, M.; Zachara, N.E.; Koestler, D.C.; et al. Sustained O-GlcNAcylation reprograms mitochondrial function to regulate energy metabolism. J. Biol. Chem. 2017, 292, 14940–14962. [Google Scholar] [CrossRef]
- Ephrame, S.J.; Cork, G.K.; Marshall, V.; Johnston, M.A.; Shawa, J.; Alghusen, I.; Qiang, A.; Denson, A.R.; Carman, M.S.; Fedosyuk, H.; et al. O-GlcNAcylation regulates extracellular signal-regulated kinase (ERK) activation in Alzheimer’s disease. Front. Aging Neurosci. 2023, 15, 1155630. [Google Scholar] [CrossRef]
- Mathys, H.; Davila-Velderrain, J.; Peng, Z.; Gao, F.; Mohammadi, S.; Young, J.Z.; Menon, M.; He, L.; Abdurrob, F.; Jiang, X.; et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 2019, 570, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.S.; Ebbert, M.T.W.; Baker, K.E.; Cook, C.; Wang, X.; Sens, J.P.; Kocher, J.P.; Petrucelli, L.; Fryer, J.D. Microglial translational profiling reveals a convergent APOE pathway from aging, amyloid, and tau. J. Exp. Med. 2018, 215, 2235–2245. [Google Scholar] [CrossRef] [PubMed]
- Raulin, A.C.; Doss, S.V.; Trottier, Z.A.; Ikezu, T.C.; Bu, G.; Liu, C.C. ApoE in Alzheimer’s disease: Pathophysiology and therapeutic strategies. Mol. Neurodegener. 2022, 17, 72. [Google Scholar] [CrossRef]
- Oliver, D.M.A.; Reddy, P.H. Molecular Basis of Alzheimer’s Disease: Focus on Mitochondria. J. Alzheimers Dis. 2019, 72, S95–S116. [Google Scholar] [CrossRef]
- Reiman, E.M.; Arboleda-Velasquez, J.F.; Quiroz, Y.T.; Huentelman, M.J.; Beach, T.G.; Caselli, R.J.; Chen, Y.; Su, Y.; Myers, A.J.; Hardy, J.; et al. Alzheimer’s Disease Genetics Consortium Exceptionally low likelihood of Alzheimer’s dementia in APOE2 homozygotes from a 5,000-person neuropathological study. Nat. Commun. 2020, 11, 667. [Google Scholar] [CrossRef]
- Safieh, M.; Korczyn, A.D.; Michaelson, D.M. ApoE4: An emerging therapeutic target for Alzheimer’s disease. BMC Med. 2019, 17, 64. [Google Scholar] [CrossRef]
- Chen, Y.; Strickland, M.R.; Soranno, A.; Holtzman, D.M. Apolipoprotein E: Structural Insights and Links to Alzheimer Disease Pathogenesis. Neuron 2021, 109, 205–221. [Google Scholar] [CrossRef]
- Lee, E.G.; Leong, L.; Chen, S.; Tulloch, J.; Yu, C.E. APOE Locus-Associated Mitochondrial Function and Its Implication in Alzheimer’s Disease and Aging. Int. J. Mol. Sci. 2023, 24, 10440. [Google Scholar] [CrossRef] [PubMed]
- Area-Gomez, E.; Schon, E.A. Mitochondria-associated ER membranes and Alzheimer disease. Curr. Opin. Genet. Dev. 2016, 38, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Mahley, R.W. Apolipoprotein E4 targets mitochondria and the mitochondria-associated membrane complex in neuropathology, including Alzheimer’s disease. Curr. Opin. Neurobiol. 2023, 79, 102684. [Google Scholar] [CrossRef] [PubMed]
- Area-Gomez, E.; Schon, E.A. On the Pathogenesis of Alzheimer’s Disease: The MAM Hypothesis. FASEB J. 2017, 31, 864–867. [Google Scholar] [CrossRef] [PubMed]
- Liang, T.; Hang, W.; Chen, J.; Wu, Y.; Wen, B.; Xu, K.; Ding, B.; Chen, J. ApoE4 (Δ272–299) induces mitochondrial-associated membrane formation and mitochondrial impairment by enhancing GRP75-modulated mitochondrial calcium overload in neuron. Cell Biosci. 2021, 11, 50. [Google Scholar] [CrossRef]
- Simonovitch, S.; Schmukler, E.; Masliah, E.; Pinkas-Kramarski, R.; Michaelson, D.M. The Effects of APOE4 on Mitochondrial Dynamics and Proteins in vivo. J. Alzheimers Dis. 2019, 70, 861–875. [Google Scholar] [CrossRef]
- Riedel, B.C.; Thompson, P.M.; Brinton, R.D. Age, APOE and sex: Triad of risk of Alzheimer’s disease. J. Steroid Biochem. Mol. Biol. 2016, 160, 134–147. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Reiman, E.M.; Beach, T.G.; Serrano, G.E.; Sabbagh, M.N.; Nielsen, M.; Caselli, R.J.; Shi, J. Effect of ApoE isoforms on mitochondria in Alzheimer disease. Neurology 2020, 94, e2404–e2411. [Google Scholar] [CrossRef] [PubMed]
- Costa-Laparra, I.; Juárez-Escoto, E.; Vicario, C.; Moratalla, R.; García-Sanz, P. APOE ε4 allele, along with G206D-PSEN1mutation, alters mitochondrial networks and their degradation in Alzheimer’s disease. Front. Aging Neurosci. 2023, 15, 1087072. [Google Scholar] [CrossRef] [PubMed]
- Orr, A.L.; Kim, C.; Jimenez-Morales, D.; Newton, B.W.; Johnson, J.R.; Krogan, N.J.; Swaney, D.L.; Mahley, R.W. Neuronal Apolipoprotein E4 Expression Results in Proteome-Wide Alterations and Compromises Bioenergetic Capacity by Disrupting Mitochondrial Function. J. Alzheimers Dis. 2019, 68, 991–1011. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Cho, S.; Kim, M.J.; Park, Y.J.; Cho, E.; Jo, Y.S.; Kim, Y.S.; Lee, J.Y.; Thoudam, T.; Woo, S.H.; et al. ApoE4-dependent lysosomal cholesterol accumulation impairs mitochondrial homeostasis and oxidative phosphorylation in human astrocytes. Cell Rep. 2023, 42, 113183. [Google Scholar] [CrossRef] [PubMed]
- Reiss, A.B.; Ahmed, S.; Dayaramani, C.; Glass, A.D.; Gomolin, I.H.; Pinkhasov, A.; Stecker, M.M.; Wisniewski, T.; De Leon, J. The role of mitochondrial dysfunction in Alzheimer’s disease: A potential pathway to treatment. Exp. Gerontol. 2022, 164, 111828. [Google Scholar] [CrossRef] [PubMed]
- Alshial, E.E.; Abdulghaney, M.I.; Wadan, A.S.; Abdellatif, M.A.; Ramadan, N.E.; Suleiman, A.M.; Waheed, N.; Abdellatif, M.; Mohammed, H.S. Mitochondrial dysfunction and neurological disorders: A narrative review and treatment overview. Life Sci. 2023, 334, 122257. [Google Scholar] [CrossRef]
- Wang, M.; Xuan, T.; Li, H.; An, J.; Hao, T.; Cheng, J. Protective effect of FXN overexpression on ferroptosis in L-Glu-induced SH-SY5Y cells. Acta Histochem. 2024, 126, 152135. [Google Scholar] [CrossRef]
- Rajkumar, M.; Govindaraj, P.; Vimala, K.; Thangaraj, R.; Kannan, S. Chitosan/PLA-loaded Magnesium oxide nanocomposite to attenuate oxidative stress, neuroinflammation and neurotoxicity in rat models of Alzheimer’s disease. Metab. Brain Dis. 2023. [Google Scholar] [CrossRef]
- Lee, D.Y.; Lee, K.M.; Um, J.H.; Kim, Y.Y.; Kim, D.H.; Yun, J. The Natural Alkaloid Palmatine Selectively Induces Mitophagy and Restores Mitochondrial Function in an Alzheimer’s Disease Mouse Model. Int. J. Mol. Sci. 2023, 24, 16542. [Google Scholar] [CrossRef]
- Jia, Y.L.; Wang, W.; Han, N.; Sun, H.L.; Dong, F.M.; Song, Y.X.; Feng, R.F.; Wang, J.H. The mitochondria-targeted small molecule SS31 delays progression of behavioral deficits by attenuating β-amyloid plaque formation and mitochondrial/synaptic deterioration in APP/PS1 mice. Biochem. Biophys. Res. Commun. 2023, 658, 36–43. [Google Scholar] [CrossRef]
- James, A.M.; Sharpley, M.S.; Manas, A.R.; Frerman, F.E.; Hirst, J.; Smith, R.A.; Murphy, M.P. Interaction of the mitochondria-targeted antioxidant MitoQ with phospholipid bilayers and ubiquinone oxidoreductases. J. Biol. Chem. 2007, 282, 14708–14718. [Google Scholar] [CrossRef]
- Ng, L.F.; Gruber, J.; Cheah, I.K.; Goo, C.K.; Cheong, W.F.; Shui, G.; Sit, K.P.; Wenk, M.R.; Halliwell, B. The mitochondria-targeted antioxidant MitoQ extends lifespan and improves healthspan of a transgenic Caenorhabditis elegans model of Alzheimer disease. Free Radic. Biol. Med. 2014, 71, 390–401. [Google Scholar] [CrossRef] [PubMed]
- McManus, M.J.; Murphy, M.P.; Franklin, J.L. The mitochondria-targeted antioxidant MitoQ prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer’s disease. J. Neurosci. 2011, 31, 15703–15715. [Google Scholar] [CrossRef] [PubMed]
- Rossman, M.J.; Santos-Parker, J.R.; Steward, C.A.C.; Bispham, N.Z.; Cuevas, L.M.; Rosenberg, H.L.; Woodward, K.A.; Chonchol, M.; Gioscia-Ryan, R.A.; Murphy, M.P.; et al. Chronic Supplementation With a Mitochondrial Antioxidant (MitoQ) Improves Vascular Function in Healthy Older Adults. Hypertension 2018, 71, 1056–1063. [Google Scholar] [CrossRef]
- Stefanova, N.A.; Ershov, N.I.; Kolosova, N.G. Suppression of Alzheimer’s Disease-Like Pathology Progression by Mitochondria-Targeted Antioxidant SkQ1: A Transcriptome Profiling Study. Oxid. Med. Cell. Longev. 2019, 2019, 3984906. [Google Scholar] [CrossRef] [PubMed]
- Langley, M.; Ghosh, A.; Charli, A.; Sarkar, S.; Ay, M.; Luo, J.; Zielonka, J.; Brenza, T.; Bennett, B.; Jin, H.; et al. Mito-Apocynin Prevents Mitochondrial Dysfunction, Microglial Activation, Oxidative Damage, and Progressive Neurodegeneration in MitoPark Transgenic Mice. Antioxid. Redox Signal. 2017, 27, 1048–1066. [Google Scholar] [CrossRef] [PubMed]
- Mahmood, A.; Bisoyi, P.; Banerjee, R.; Yousuf, M.; Goswami, S.K. Mitoapocynin, a mitochondria targeted derivative of apocynin induces mitochondrial ROS generation and apoptosis in multiple cell types including cardiac myoblasts: A potential constraint to its therapeutic use. Mol. Cell. Biochem. 2021, 476, 2047–2059. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Lyu, X.; Zhang, X.; Zhang, F.; Chen, Y.; Li, G. Astaxanthin attenuates cognitive deficits in Alzheimer’s disease models by reducing oxidative stress via the SIRT1/PGC-1α signaling pathway. Cell Biosci. 2023, 13, 173. [Google Scholar] [CrossRef]
- Oliyaei, N.; Moosavi-Nasab, M.; Tanideh, N.; Iraji, A. Multiple roles of fucoxanthin and astaxanthin against Alzheimer’s disease: Their pharmacological potential and therapeutic insights. Brain Res. Bull. 2023, 193, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Nishida, Y.; Nawaz, A.; Hecht, K.; Tobe, K. Astaxanthin as a Novel Mitochondrial Regulator: A New Aspect of Carotenoids, beyond Antioxidants. Nutrients 2021, 14, 107. [Google Scholar] [CrossRef]
- Manczak, M.; Reddy, P.H. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: Implications for mitochondrial dysfunction and neuronal damage. Hum. Mol. Genet. 2012, 21, 2538–2547. [Google Scholar] [CrossRef]
- Salman, M.; Akram, M.; Shahrukh, M.; Ishrat, T.; Parvez, S. Effects of pramipexole on beta-amyloid1-42 memory deficits and evaluation of oxidative stress and mitochondrial function markers in the hippocampus of Wistar rat. Neurotoxicology 2022, 92, 91–101. [Google Scholar] [CrossRef]
- Baek, S.H.; Park, S.J.; Jeong, J.I.; Kim, S.H.; Han, J.; Kyung, J.W.; Baik, S.H.; Choi, Y.; Choi, B.Y.; Park, J.S.; et al. Inhibition of Drp1 Ameliorates Synaptic Depression, Aβ Deposition, and Cognitive Impairment in an Alzheimer’s Disease Model. J. Neurosci. 2017, 37, 5099–5110. [Google Scholar] [CrossRef] [PubMed]
- Cassidy-Stone, A.; Chipuk, J.E.; Ingerman, E.; Song, C.; Yoo, C.; Kuwana, T.; Kurth, M.J.; Shaw, J.T.; Hinshaw, J.E.; Green, D.R.; et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev. Cell 2008, 14, 193–204. [Google Scholar] [CrossRef]
- Sbai, O.; Bazzani, V.; Tapaswi, S.; McHale, J.; Vascotto, C.; Perrone, L. Is Drp1 a link between mitochondrial dysfunction and inflammation in Alzheimer’s disease? Front. Mol. Neurosci. 2023, 16, 1166879. [Google Scholar] [CrossRef] [PubMed]
- Bordt, E.A.; Clerc, P.; Roelofs, B.A.; Saladino, A.J.; Tretter, L.; Adam-Vizi, V.; Cherok, E.; Khalil, A.; Yadava, N.; Ge, S.X.; et al. The Putative Drp1 Inhibitor mdivi-1 Is a Reversible Mitochondrial Complex I Inhibitor that Modulates Reactive Oxygen Species. Dev. Cell 2017, 40, 583–594.e6. [Google Scholar] [CrossRef]
- Bhatti, J.S.; Kaur, S.; Mishra, J.; Dibbanti, H.; Singh, A.; Reddy, A.P.; Bhatti, G.K.; Reddy, P.H. Targeting dynamin-related protein-1 as a potential therapeutic approach for mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta (BBA)–Mol. Basis Dis. 2023, 1869, 166798. [Google Scholar] [CrossRef]
- Liu, X.; Song, L.; Yu, J.; Huang, F.; Li, Y.; Ma, C. Mdivi-1: A promising drug and its underlying mechanisms in the treatment of neurodegenerative diseases. Histol. Histopathol. 2022, 37, 505–512. [Google Scholar] [CrossRef]
- Rosdah, A.A.; Abbott, B.M.; Langendorf, C.G.; Deng, Y.; Truong, J.Q.; Waddell, H.M.M.; Ling, N.X.Y.; Smiles, W.J.; Delbridge, L.M.D.; Liu, G.S.; et al. A novel small molecule inhibitor of human Drp1. Sci. Rep. 2022, 12, 21531. [Google Scholar] [CrossRef] [PubMed]
- Van Bulck, M.; Sierra-Magro, A.; Alarcon-Gil, J.; Perez-Castillo, A.; Morales-Garcia, J.A. Novel approaches for the treatment of Alzheimer’s and Parkinson’s disease. Int. J. Mol. Sci. 2019, 20, 719. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, M.; Bose, C.; Reddy, P.H. Protective effects of a small molecule inhibitor, DDQ against amyloid beta in Alzheimer’s disease. Mitochondrion 2021, 59, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Kuruva, C.S.; Manczak, M.; Yin, X.; Ogunmokun, G.; Reddy, A.P.; Reddy, P.H. Aqua-soluble DDQ reduces the levels of Drp1 and Aβ and inhibits abnormal interactions between Aβ and Drp1 and protects Alzheimer’s disease neurons from Aβ- and Drp1-induced mitochondrial and synaptic toxicities. Hum. Mol. Genet. 2017, 26, 3375–3395. [Google Scholar] [CrossRef] [PubMed]
- Mi, Y.; Qi, G.; Brinton, R.D.; Yin, F. Mitochondria-Targeted Therapeutics for Alzheimer’s Disease: The Good, the Bad, the Potential. Antioxid. Redox. Signal. 2021, 34, 611–630. [Google Scholar] [CrossRef]
- Misrani, A.; Tabassum, S.; Huo, Q.; Tabassum, S.; Jiang, J.; Ahmed, A.; Chen, X.; Zhou, J.; Zhang, J.; Liu, S.; et al. Mitochondrial Deficits With Neural and Social Damage in Early Stage Alzheimer’s Disease Model Mice. Front. Aging Neurosci. 2021, 13, 748388. [Google Scholar] [CrossRef]
- Wu, W.; Yuan, S.; Tang, Y.; Meng, X.; Peng, M.; Hu, Z.; Liu, W. Effect of Exercise and Oral Niacinamide Mononucleotide on Improving Mitochondrial Autophagy in Alzheimer’s Disease. Nutrients 2023, 15, 2851. [Google Scholar] [CrossRef]
- Hosseini, L.; Mahmoudi, J.; Pashazadeh, F.; Salehi-Pourmehr, H.; Sadigh-Eteghad, S. Protective Effects of Nicotinamide Adenine Dinucleotide and Related Precursors in Alzheimer’s Disease: A Systematic Review of Preclinical Studies. J. Mol. Neurosci. 2021, 71, 1425–1435. [Google Scholar] [CrossRef]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Primers 2016, 2, 16080. [Google Scholar] [CrossRef]
- Jurkute, N.; Yu-Wai-Man, P. Leber hereditary optic neuropathy: Bridging the translational gap. Curr. Opin. Ophthalmol. 2017, 28, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Arnaldi, D.; Donniaquio, A.; Mattioli, P.; Massa, F.; Grazzini, M.; Meli, R.; Filippi, L.; Grisanti, S.; Famà, F.; Terzaghi, M.; et al. Epilepsy in Neurodegenerative Dementias: A Clinical, Epidemiological, and EEG Study. J. Alzheimers Dis. 2020, 74, 865–874. [Google Scholar] [CrossRef] [PubMed]
- Bature, F.; Guinn, B.A.; Pang, D.; Pappas, Y. Signs and symptoms preceding the diagnosis of Alzheimer’s disease: A systematic scoping review of literature from 1937 to 2016. BMJ Open 2017, 7, e015746. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.; Xu, J.; Chen, Y.; Ip, N.Y. Retinal Dysfunction in Alzheimer’s Disease and Implications for Biomarkers. Biomolecules 2021, 11, 1215. [Google Scholar] [CrossRef] [PubMed]
- Gropman, A.L. Neuroimaging in mitochondrial disorders. Neurotherapeutics 2013, 10, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, H.I.L.; Hopkins, D.A.; Mayrhofer, H.C.; Bruner, E.; van Leeuwen, F.W.; Raaijmakers, W.; Schmahmann, J.D. The cerebellum in Alzheimer’s disease: Evaluating its role in cognitive decline. Brain 2018, 141, 37–47. [Google Scholar] [CrossRef]
- Nasrabady, S.E.; Rizvi, B.; Goldman, J.E.; Brickman, A.M. White matter changes in Alzheimer’s disease: A focus on myelin and oligodendrocytes. Acta Neuropathol. Commun. 2018, 6, 22. [Google Scholar] [CrossRef] [PubMed]
- Eckert, A.; Schmitt, K.; Götz, J. Mitochondrial dysfunction–the beginning of the end in Alzheimer’s disease? Separate and synergistic modes of tau and amyloid-β toxicity. Alzheimers Res. Ther. 2011, 3, 15. [Google Scholar] [CrossRef]
- Sandberg, A.A.; Manning, E.; Wilkins, H.M.; Mazzarino, R.; Minckley, T.; Swerdlow, R.H.; Patterson, D.; Qin, Y.; Linseman, D.A. Mitochondrial Targeting of Amyloid-β Protein Precursor Intracellular Domain Induces Hippocampal Cell Death via a Mechanism Distinct from Amyloid-β. J. Alzheimers Dis. 2022, 86, 1727–1744. [Google Scholar] [CrossRef]
- Volloch, V.; Rits-Volloch, S. Next Generation Therapeutic Strategy for Treatment and Prevention of Alzheimer’s Disease and Aging-Associated Cognitive Decline: Transient, Once-in-a-Lifetime-Only Depletion of Intraneuronal Aβ (iAβ) by Its Targeted Degradation via Augmentation of Intra-iAβ-Cleaving Activities of BACE1 and/or BACE2. Int. J. Mol. Sci. 2023, 24, 17586. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Treatment | Effect on mitochondria and neuron | References |
|---|---|---|
| MitoQ | Prevention of ↑ ROS production, ↓ Aβ accumulation, ↓ astrogliosis, minimize synaptic loss | [184,185,186] |
| SkQ1 | ↓ Aβ accumulation and tau hyperphosphorylation in hippocampus in rat AD model | [188] |
| Mito-apocynin | NADPH oxidase inhibitor that acts as an anti-inflammatory and antioxidant. | [189,190] |
| Astaxanthin | Carotenoid and dietary supplement with neuroprotective and antioxidant effects. Maintains mitochondrial membrane potential. | [191,192,193] |
| Mdivi1 | ↓ in the fission proteins Drp1 and Fis1, ↑ in the fusion proteins Mfn1 and Mfn2, ↓ excessive fragmentation of mitochondria, inhibition of Aβ-DRP1 complex formation | [197,198,199,200,201] |
| Nicotinamide compounds: nicotinamide mononucleoside, nicotinamide mononucleotide, nicotinamide riboside | ↓ DNA damage, ↓ neuroinflammation, and ↓apoptosis of hippocampal neurons | [209] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Reiss, A.B.; Gulkarov, S.; Jacob, B.; Srivastava, A.; Pinkhasov, A.; Gomolin, I.H.; Stecker, M.M.; Wisniewski, T.; De Leon, J. Mitochondria in Alzheimer’s Disease Pathogenesis. Life 2024, 14, 196. https://doi.org/10.3390/life14020196
Reiss AB, Gulkarov S, Jacob B, Srivastava A, Pinkhasov A, Gomolin IH, Stecker MM, Wisniewski T, De Leon J. Mitochondria in Alzheimer’s Disease Pathogenesis. Life. 2024; 14(2):196. https://doi.org/10.3390/life14020196
Chicago/Turabian StyleReiss, Allison B., Shelly Gulkarov, Benna Jacob, Ankita Srivastava, Aaron Pinkhasov, Irving H. Gomolin, Mark M. Stecker, Thomas Wisniewski, and Joshua De Leon. 2024. "Mitochondria in Alzheimer’s Disease Pathogenesis" Life 14, no. 2: 196. https://doi.org/10.3390/life14020196
APA StyleReiss, A. B., Gulkarov, S., Jacob, B., Srivastava, A., Pinkhasov, A., Gomolin, I. H., Stecker, M. M., Wisniewski, T., & De Leon, J. (2024). Mitochondria in Alzheimer’s Disease Pathogenesis. Life, 14(2), 196. https://doi.org/10.3390/life14020196