
Influence of Alcohol on Intracerebral Hemorrhage: From Oxidative Stress to Glial Cell Activation
 ,
,
Abstract
:1. Introduction
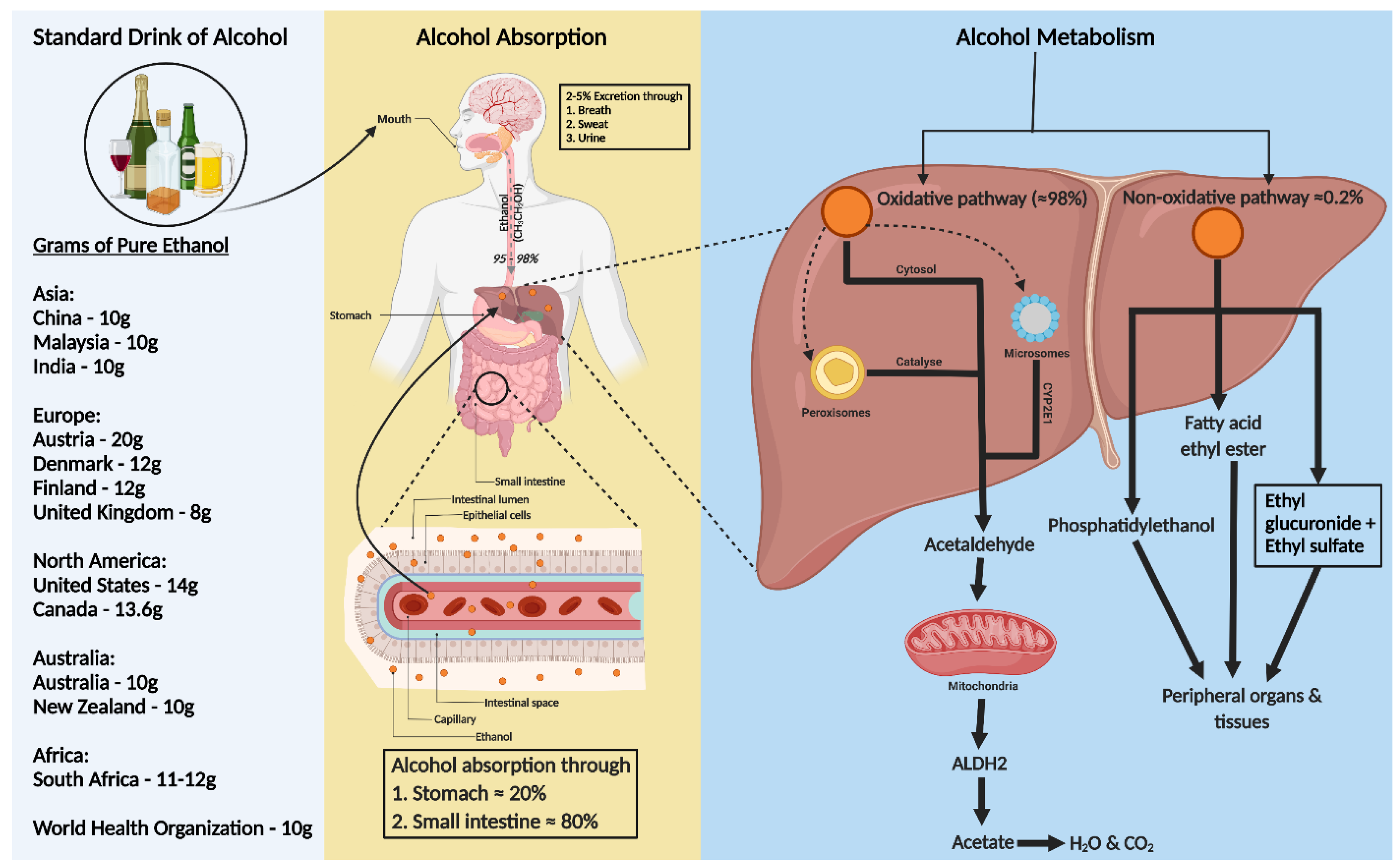
2. Standard Drink of Alcohol
3. Alcohol Absorption and Metabolism
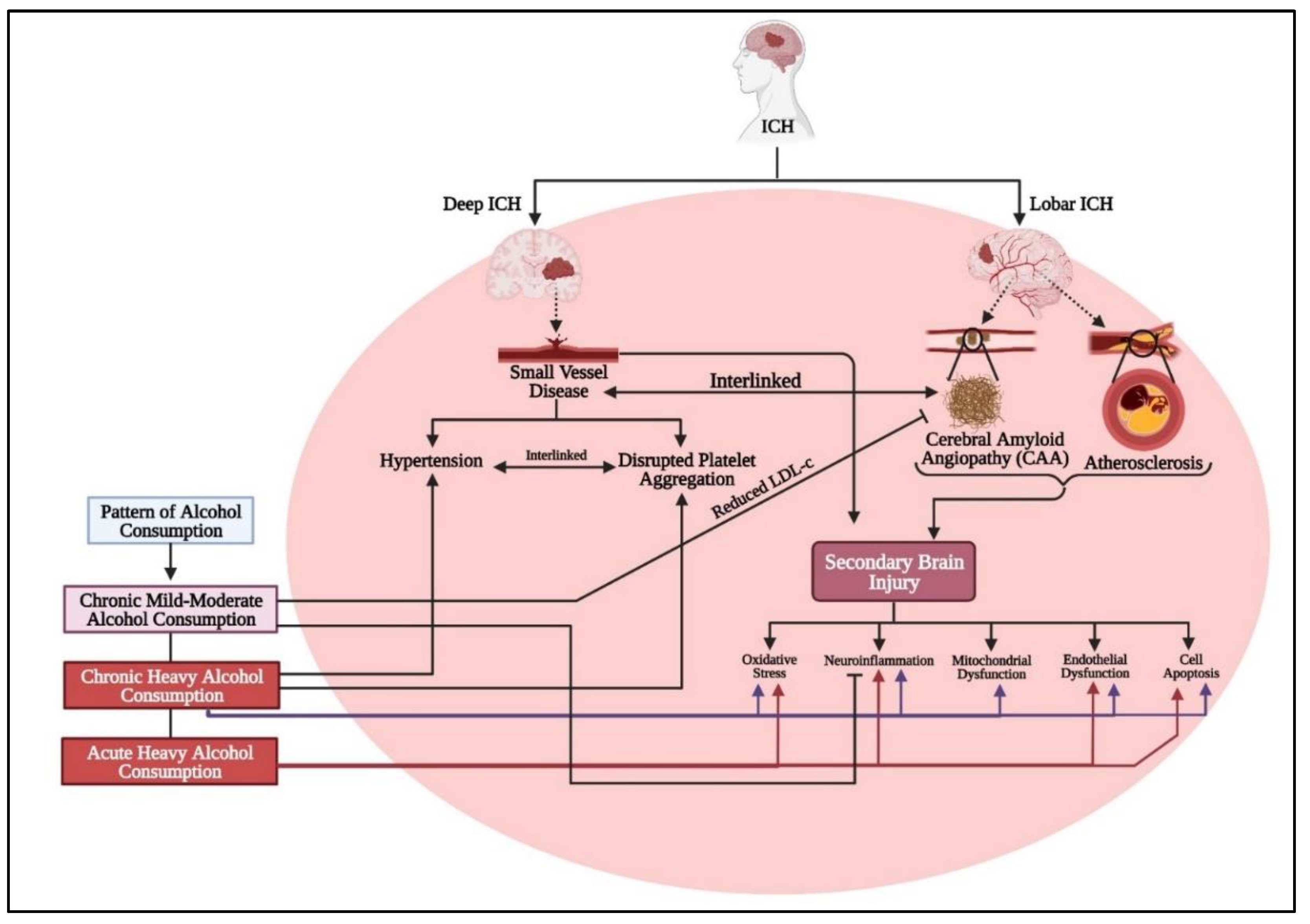
4. Alcohol Induced Hypertension
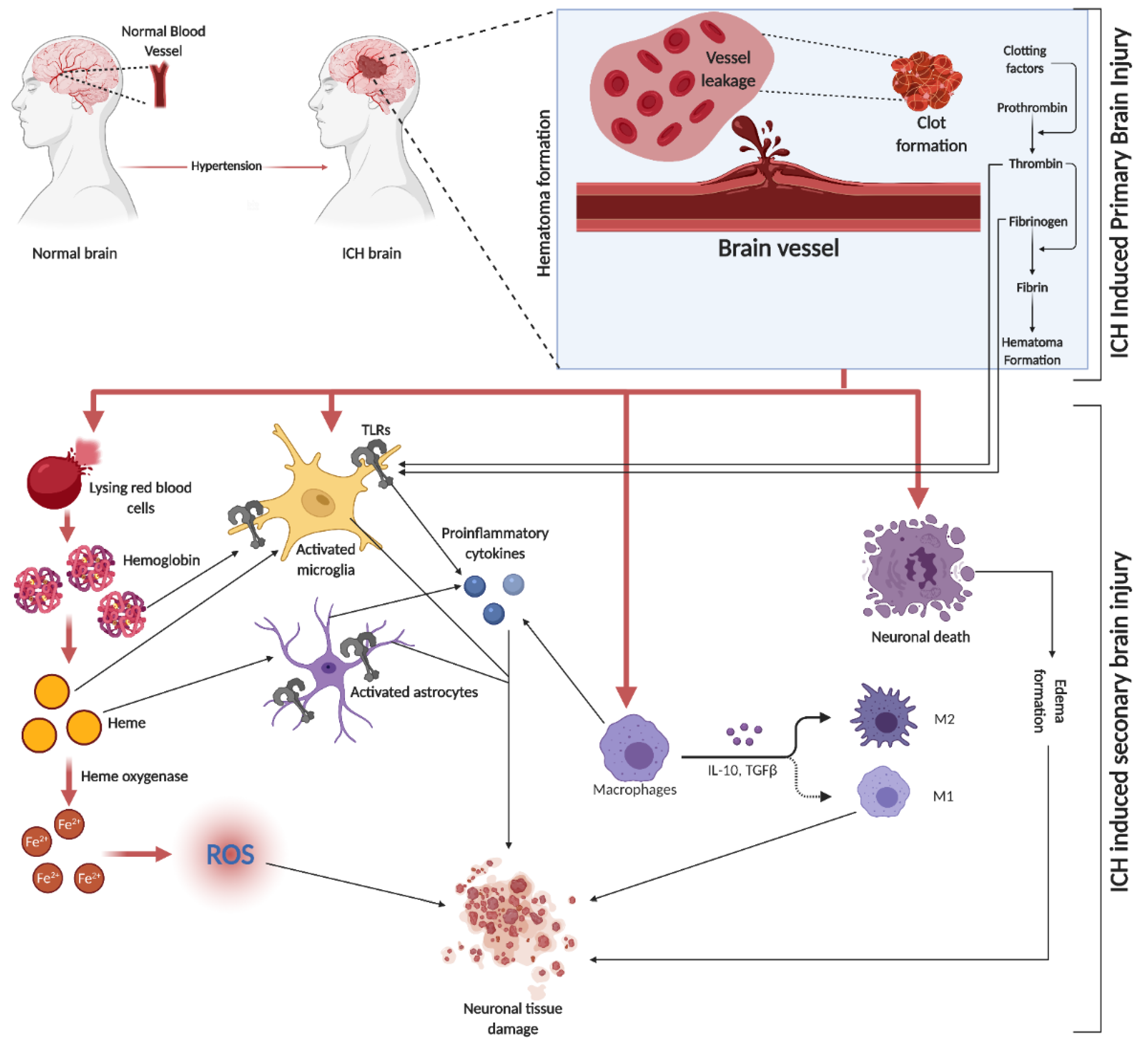
5. ICH
6. SBI in ICH
7. Role of Alcohol on SBI-Induced Toxic Products
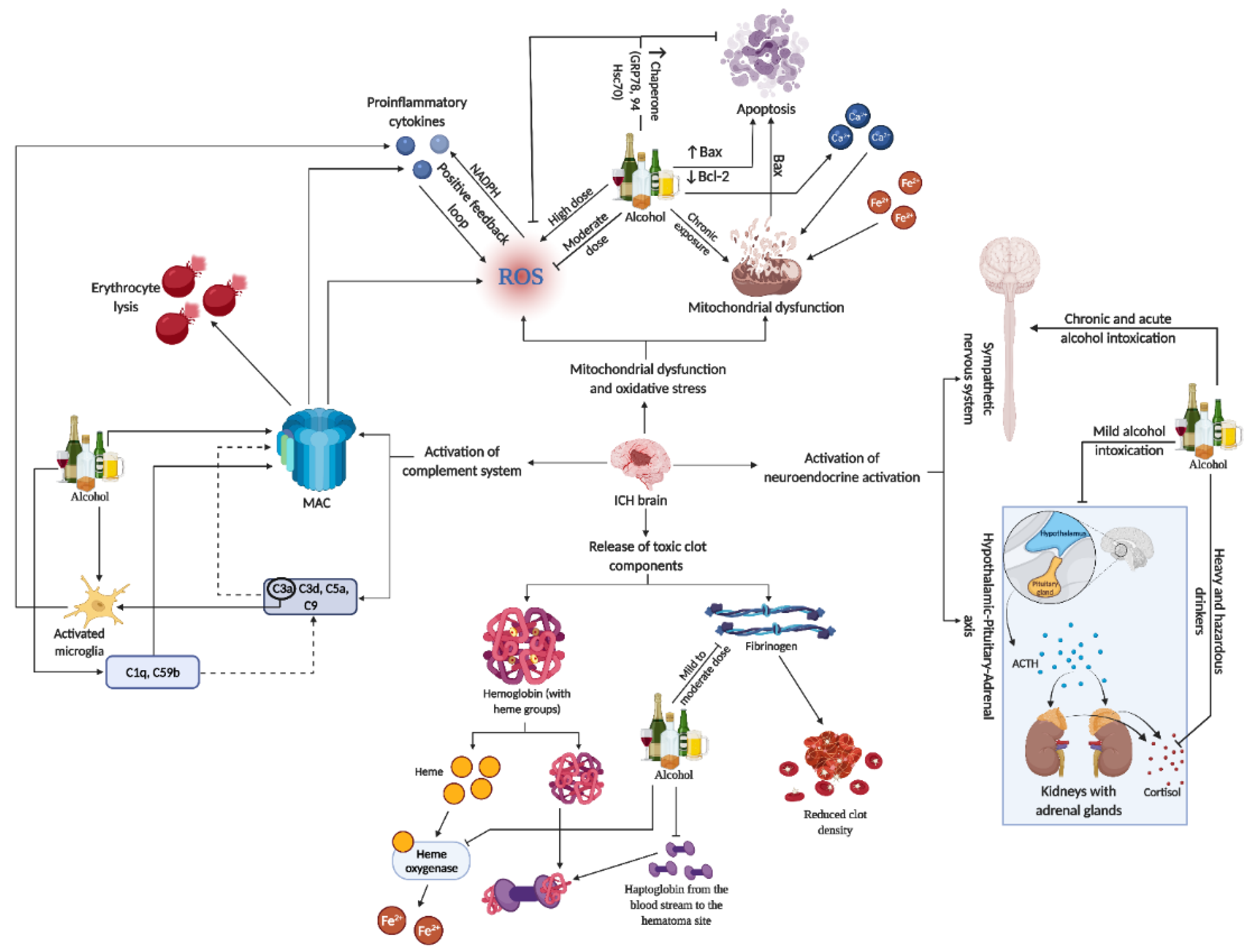
8. Effect of Alcohol on Mitochondrial Dysfunction and Oxidative Stress in Brain Injury
9. Effect of Alcohol on Neuroendocrine Axis Activation
10. Alcohol and Activation of Complement System during SBI
11. Alcohol and Neuroinflammatory Mechanism Activated in SBI
11.1. Influence of Alcohol on Microglial Activation and Polarization
11.2. Influence of Alcohol on Astrocyte Activation
11.3. Relationship between and Neurotrophic Factors (NTFs) and Influence of Alcohol on NTFs
11.4. Beneficial Effects of Antioxidant Polyphenols
11.5. Epidemiological Evidence Linking Alcohol and ICH
12. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rosso, A.M. Beer and wine in antiquity: Beneficial remedy or punishment imposed by the Gods? Acta Med. Hist. Adriat. 2012, 10, 237–262. [Google Scholar] [PubMed]
- McGinnis, J.M.; Foege, W.H. Actual causes of death in the United States. JAMA 1993, 270, 2207–2212. [Google Scholar] [CrossRef] [PubMed]
- Li, T.K.; Hewitt, B.G.; Grant, B.F. Alcohol use disorders and mood disorders: A National Institute on Alcohol Abuse and Alcoholism perspective. Biol. Psychiatry 2004, 56, 718–720. [Google Scholar] [CrossRef] [PubMed]
- Room, R.; Babor, T.; Rehm, J. Alcohol and public health. Lancet 2005, 365, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.B.; Wang, P.K.; Pang, C.Y.; Hu, W.F.; Tsai, A.P.; Oblak, A.L.; Liew, H.K. Moderate Ethanol Pre-treatment Mitigates ICH-Induced Injury via ER Stress Modulation in Rats. Front. Mol. Neurosci. 2021, 14, 682775. [Google Scholar] [CrossRef]
- Beilin, L.J.; Puddey, I.B. Alcohol and hypertension: An update. Hypertension 2006, 47, 1035–1038. [Google Scholar] [CrossRef]
- Klatsky, A.L. Alcohol-associated hypertension: When one drinks makes a difference. Hypertension 2004, 44, 805–806. [Google Scholar] [CrossRef]
- Sesso, H.D.; Cook, N.R.; Buring, J.E.; Manson, J.E.; Gaziano, J.M. Alcohol consumption and the risk of hypertension in women and men. Hypertension 2008, 51, 1080–1087. [Google Scholar] [CrossRef]
- Pletsch, G.R.; Boehme, A.K.; Albright, K.C.; Burns, C.; Beasley, T.M.; Martin-Schild, S. Low-density lipoprotein and intracerebral hematoma expansion in daily alcohol users. Cerebrovasc. Dis. Extra 2014, 4, 1–8. [Google Scholar] [CrossRef]
- Rehm, J.; Rehn, N.; Room, R.; Monteiro, M.; Gmel, G.; Jernigan, D.; Frick, U. The global distribution of average volume of alcohol consumption and patterns of drinking. Eur. Addict. Res. 2003, 9, 147–156. [Google Scholar] [CrossRef]
- Rehm, J.; Room, R.; Graham, K.; Monteiro, M.; Gmel, G.; Sempos, C.T. The relationship of average volume of alcohol consumption and patterns of drinking to burden of disease: An overview. Addiction 2003, 98, 1209–1228. [Google Scholar] [CrossRef] [PubMed]
- Rehm, J.; Sempos, C.T.; Trevisan, M. Alcohol and cardiovascular disease—More than one paradox to consider. Average volume of alcohol consumption, patterns of drinking and risk of coronary heart disease—A review. J. Cardiovasc. Risk 2003, 10, 15–20. [Google Scholar] [CrossRef]
- Collins, M.A.; Neafsey, E.J.; Mukamal, K.J.; Gray, M.O.; Parks, D.A.; Das, D.K.; Korthuis, R.J. Alcohol in moderation, cardioprotection, and neuroprotection: Epidemiological considerations and mechanistic studies. Alcohol. Clin. Exp. Res. 2009, 33, 206–219. [Google Scholar] [CrossRef] [PubMed]
- Mukamal, K.J.; Chung, H.; Jenny, N.S.; Kuller, L.H.; Longstreth, W.T., Jr.; Mittleman, M.A.; Burke, G.L.; Cushman, M.; Beauchamp, N.J., Jr.; Siscovick, D.S. Alcohol use and risk of ischemic stroke among older adults: The cardiovascular health study. Stroke 2005, 36, 1830–1834. [Google Scholar] [CrossRef]
- Huang, F.P.; Xi, G.; Keep, R.F.; Hua, Y.; Nemoianu, A.; Hoff, J.T. Brain edema after experimental intracerebral hemorrhage: Role of hemoglobin degradation products. J. Neurosurg. 2002, 96, 287–293. [Google Scholar] [CrossRef]
- Monforte, R.; Estruch, R.; Graus, F.; Nicolas, J.M.; Urbano-Marquez, A. High ethanol consumption as risk factor for intracerebral hemorrhage in young and middle-aged people. Stroke 1990, 21, 1529–1532. [Google Scholar] [CrossRef]
- Taylor, J.R.; Combs-Orme, T. Alcohol and strokes in young adults. Am. J. Psychiatry 1985, 142, 116–118. [Google Scholar]
- Kalinowski, A.; Humphreys, K. Governmental standard drink definitions and low-risk alcohol consumption guidelines in 37 countries. Addiction 2016, 111, 1293–1298. [Google Scholar] [CrossRef] [PubMed]
- Paton, A. Alcohol in the body. BMJ 2005, 330, 85–87. [Google Scholar] [CrossRef]
- Jones, A.W.; Jonsson, K.A.; Kechagias, S. Effect of high-fat, high-protein, and high-carbohydrate meals on the pharmacokinetics of a small dose of ethanol. Br. J. Clin. Pharmacol. 1997, 44, 521–526. [Google Scholar] [CrossRef]
- Holford, N.H. Clinical pharmacokinetics of ethanol. Clin. Pharmacokinet. 1987, 13, 273–292. [Google Scholar] [CrossRef] [PubMed]
- Zakhari, S. Overview: How is alcohol metabolized by the body? Alcohol Res. Health 2006, 29, 245–254. [Google Scholar] [PubMed]
- Zakhari, S. Special remarks from the National Institute on Alcohol Abuse and Alcoholism. J. Gastroenterol. Hepatol. 2006, 21 (Suppl. 3), S2. [Google Scholar] [CrossRef]
- Edenberg, H.J. The genetics of alcohol metabolism: Role of alcohol dehydrogenase and aldehyde dehydrogenase variants. Alcohol Res. Health 2007, 30, 5–13. [Google Scholar]
- Lieber, C.S. Hepatic, metabolic and toxic effects of ethanol: 1991 update. Alcohol. Clin. Exp. Res. 1991, 15, 573–592. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, E.; Terada, M.; Misawa, S. Cytochrome P450 2E1: Its clinical and toxicological role. J. Clin. Pharm. Ther. 2000, 25, 165–175. [Google Scholar] [CrossRef]
- Tsutsumi, M.; Lasker, J.M.; Takahashi, T.; Lieber, C.S. In vivo induction of hepatic P4502E1 by ethanol: Role of increased enzyme synthesis. Arch. Biochem. Biophys. 1993, 304, 209–218. [Google Scholar] [CrossRef]
- Palmer, R.B. A review of the use of ethyl glucuronide as a marker for ethanol consumption in forensic and clinical medicine. Semin. Diagn. Pathol. 2009, 26, 18–27. [Google Scholar] [CrossRef]
- Staufer, K.; Yegles, M. Biomarkers for detection of alcohol consumption in liver transplantation. World J. Gastroenterol. 2016, 22, 3725–3734. [Google Scholar] [CrossRef]
- Gyntelberg, F.; Meyer, J. Relationship between blood pressure and physical fitness, smoking and alcohol consumption in Copenhagen males aged 40–59. Acta Med. Scand. 1974, 195, 375–380. [Google Scholar] [CrossRef]
- Bos, S.; Grobbee, D.E.; Boer, J.M.; Verschuren, W.M.; Beulens, J.W. Alcohol consumption and risk of cardiovascular disease among hypertensive women. Eur. J. Cardiovasc. Prev. Rehabil. 2010, 17, 119–126. [Google Scholar] [CrossRef]
- Husain, K.; Ansari, R.A.; Ferder, L. Alcohol-induced hypertension: Mechanism and prevention. World J. Cardiol. 2014, 6, 245–252. [Google Scholar] [CrossRef]
- McFadden, C.B.; Brensinger, C.M.; Berlin, J.A.; Townsend, R.R. Systematic review of the effect of daily alcohol intake on blood pressure. Am. J. Hypertens. 2005, 18 Pt 1, 276–286. [Google Scholar] [CrossRef]
- Rupp, H.; Brilla, C.G.; Maisch, B. [Hypertension and alcohol: Central and peripheral mechanisms]. Herz 1996, 21, 258–264. [Google Scholar]
- Jing, L.; Li, W.M.; Zhou, L.J.; Li, S.; Kou, J.J.; Song, J. Expression of renin-angiotensin system and peroxisome proliferator-activated receptors in alcoholic cardiomyopathy. Alcohol. Clin. Exp. Res. 2008, 32, 1999–2007. [Google Scholar] [CrossRef]
- Potter, J.F.; Watson, R.D.; Skan, W.; Beevers, D.G. The pressor and metabolic effects of alcohol in normotensive subjects. Hypertension 1986, 8, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Altura, B.M.; Altura, B.T. Microvascular and vascular smooth muscle actions of ethanol, acetaldehyde, and acetate. Fed. Proc. 1982, 41, 2447–2451. [Google Scholar] [PubMed]
- Slomiany, B.L.; Piotrowski, J.; Slomiany, A. Alterations in buccal mucosal endothelin-1 and nitric oxide synthase with chronic alcohol ingestion. Biochem. Mol. Biol. Int. 1998, 45, 681–688. [Google Scholar] [CrossRef] [PubMed]
- Puddey, I.B.; Zilkens, R.R.; Croft, K.D.; Beilin, L.J. Alcohol and endothelial function: A brief review. Clin. Exp. Pharmacol. Physiol. 2001, 28, 1020–1024. [Google Scholar] [CrossRef]
- MacMahon, S. Alcohol consumption and hypertension. Hypertension 1987, 9, 111–121. [Google Scholar] [CrossRef]
- Roerecke, M.; Kaczorowski, J.; Tobe, S.W.; Gmel, G.; Hasan, O.S.M.; Rehm, J. The effect of a reduction in alcohol consumption on blood pressure: A systematic review and meta-analysis. Lancet Public Health 2017, 2, e108–e120. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthi, R.V.; Feigin, V.L.; Forouzanfar, M.H.; Mensah, G.A.; Connor, M.; Bennett, D.A.; Moran, A.E.; Sacco, R.L.; Anderson, L.M.; Truelsen, T.; et al. Global and regional burden of first-ever ischaemic and haemorrhagic stroke during 1990–2010: Findings from the Global Burden of Disease Study 2010. Lancet Glob. Health 2013, 1, e259–e281. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, A.I.; Tuhrim, S.; Broderick, J.P.; Batjer, H.H.; Hondo, H.; Hanley, D.F. Spontaneous intracerebral hemorrhage. N. Engl. J. Med. 2001, 344, 1450–1460. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, A.I.; Mendelow, A.D.; Hanley, D.F. Intracerebral haemorrhage. Lancet 2009, 373, 1632–1644. [Google Scholar] [CrossRef] [PubMed]
- Xi, G.; Keep, R.F.; Hoff, J.T. Mechanisms of brain injury after intracerebral haemorrhage. Lancet Neurol. 2006, 5, 53–63. [Google Scholar] [CrossRef]
- Takebayashi, S.; Kaneko, M. Electron microscopic studies of ruptured arteries in hypertensive intracerebral hemorrhage. Stroke 1983, 14, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wang, Z.; Yu, J.; Yang, X.; He, F.; Liu, Z.; Che, F.; Chen, X.; Ren, H.; Hong, M.; et al. Role and mechanisms of cytokines in the secondary brain injury after intracerebral hemorrhage. Prog. Neurobiol. 2019, 178, 101610. [Google Scholar] [CrossRef]
- Appelboom, G.; Bruce, S.S.; Hickman, Z.L.; Zacharia, B.E.; Carpenter, A.M.; Vaughan, K.A.; Duren, A.; Hwang, R.Y.; Piazza, M.; Lee, K.; et al. Volume-dependent effect of perihaematomal oedema on outcome for spontaneous intracerebral haemorrhages. J. Neurol. Neurosurg. Psychiatry 2013, 84, 488–493. [Google Scholar] [CrossRef]
- Dowlatshahi, D.; Demchuk, A.M.; Flaherty, M.L.; Ali, M.; Lyden, P.L.; Smith, E.E.; Collaboration, V. Defining hematoma expansion in intracerebral hemorrhage: Relationship with patient outcomes. Neurology 2011, 76, 1238–1244. [Google Scholar] [CrossRef]
- LoPresti, M.A.; Bruce, S.S.; Camacho, E.; Kunchala, S.; Dubois, B.G.; Bruce, E.; Appelboom, G.; Connolly, E.S., Jr. Hematoma volume as the major determinant of outcomes after intracerebral hemorrhage. J. Neurol. Sci. 2014, 345, 3–7. [Google Scholar] [CrossRef]
- Mayer, S.A.; Lignelli, A.; Fink, M.E.; Kessler, D.B.; Thomas, C.E.; Swarup, R.; Van Heertum, R.L. Perilesional blood flow and edema formation in acute intracerebral hemorrhage: A SPECT study. Stroke 1998, 29, 1791–1798. [Google Scholar] [CrossRef]
- Li, Q.; Han, X.; Lan, X.; Gao, Y.; Wan, J.; Durham, F.; Cheng, T.; Yang, J.; Wang, Z.; Jiang, C.; et al. Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight 2017, 2, e90777. [Google Scholar] [CrossRef]
- Keep, R.F.; Hua, Y.; Xi, G. Intracerebral haemorrhage: Mechanisms of injury and therapeutic targets. Lancet Neurol. 2012, 11, 720–731. [Google Scholar] [CrossRef]
- Madangarli, N.; Bonsack, F.; Dasari, R.; Sukumari-Ramesh, S. Intracerebral Hemorrhage: Blood Components and Neurotoxicity. Brain Sci. 2019, 9, 316. [Google Scholar] [CrossRef] [PubMed]
- Senn, R.; Elkind, M.S.; Montaner, J.; Christ-Crain, M.; Katan, M. Potential role of blood biomarkers in the management of nontraumatic intracerebral hemorrhage. Cerebrovasc. Dis. 2014, 38, 395–409. [Google Scholar] [CrossRef] [PubMed]
- Gurley, C.; Nichols, J.; Liu, S.; Phulwani, N.K.; Esen, N.; Kielian, T. Microglia and Astrocyte Activation by Toll-Like Receptor Ligands: Modulation by PPAR-gamma Agonists. PPAR Res. 2008, 2008, 453120. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Brown, W.M.; Moomaw, C.J.; Langefeld, C.D.; Osborne, J.; Worrall, B.B.; Woo, D.; Koch, S.; Investigators, E. Alcohol use and risk of intracerebral hemorrhage. Neurology 2017, 88, 2043–2051. [Google Scholar] [CrossRef]
- Aronowski, J.; Zhao, X. Molecular pathophysiology of cerebral hemorrhage: Secondary brain injury. Stroke 2011, 42, 1781–1786. [Google Scholar] [CrossRef] [PubMed]
- Bai, Q.; Liu, J.; Wang, G. Ferroptosis, a Regulated Neuronal Cell Death Type After Intracerebral Hemorrhage. Front. Cell Neurosci. 2020, 14, 591874. [Google Scholar] [CrossRef]
- Rautenbach, P.H.; Nienaber-Rousseau, C.; Pieters, M. The association of alcohol with circulating total fibrinogen and plasma clot density is mediated by fibrinogen and FXIII genotypes. Thromb. J. 2020, 18, 35. [Google Scholar] [CrossRef]
- el-Sayed, M.S.; Eastland, P.; Lin, X.; Rattu, A.M. The effect of moderate alcohol ingestion on blood coagulation and fibrinolysis at rest and in response to exercise. J. Sports Sci. 1999, 17, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Gerjevic, L.N.; Lu, S.; Chaky, J.P.; Harrison-Findik, D.D. Regulation of heme oxygenase expression by alcohol, hypoxia and oxidative stress. World J. Biol. Chem. 2011, 2, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Bertero, E.; Maack, C. Calcium Signaling and Reactive Oxygen Species in Mitochondria. Circ. Res. 2018, 122, 1460–1478. [Google Scholar] [CrossRef] [PubMed]
- Zille, M.; Karuppagounder, S.S.; Chen, Y.; Gough, P.J.; Bertin, J.; Finger, J.; Milner, T.A.; Jonas, E.A.; Ratan, R.R. Neuronal Death After Hemorrhagic Stroke In Vitro and In Vivo Shares Features of Ferroptosis and Necroptosis. Stroke 2017, 48, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Sinha, K.; Das, J.; Pal, P.B.; Sil, P.C. Oxidative stress: The mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch. Toxicol. 2013, 87, 1157–1180. [Google Scholar] [CrossRef]
- Nakamura, T.; Keep, R.F.; Hua, Y.; Nagao, S.; Hoff, J.T.; Xi, G. Iron-induced oxidative brain injury after experimental intracerebral hemorrhage. Acta Neurochir. Suppl. 2006, 96, 194–198. [Google Scholar]
- Nakamura, T.; Keep, R.F.; Hua, Y.; Hoff, J.T.; Xi, G. Oxidative DNA injury after experimental intracerebral hemorrhage. Brain Res. 2005, 1039, 30–36. [Google Scholar] [CrossRef]
- Mates, J.M.; Segura, J.A.; Alonso, F.J.; Marquez, J. Oxidative stress in apoptosis and cancer: An update. Arch. Toxicol. 2012, 86, 1649–1665. [Google Scholar] [CrossRef]
- Zia, M.T.; Csiszar, A.; Labinskyy, N.; Hu, F.; Vinukonda, G.; LaGamma, E.F.; Ungvari, Z.; Ballabh, P. Oxidative-nitrosative stress in a rabbit pup model of germinal matrix hemorrhage: Role of NAD(P)H oxidase. Stroke 2009, 40, 2191–2198. [Google Scholar] [CrossRef]
- Qi, J.; Kim, J.W.; Zhou, Z.; Lim, C.W.; Kim, B. Ferroptosis Affects the Progression of Nonalcoholic Steatohepatitis via the Modulation of Lipid Peroxidation-Mediated Cell Death in Mice. Am. J. Pathol. 2020, 190, 68–81. [Google Scholar] [CrossRef]
- Duan, X.; Wen, Z.; Shen, H.; Shen, M.; Chen, G. Intracerebral Hemorrhage, Oxidative Stress, and Antioxidant Therapy. Oxid. Med. Cell Longev. 2016, 2016, 1203285. [Google Scholar] [CrossRef]
- Montoliu, C.; Valles, S.; Renau-Piqueras, J.; Guerri, C. Ethanol-induced oxygen radical formation and lipid peroxidation in rat brain: Effect of chronic alcohol consumption. J. Neurochem. 1994, 63, 1855–1862. [Google Scholar] [CrossRef]
- Chen, W.; Guo, C.; Feng, H.; Chen, Y. Mitochondria: Novel Mechanisms and Therapeutic Targets for Secondary Brain Injury After Intracerebral Hemorrhage. Front. Aging Neurosci. 2020, 12, 615451. [Google Scholar] [CrossRef]
- Mariani, E.; Polidori, M.C.; Cherubini, A.; Mecocci, P. Oxidative stress in brain aging, neurodegenerative and vascular diseases: An overview. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2005, 827, 65–75. [Google Scholar] [CrossRef]
- Sidlauskaite, E.; Gibson, J.W.; Megson, I.L.; Whitfield, P.D.; Tovmasyan, A.; Batinic-Haberle, I.; Murphy, M.P.; Moult, P.R.; Cobley, J.N. Mitochondrial ROS cause motor deficits induced by synaptic inactivity: Implications for synapse pruning. Redox Biol. 2018, 16, 344–351. [Google Scholar] [CrossRef]
- Zheng, J.; Shi, L.; Liang, F.; Xu, W.; Li, T.; Gao, L.; Sun, Z.; Yu, J.; Zhang, J. Sirt3 Ameliorates Oxidative Stress and Mitochondrial Dysfunction After Intracerebral Hemorrhage in Diabetic Rats. Front. Neurosci. 2018, 12, 414. [Google Scholar] [CrossRef]
- Liew, H.K.; Cheng, H.Y.; Huang, L.C.; Li, K.W.; Peng, H.F.; Yang, H.I.; Lin, P.B.; Kuo, J.S.; Pang, C.Y. Acute Alcohol Intoxication Aggravates Brain Injury Caused by Intracerebral Hemorrhage in Rats. J. Stroke Cerebrovasc. Dis. 2016, 25, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Hoek, J.B.; Cahill, A.; Pastorino, J.G. Alcohol and mitochondria: A dysfunctional relationship. Gastroenterology 2002, 122, 2049–2063. [Google Scholar] [CrossRef] [PubMed]
- Manzo-Avalos, S.; Saavedra-Molina, A. Cellular and mitochondrial effects of alcohol consumption. Int. J. Environ. Res. Public Health 2010, 7, 4281–4304. [Google Scholar] [CrossRef] [PubMed]
- Abdul Muneer, P.M.; Alikunju, S.; Szlachetka, A.M.; Haorah, J. The mechanisms of cerebral vascular dysfunction and neuroinflammation by MMP-mediated degradation of VEGFR-2 in alcohol ingestion. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1167–1177. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Chen, M.; Zhao, L.; Zhao, Q.; Hu, R.; Zhu, J.; Yan, R.; Dai, K. Ethanol Induces Platelet Apoptosis. Alcohol. Clin. Exp. Res. 2017, 41, 291–298. [Google Scholar] [CrossRef]
- Elenkov, I.J.; Wilder, R.L.; Chrousos, G.P.; Vizi, E.S. The sympathetic nerve—An integrative interface between two supersystems: The brain and the immune system. Pharmacol. Rev. 2000, 52, 595–638. [Google Scholar]
- Liesz, A.; Middelhoff, M.; Zhou, W.; Karcher, S.; Illanes, S.; Veltkamp, R. Comparison of humoral neuroinflammation and adhesion molecule expression in two models of experimental intracerebral hemorrhage. Exp. Transl. Stroke Med. 2011, 3, 11. [Google Scholar] [CrossRef] [PubMed]
- Olsson, T.; Marklund, N.; Gustafson, Y.; Nasman, B. Abnormalities at different levels of the hypothalamic-pituitary-adrenocortical axis early after stroke. Stroke 1992, 23, 1573–1576. [Google Scholar] [CrossRef] [PubMed]
- Pennypacker, K.R. Targeting the peripheral inflammatory response to stroke: Role of the spleen. Transl. Stroke Res. 2014, 5, 635–637. [Google Scholar] [CrossRef]
- Zhang, J.; Shi, K.; Li, Z.; Li, M.; Han, Y.; Wang, L.; Zhang, Z.; Yu, C.; Zhang, F.; Song, L.; et al. Organ- and cell-specific immune responses are associated with the outcomes of intracerebral hemorrhage. FASEB J. 2018, 32, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Waltman, C.; Blevins, L.S., Jr.; Boyd, G.; Wand, G.S. The effects of mild ethanol intoxication on the hypothalamic-pituitary-adrenal axis in nonalcoholic men. J. Clin. Endocrinol. Metab. 1993, 77, 518–522. [Google Scholar]
- Dai, X.; Thavundayil, J.; Santella, S.; Gianoulakis, C. Response of the HPA-axis to alcohol and stress as a function of alcohol dependence and family history of alcoholism. Psychoneuroendocrinology 2007, 32, 293–305. [Google Scholar] [CrossRef]
- Schepis, T.S.; Rao, U.; Yadav, H.; Adinoff, B. The limbic-hypothalamic-pituitary-adrenal axis and the development of alcohol use disorders in youth. Alcohol. Clin. Exp. Res. 2011, 35, 595–605. [Google Scholar] [CrossRef]
- Clarke, T.K.; Treutlein, J.; Zimmermann, U.S.; Kiefer, F.; Skowronek, M.H.; Rietschel, M.; Mann, K.; Schumann, G. HPA-axis activity in alcoholism: Examples for a gene-environment interaction. Addict. Biol. 2008, 13, 1–14. [Google Scholar] [CrossRef]
- Du, W.; Chen, X.; Shi, M.; Bian, F.; Zhao, Z. Ethanol affects behavior and HPA axis activity during development in zebrafish larvae. Sci. Rep. 2020, 10, 21402. [Google Scholar] [CrossRef]
- van de Borne, P.; Mark, A.L.; Montano, N.; Mion, D.; Somers, V.K. Effects of alcohol on sympathetic activity, hemodynamics, and chemoreflex sensitivity. Hypertension 1997, 29, 1278–1283. [Google Scholar] [CrossRef]
- Johnson, R.H.; Eisenhofer, G.; Lambie, D.G. The effects of acute and chronic ingestion of ethanol on the autonomic nervous system. Drug Alcohol Depend. 1986, 18, 319–328. [Google Scholar] [CrossRef]
- Xi, G.; Keep, R.F.; Hoff, J.T. Erythrocytes and delayed brain edema formation following intracerebral hemorrhage in rats. J. Neurosurg. 1998, 89, 991–996. [Google Scholar] [CrossRef] [PubMed]
- Hansch, G.M. The complement attack phase: Control of lysis and non-lethal effects of C5b-9. Immunopharmacology 1992, 24, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.; Botto, M.; Walport, M. The complement system. Curr. Biol. 1998, 8, R259–R261. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Xia, F.; Wan, S.; Hua, Y.; Keep, R.F.; Xi, G. Role of Complement Component 3 in Early Erythrolysis in the Hematoma After Experimental Intracerebral Hemorrhage. Stroke 2021, 52, 2649–2660. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Nakamura, T.; Hua, Y.; Keep, R.F.; Younger, J.G.; He, Y.; Hoff, J.T.; Xi, G. The role of complement C3 in intracerebral hemorrhage-induced brain injury. J. Cereb. Blood Flow. Metab. 2006, 26, 1490–1495. [Google Scholar] [CrossRef] [PubMed]
- Hua, Y.; Xi, G.; Keep, R.F.; Hoff, J.T. Complement activation in the brain after experimental intracerebral hemorrhage. J. Neurosurg. 2000, 92, 1016–1022. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Cabrera, M.A.; Boyadjieva, N.I.; Berger, G.; Rousseau, B.; Sarkar, D.K. Alcohol Increases Exosome Release from Microglia to Promote Complement C1q-Induced Cellular Death of Proopiomelanocortin Neurons in the Hypothalamus in a Rat Model of Fetal Alcohol Spectrum Disorders. J. Neurosci. 2020, 40, 7965–7979. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Yong, V.W. Neuroinflammation in intracerebral haemorrhage: Immunotherapies with potential for translation. Lancet Neurol. 2020, 19, 1023–1032. [Google Scholar] [CrossRef]
- Wang, J.; Dore, S. Inflammation after intracerebral hemorrhage. J. Cereb. Blood Flow Metab. 2007, 27, 894–908. [Google Scholar] [CrossRef] [PubMed]
- Wang, J. Preclinical and clinical research on inflammation after intracerebral hemorrhage. Prog. Neurobiol. 2010, 92, 463–477. [Google Scholar] [CrossRef]
- Mracsko, E.; Veltkamp, R. Neuroinflammation after intracerebral hemorrhage. Front. Cell Neurosci. 2014, 8, 388. [Google Scholar] [CrossRef]
- Lin, S.; Yin, Q.; Zhong, Q.; Lv, F.L.; Zhou, Y.; Li, J.Q.; Wang, J.Z.; Su, B.Y.; Yang, Q.W. Heme activates TLR4-mediated inflammatory injury via MyD88/TRIF signaling pathway in intracerebral hemorrhage. J. Neuroinflamm. 2012, 9, 46. [Google Scholar] [CrossRef]
- Smiley, S.T.; King, J.A.; Hancock, W.W. Fibrinogen stimulates macrophage chemokine secretion through toll-like receptor 4. J. Immunol. 2001, 167, 2887–2894. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Sun, G.; Ting, S.M.; Song, S.; Zhang, J.; Edwards, N.J.; Aronowski, J. Cleaning up after ICH: The role of Nrf2 in modulating microglia function and hematoma clearance. J. Neurochem. 2015, 133, 144–152. [Google Scholar] [CrossRef]
- Chang, C.F.; Wan, J.; Li, Q.; Renfroe, S.C.; Heller, N.M.; Wang, J. Alternative activation-skewed microglia/macrophages promote hematoma resolution in experimental intracerebral hemorrhage. Neurobiol. Dis. 2017, 103, 54–69. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Lan, X.; Han, X.; Durham, F.; Wan, J.; Weiland, A.; Koehler, R.C.; Wang, J. Microglia-derived interleukin-10 accelerates post-intracerebral hemorrhage hematoma clearance by regulating CD36. Brain Behav. Immun. 2021, 94, 437–457. [Google Scholar] [CrossRef]
- Guo, S.; Wang, H.; Yin, Y. Microglia Polarization From M1 to M2 in Neurodegenerative Diseases. Front. Aging Neurosci. 2022, 14, 815347. [Google Scholar] [CrossRef]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef]
- Peng, H.; Geil Nickell, C.R.; Chen, K.Y.; McClain, J.A.; Nixon, K. Increased expression of M1 and M2 phenotypic markers in isolated microglia after four-day binge alcohol exposure in male rats. Alcohol. 2017, 62, 29–40. [Google Scholar] [CrossRef]
- Alfonso-Loeches, S.; Pascual-Lucas, M.; Blanco, A.M.; Sanchez-Vera, I.; Guerri, C. Pivotal role of TLR4 receptors in alcohol-induced neuroinflammation and brain damage. J. Neurosci. 2010, 30, 8285–8295. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Lizarbe, S.; Montesinos, J.; Guerri, C. Ethanol induces TLR4/TLR2 association, triggering an inflammatory response in microglial cells. J. Neurochem. 2013, 126, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Lizarbe, S.; Pascual, M.; Guerri, C. Critical role of TLR4 response in the activation of microglia induced by ethanol. J. Immunol. 2009, 183, 4733–4744. [Google Scholar] [CrossRef] [PubMed]
- Qin, L.; Crews, F.T. NADPH oxidase and reactive oxygen species contribute to alcohol-induced microglial activation and neurodegeneration. J. Neuroinflamm. 2012, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Siracusa, R.; Fusco, R.; Cuzzocrea, S. Astrocytes: Role and Functions in Brain Pathologies. Front. Pharmacol. 2019, 10, 1114. [Google Scholar] [CrossRef]
- Scimemi, A. Astrocytes and the Warning Signs of Intracerebral Hemorrhagic Stroke. Neural Plast. 2018, 2018, 7301623. [Google Scholar] [CrossRef]
- Gomez, G.I.; Falcon, R.V.; Maturana, C.J.; Labra, V.C.; Salgado, N.; Rojas, C.A.; Oyarzun, J.E.; Cerpa, W.; Quintanilla, R.A.; Orellana, J.A. Heavy Alcohol Exposure Activates Astroglial Hemichannels and Pannexons in the Hippocampus of Adolescent Rats: Effects on Neuroinflammation and Astrocyte Arborization. Front. Cell Neurosci. 2018, 12, 472. [Google Scholar] [CrossRef]
- Dalcik, H.; Yardimoglu, M.; Filiz, S.; Gonca, S.; Dalcik, C.; Erden, B.F. Chronic ethanol-induced glial fibrillary acidic protein (GFAP) immunoreactivity: An immunocytochemical observation in various regions of adult rat brain. Int. J. Neurosci. 2009, 119, 1303–1318. [Google Scholar] [CrossRef]
- Miguel-Hidalgo, J.J. Withdrawal from free-choice ethanol consumption results in increased packing density of glutamine synthetase-immunoreactive astrocytes in the prelimbic cortex of alcohol-preferring rats. Alcohol Alcohol. 2006, 41, 379–385. [Google Scholar] [CrossRef]
- Blanco, A.M.; Pascual, M.; Valles, S.L.; Guerri, C. Ethanol-induced iNOS and COX-2 expression in cultured astrocytes via NF-kappa B. Neuroreport 2004, 15, 681–685. [Google Scholar] [CrossRef]
- Temel, S.G.; Kahveci, Z. Cyclooxygenase-2 expression in astrocytes and microglia in human oligodendroglioma and astrocytoma. J. Mol. Histol. 2009, 40, 369–377. [Google Scholar] [CrossRef]
- Poyhonen, S.; Er, S.; Domanskyi, A.; Airavaara, M. Effects of Neurotrophic Factors in Glial Cells in the Central Nervous System: Expression and Properties in Neurodegeneration and Injury. Front. Physiol. 2019, 10, 486. [Google Scholar] [CrossRef]
- Razavi, S.; Nazem, G.; Mardani, M.; Esfandiari, E.; Salehi, H.; Esfahani, S.H. Neurotrophic factors and their effects in the treatment of multiple sclerosis. Adv. Biomed. Res. 2015, 4, 53. [Google Scholar] [CrossRef]
- Bothwell, M. NGF, BDNF, NT3, and NT4. Handb. Exp. Pharmacol. 2014, 220, 3–15. [Google Scholar]
- Castilla-Cortazar, I.; Iturrieta, I.; Garcia-Magarino, M.; Puche, J.E.; Martin-Estal, I.; Aguirre, G.A.; Femat-Roldan, G.; Cantu-Martinez, L.; Munoz, U. Neurotrophic Factors and Their Receptors Are Altered by the Mere Partial IGF-1 Deficiency. Neuroscience 2019, 404, 445–458. [Google Scholar] [CrossRef]
- Sousa-Victor, P.; Jasper, H.; Neves, J. Trophic Factors in Inflammation and Regeneration: The Role of MANF and CDNF. Front. Physiol. 2018, 9, 1629. [Google Scholar] [CrossRef]
- Bi, R.; Fang, Z.; You, M.; He, Q.; Hu, B. Microglia Phenotype and Intracerebral Hemorrhage: A Balance of Yin and Yang. Front. Cell Neurosci. 2021, 15, 765205. [Google Scholar] [CrossRef]
- Li, J.; Xiao, L.; He, D.; Luo, Y.; Sun, H. Mechanism of White Matter Injury and Promising Therapeutic Strategies of MSCs After Intracerebral Hemorrhage. Front. Aging Neurosci. 2021, 13, 632054. [Google Scholar] [CrossRef]
- Lin, T.C.; Tsai, Y.C.; Chen, Y.A.; Young, T.H.; Wu, C.C.; Chiang, Y.H.; Kao, C.H.; Huang, A.P.; Hsu, Y.H.; Chen, K.Y.; et al. Brain-derived neurotrophic factor contributes to neurogenesis after intracerebral hemorrhage: A rodent model and human study. Front. Cell Neurosci. 2023, 17, 1170251. [Google Scholar] [CrossRef]
- Bahlakeh, G.; Rahbarghazi, R.; Mohammadnejad, D.; Abedelahi, A.; Karimipour, M. Current knowledge and challenges associated with targeted delivery of neurotrophic factors into the central nervous system: Focus on available approaches. Cell Biosci. 2021, 11, 181. [Google Scholar] [CrossRef]
- Barker, P.A.; Mantyh, P.; Arendt-Nielsen, L.; Viktrup, L.; Tive, L. Nerve Growth Factor Signaling and Its Contribution to Pain. J. Pain Res. 2020, 13, 1223–1241. [Google Scholar] [CrossRef] [PubMed]
- Minnone, G.; De Benedetti, F.; Bracci-Laudiero, L. NGF and Its Receptors in the Regulation of Inflammatory Response. Int. J. Mol. Sci. 2017, 18, 1028. [Google Scholar] [CrossRef]
- Malik, S.C.; Sozmen, E.G.; Baeza-Raja, B.; Le Moan, N.; Akassoglou, K.; Schachtrup, C. In vivo functions of p75(NTR): Challenges and opportunities for an emerging therapeutic target. Trends Pharmacol. Sci. 2021, 42, 772–788. [Google Scholar] [CrossRef]
- Li, Q.; Hu, Y.Z.; Gao, S.; Wang, P.F.; Hu, Z.L.; Dai, R.P. ProBDNF and its receptors in immune-mediated inflammatory diseases: Novel insights into the regulation of metabolism and mitochondria. Front. Immunol. 2023, 14, 1155333. [Google Scholar] [CrossRef]
- Carito, V.; Ceccanti, M.; Ferraguti, G.; Coccurello, R.; Ciafre, S.; Tirassa, P.; Fiore, M. NGF and BDNF Alterations by Prenatal Alcohol Exposure. Curr. Neuropharmacol. 2019, 17, 308–317. [Google Scholar] [CrossRef]
- Ceci, F.M.; Ferraguti, G.; Petrella, C.; Greco, A.; Ralli, M.; Iannitelli, A.; Carito, V.; Tirassa, P.; Chaldakov, G.N.; Messina, M.P.; et al. Nerve Growth Factor in Alcohol Use Disorders. Curr. Neuropharmacol. 2021, 19, 45–60. [Google Scholar] [CrossRef]
- Zhou, L.; Xiong, J.; Ruan, C.S.; Ruan, Y.; Liu, D.; Bao, J.J.; Zhou, X.F. ProBDNF/p75NTR/sortilin pathway is activated in peripheral blood of patients with alcohol dependence. Transl. Psychiatry 2018, 7, 2. [Google Scholar] [CrossRef]
- Darcq, E.; Morisot, N.; Phamluong, K.; Warnault, V.; Jeanblanc, J.; Longo, F.M.; Massa, S.M.; Ron, D. The Neurotrophic Factor Receptor p75 in the Rat Dorsolateral Striatum Drives Excessive Alcohol Drinking. J. Neurosci. 2016, 36, 10116–10127. [Google Scholar] [CrossRef]
- Wang, X.; Qi, Y.; Zheng, H. Dietary Polyphenol, Gut Microbiota, and Health Benefits. Antioxidants 2022, 11, 1212. [Google Scholar] [CrossRef]
- Iqbal, I.; Wilairatana, P.; Saqib, F.; Nasir, B.; Wahid, M.; Latif, M.F.; Iqbal, A.; Naz, R.; Mubarak, M.S. Plant Polyphenols and Their Potential Benefits on Cardiovascular Health: A Review. Molecules 2023, 28, 6403. [Google Scholar] [CrossRef]
- Santhiravel, S.; Bekhit, A.E.A.; Mendis, E.; Jacobs, J.L.; Dunshea, F.R.; Rajapakse, N.; Ponnampalam, E.N. The Impact of Plant Phytochemicals on the Gut Microbiota of Humans for a Balanced Life. Int. J. Mol. Sci. 2022, 23, 8124. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhang, S.; Fan, X. Role of Polyphenols as Antioxidant Supplementation in Ischemic Stroke. Oxid. Med. Cell Longev. 2021, 2021, 5471347. [Google Scholar] [CrossRef] [PubMed]
- Lan, X.; Han, X.; Li, Q.; Wang, J. (−)-Epicatechin, a Natural Flavonoid Compound, Protects Astrocytes Against Hemoglobin Toxicity via Nrf2 and AP-1 Signaling Pathways. Mol. Neurobiol. 2017, 54, 7898–7907. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yi, B.; Ma, J.; Zhang, L.; Zhang, H.; Yang, Y.; Dai, Y. Quercetin promotes neuronal and behavioral recovery by suppressing inflammatory response and apoptosis in a rat model of intracerebral hemorrhage. Neurochem. Res. 2015, 40, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; He, J.; Huang, Y.; Hu, Z. Resveratrol has an Overall Neuroprotective Role in Ischemic Stroke: A Meta-Analysis in Rodents. Front. Pharmacol. 2021, 12, 795409. [Google Scholar] [CrossRef] [PubMed]
- Bonsack, F.; Alleyne, C.H., Jr.; Sukumari-Ramesh, S. Resveratrol Attenuates Neurodegeneration and Improves Neurological Outcomes after Intracerebral Hemorrhage in Mice. Front. Cell Neurosci. 2017, 11, 228. [Google Scholar] [CrossRef]
- Pacifici, F.; Rovella, V.; Pastore, D.; Bellia, A.; Abete, P.; Donadel, G.; Santini, S.; Beck, H.; Ricordi, C.; Daniele, N.D.; et al. Polyphenols and Ischemic Stroke: Insight into One of the Best Strategies for Prevention and Treatment. Nutrients 2021, 13, 1967. [Google Scholar] [CrossRef]
- Panickar, K.S.; Anderson, R.A. Effect of polyphenols on oxidative stress and mitochondrial dysfunction in neuronal death and brain edema in cerebral ischemia. Int. J. Mol. Sci. 2011, 12, 8181–8207. [Google Scholar] [CrossRef]
- Carito, V.; Venditti, A.; Bianco, A.; Ceccanti, M.; Serrilli, A.M.; Chaldakov, G.; Tarani, L.; De Nicolo, S.; Fiore, M. Effects of olive leaf polyphenols on male mouse brain NGF, BDNF and their receptors TrkA, TrkB and p75. Nat. Prod. Res. 2014, 28, 1970–1984. [Google Scholar] [CrossRef]
- Ferraguti, G.; Terracina, S.; Petrella, C.; Greco, A.; Minni, A.; Lucarelli, M.; Agostinelli, E.; Ralli, M.; de Vincentiis, M.; Raponi, G.; et al. Alcohol and Head and Neck Cancer: Updates on the Role of Oxidative Stress, Genetic, Epigenetics, Oral Microbiota, Antioxidants, and Alkylating Agents. Antioxidants 2022, 11, 145. [Google Scholar] [CrossRef] [PubMed]
- Carito, V.; Ceccanti, M.; Cestari, V.; Natella, F.; Bello, C.; Coccurello, R.; Mancinelli, R.; Fiore, M. Olive polyphenol effects in a mouse model of chronic ethanol addiction. Nutrition 2017, 33, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Nabavi, S.F.; Dean, O.M.; Turner, A.; Sureda, A.; Daglia, M.; Nabavi, S.M. Oxidative stress and post-stroke depression: Possible therapeutic role of polyphenols? Curr. Med. Chem. 2015, 22, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Abdelsalam, S.A.; Renu, K.; Zahra, H.A.; Abdallah, B.M.; Ali, E.M.; Veeraraghavan, V.P.; Sivalingam, K.; Ronsard, L.; Ammar, R.B.; Vidya, D.S.; et al. Polyphenols Mediate Neuroprotection in Cerebral Ischemic Stroke-An Update. Nutrients 2023, 15, 1107. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhao, T.; Shi, M.; Wei, Y.; Huang, X.; Shen, J.; Zhang, X.; Xie, Z.; Huang, P.; Yuan, K.; et al. Polyphenols: Natural food grade biomolecules for treating neurodegenerative diseases from a multi-target perspective. Front. Nutr. 2023, 10, 1139558. [Google Scholar] [CrossRef] [PubMed]
- Beseler, C.L.; Taylor, L.A.; Kraemer, D.T.; Leeman, R.F. A latent class analysis of DSM-IV alcohol use disorder criteria and binge drinking in undergraduates. Alcohol. Clin. Exp. Res. 2012, 36, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Beseler, C.L.; Taylor, L.A.; Leeman, R.F. An item-response theory analysis of DSM-IV alcohol-use disorder criteria and “binge” drinking in undergraduates. J. Stud. Alcohol. Drugs 2010, 71, 418–423. [Google Scholar] [CrossRef]
- Bhochhibhoya, A.; Hayes, L.; Branscum, P.; Taylor, L. The Use of the Internet for Prevention of Binge Drinking Among the College Population: A Systematic Review of Evidence. Alcohol Alcohol. 2015, 50, 526–535. [Google Scholar] [CrossRef]
- Viner, R.M.; Taylor, B. Adult outcomes of binge drinking in adolescence: Findings from a UK national birth cohort. J. Epidemiol. Community Health 2007, 61, 902–907. [Google Scholar] [CrossRef]
- Woo, D.; Rosand, J.; Kidwell, C.; McCauley, J.L.; Osborne, J.; Brown, M.W.; West, S.E.; Rademacher, E.W.; Waddy, S.; Roberts, J.N.; et al. The Ethnic/Racial Variations of Intracerebral Hemorrhage (ERICH) study protocol. Stroke 2013, 44, e120–e125. [Google Scholar] [CrossRef] [PubMed]
- Poli, L.; Grassi, M.; Zedde, M.; Marcheselli, S.; Silvestrelli, G.; Sessa, M.; Zini, A.; Paciaroni, M.; Azzini, C.; Gamba, M.; et al. Anticoagulants Resumption after Warfarin-Related Intracerebral Haemorrhage: The Multicenter Study on Cerebral Hemorrhage in Italy (MUCH-Italy). Thromb. Haemost. 2018, 118, 572–580. [Google Scholar] [CrossRef]
- Larsson, S.C.; Wallin, A.; Wolk, A.; Markus, H.S. Differing association of alcohol consumption with different stroke types: A systematic review and meta-analysis. BMC Med. 2016, 14, 178. [Google Scholar] [CrossRef]
- Fewel, M.E.; Thompson, B.G., Jr.; Hoff, J.T. Spontaneous intracerebral hemorrhage: A review. Neurosurg. Focus 2003, 15, E1. [Google Scholar] [CrossRef] [PubMed]
- Juvela, S.; Hillbom, M.; Palomaki, H. Risk factors for spontaneous intracerebral hemorrhage. Stroke 1995, 26, 1558–1564. [Google Scholar] [CrossRef] [PubMed]
- Lyden, P.D.; Zivin, J.A.; Soll, M.; Sitzer, M.; Rothrock, J.F.; Alksne, J. Intracerebral hemorrhage after experimental embolic infarction. Anticoagulation. Arch. Neurol. 1987, 44, 848–850. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Quinones, P.; McCarthy, C.G.; Watts, S.W.; Klee, N.S.; Komic, A.; Calmasini, F.B.; Priviero, F.; Warner, A.; Chenghao, Y.; Wenceslau, C.F. Hypertension Induced Morphological and Physiological Changes in Cells of the Arterial Wall. Am. J. Hypertens. 2018, 31, 1067–1078. [Google Scholar] [CrossRef] [PubMed]
- Piano, M.R. Alcohol’s Effects on the Cardiovascular System. Alcohol. Res. 2017, 38, 219–241. [Google Scholar]
- Briasoulis, A.; Agarwal, V.; Messerli, F.H. Alcohol consumption and the risk of hypertension in men and women: A systematic review and meta-analysis. J. Clin. Hypertens. (Greenwich) 2012, 14, 792–798. [Google Scholar] [CrossRef]
- Piano, M.R.; Thur, L.A.; Hwang, C.L.; Phillips, S.A. Effects of Alcohol on the Cardiovascular System in Women. Alcohol Res. 2020, 40, 12. [Google Scholar] [CrossRef]
- Mori, T.A.; Burke, V.; Beilin, L.J.; Puddey, I.B. Randomized Controlled Intervention of the Effects of Alcohol on Blood Pressure in Premenopausal Women. Hypertension 2015, 66, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Dalle-Donne, I.; Rossi, R.; Colombo, R.; Giustarini, D.; Milzani, A. Biomarkers of oxidative damage in human disease. Clin. Chem. 2006, 52, 601–623. [Google Scholar] [CrossRef] [PubMed]
- Ceni, E.; Mello, T.; Galli, A. Pathogenesis of alcoholic liver disease: Role of oxidative metabolism. World J. Gastroenterol. 2014, 20, 17756–17772. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Li, X.; Prabhu, S.D.; Brittian, K.R.; Chen, Q.; Yin, X.; McClain, C.J.; Zhou, Z.; Cai, L. Angiotensin II plays a critical role in alcohol-induced cardiac nitrative damage, cell death, remodeling, and cardiomyopathy in a protein kinase C/nicotinamide adenine dinucleotide phosphate oxidase-dependent manner. J. Am. Coll. Cardiol. 2012, 59, 1477–1486. [Google Scholar] [CrossRef]
- Huang, L.C.; Liew, H.K.; Cheng, H.Y.; Kuo, J.S.; Hsu, W.L.; Pang, C.Y. Brain Magnetic Resonance Imaging of Intracerebral Hemorrhagic Rats after Alcohol Consumption. J. Stroke Cerebrovasc. Dis. 2018, 27, 3493–3502. [Google Scholar] [CrossRef]
- Cheng, H.Y.; Huang, L.C.; Peng, H.F.; Kuo, J.S.; Liew, H.K.; Pang, C.Y. Delayed formation of hematomas with ethanol preconditioning in experimental intracerebral hemorrhage rats. Ci Ji Yi Xue Za Zhi 2018, 30, 5–9. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Relationship with SBI | SBI Components | Effects | Ref. |
|---|---|---|---|
| Direct relationship with neuroinflammation | ↓ HO-1 | Reduced heme catabolism thereby increasing the free heme in the brain parenchyma and exacerbating the injury | [59] |
| ↑ Intracellular Calcium | Mitochondrial dysfunction to neuronal apoptosis | [76,77] | |
| ↓ ATP | Increased oxidative stress, mitochondrial dysfunction, and neuronal apoptosis | ||
| ↑ MMP-9 | BBB dysfunction | [78] | |
| Direct relationship wtih neuroinflammation | ↑ ROS-Increased oxidative stress | Increased neuronal tissue damage | [75] |
| ↑ Bax-Pro-apoptotic | Increased platelet apoptosis | [80] | |
| ↓ Bcl-2-Anti-apototic | |||
| Neuroendocrine Axis | ↓ Cortisol Reactivity | Reduced HPA activity to mitigate the ICH injury | [88,89,90] |
| Complement System | ↑ Complement C1q, C5b9 and MAC | Neuronal apoptosis and necrosis at the site of ICH injury | [100] |
| Inflammatory Mediators | ↑ M1 and M2 microglial phenotypes | M1-Pro-inflammatory activities and M2-Anti-inflammatory activities | [112] |
| ↑ Pro-inflammatory cytokines (TNF-α, IL-1β) | Increased neuroinflammation | [113] | |
| ↑ TLR-4 and TLR-2 interaction | Release inflammatory mediators | [115] | |
| ↑ NF-κB signaling and NADPH oxidase | Increased ROS production and changes in microglial morphology thereby increasing neuroinflammation | [116] | |
| ↑ iNOS ↑ COX-2 | Activated immune response | [112] | |
| ↑ Pro-inflammatory cytokines (TNF-α, IL-1β & IL-6), reduced chaperone proteins and diminished anti-inflammatory cytokines | Increased neuroinflammation | [79] | |
| Direct relationship with neuroinflammation | ↓ Fibrinogen | Reduces the clot density | [58] |
| ↓ Oxidative stress | Ameliorated neurological deficits | [79] | |
| Neuroendocrine Axis | ↓ HPA Acvitity | Compromised HPA activity dampens the stress mitigation caused by ICH | [86] |
| ↑ SNS Activity | Exerts already existing SNS activity thereby compromising the SNS activity needed to accord the stress caused by ICH | [92,93] | |
| Inflammatory mediators and stressors | ↓ ER stress | Increased ER homeostasis | [79] |
| ↑ Chaperone proteins (GRP78, GRP94 & Hsc70) | Reduced ER stress thereby reducing oxidative stress and neuronal apoptosis | ||
| ↓ Pro-inflammatory cytokines (TNF-α, IL-1β & IL-6) | Reduced neuroinflammation | ||
| Restored anti-inflammatory cytokine (IL-10) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thangameeran, S.I.M.; Wang, P.-K.; Liew, H.-K.; Pang, C.-Y. Influence of Alcohol on Intracerebral Hemorrhage: From Oxidative Stress to Glial Cell Activation. Life 2024, 14, 311. https://doi.org/10.3390/life14030311
Thangameeran SIM, Wang P-K, Liew H-K, Pang C-Y. Influence of Alcohol on Intracerebral Hemorrhage: From Oxidative Stress to Glial Cell Activation. Life. 2024; 14(3):311. https://doi.org/10.3390/life14030311
Chicago/Turabian StyleThangameeran, Shaik Ismail Mohammed, Po-Kai Wang, Hock-Kean Liew, and Cheng-Yoong Pang. 2024. "Influence of Alcohol on Intracerebral Hemorrhage: From Oxidative Stress to Glial Cell Activation" Life 14, no. 3: 311. https://doi.org/10.3390/life14030311