

Novel Therapeutic Approaches for the Management of Elevated Lipoprotein(a): From Traditional Agents to Future Treatment Options

 ,
,  ,
, 
Abstract
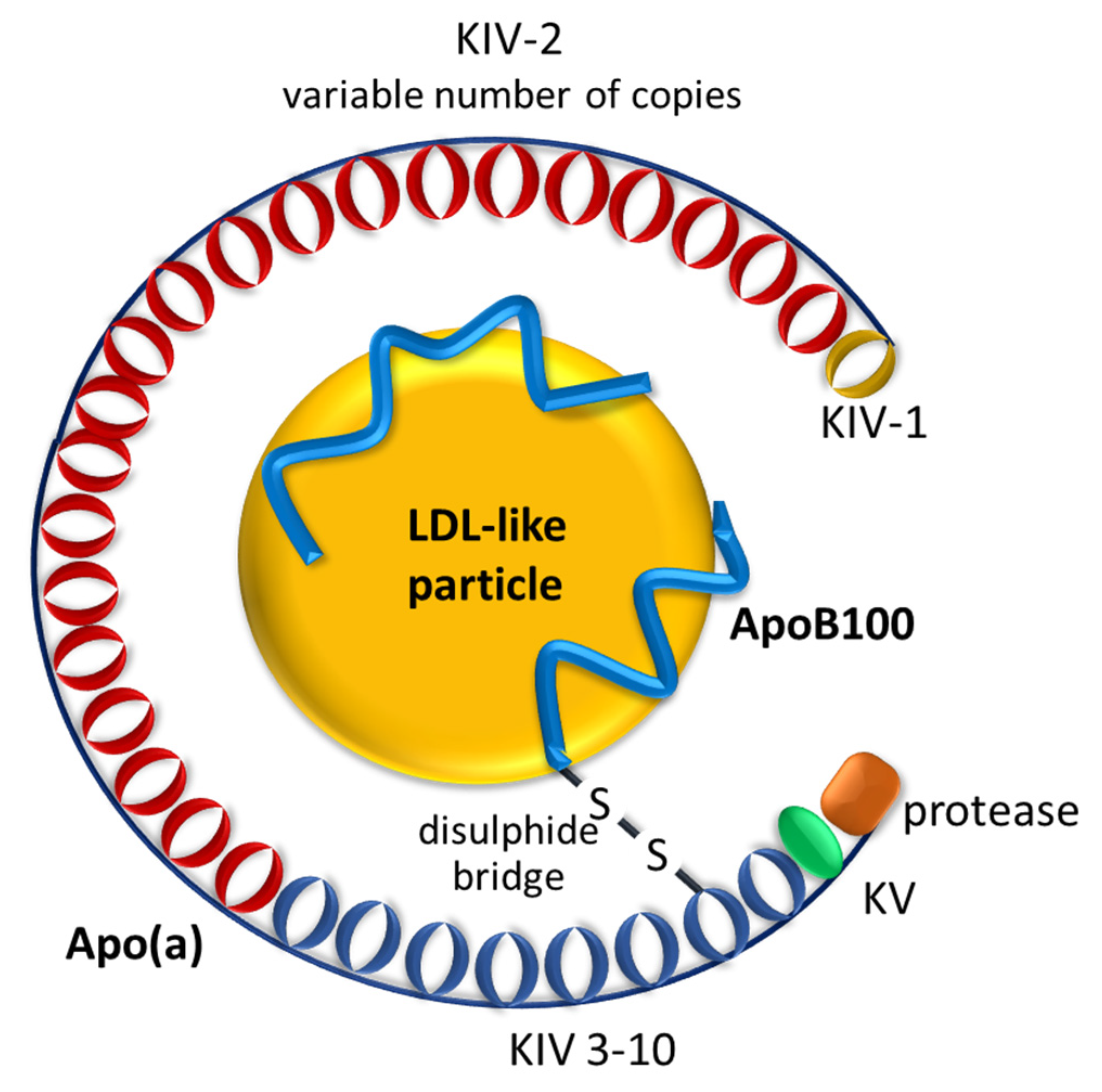
:1. Introduction
2. Agents That Increase LDL Receptor Expression
2.1. Statins
2.2. Ezetimibe
2.3. PCSK9 Inhibitors
3. LDL Production Inhibitors
4. Other Treatment Options to Reduce Lp(a) Concentration
4.1. Cholesteryl Ester Transfer Protein (CETP) Inhibitors
4.2. Nicotinic Acid
4.3. Aspirin
4.4. Thyroid Hormone Mimetics
4.5. Lipoprotein Apheresis
4.6. Apo(a)-Reducing ASO and SiRNA Formulations
5. Future Perspectives
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kamstrup, P.R.; Benn, M.; Tybjaerg-Hansen, A.; Nordestgaard, B.G. Extreme lipoprotein(a) levels and risk of myocardial infarction in the general population: The Copenhagen City Heart Study. Circulation 2008, 117, 176–184. [Google Scholar] [CrossRef]
- Erqou, S.; Kaptoge, S.; Perry, P.L.; Di Angelantonio, E.; Thompson, A.; White, I.R.; Marcovina, S.M.; Collins, R.; Thompson, S.G.; Danesh, J.; et al. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA 2009, 302, 412–423. [Google Scholar] [CrossRef]
- Khera, A.V.; Everett, B.M.; Caulfield, M.P.; Hantash, F.M.; Wohlgemuth, J.; Ridker, P.M.; Mora, S. Lipoprotein(a) concentrations, rosuvastatin therapy, and residual vascular risk: An analysis from the JUPITER Trial (Justification for the Use of Statins in Prevention: An Intervention Trial Evaluating Rosuvastatin). Circulation 2014, 129, 635–642. [Google Scholar] [CrossRef]
- Gurdasani, D.; Sjouke, B.; Tsimikas, S.; Hovingh, G.K.; Luben, R.N.; Wainwright, N.W.; Pomilla, C.; Wareham, N.J.; Khaw, K.T.; Boekholdt, S.M.; et al. Lipoprotein(a) and risk of coronary, cerebrovascular, and peripheral artery disease: The EPIC-Norfolk prospective population study. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 3058–3065. [Google Scholar] [CrossRef] [PubMed]
- Bonaca, M.P.; Nault, P.; Giugliano, R.P.; Keech, A.C.; Pineda, A.L.; Kanevsky, E.; Kuder, J.; Murphy, S.A.; Jukema, J.W.; Lewis, B.S.; et al. Low-Density Lipoprotein Cholesterol Lowering With Evolocumab and Outcomes in Patients With Peripheral Artery Disease: Insights From the FOURIER Trial (Further Cardiovascular Outcomes Research With PCSK9 Inhibition in Subjects With Elevated Risk). Circulation 2018, 137, 338–350. [Google Scholar] [CrossRef] [PubMed]
- Peeters, F.E.C.M.; Meex, S.J.R.; Dweck, M.R.; Aikawa, E.; Crijns, H.J.G.M.; Schurgers, L.J.; Kietselaer, B.L.J.H. Calcific aortic valve stenosis: Hard disease in the heart: A biomolecular approach towards diagnosis and treatment. Eur. Heart J. 2018, 39, 2618–2624. [Google Scholar] [CrossRef] [PubMed]
- Schnitzler, J.G.; Ali, L.; Groenen, A.G.; Kaiser, Y.; Kroon, J. Lipoprotein(a) as Orchestrator of Calcific Aortic Valve Stenosis. Biomolecules 2019, 9, 760. [Google Scholar] [CrossRef] [PubMed]
- Maranhão, R.C.; Carvalho, P.O.; Strunz, C.C.; Pileggi, F. Lipoprotein (a): Structure, pathophysiology and clinical implications. Arq. Bras. Cardiol. 2014, 103, 76–84. [Google Scholar] [CrossRef] [PubMed]
- Ugovšek, S.; Šebeštjen, M. Lipoprotein(a)-The Crossroads of Atherosclerosis, Atherothrombosis and Inflammation. Biomolecules 2021, 12, 26. [Google Scholar] [CrossRef] [PubMed]
- Kronenberg, F. Lipoprotein(a) measurement issues: Are we making a mountain out of a molehill? Atherosclerosis 2022, 349, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Scharnagl, H.; Stojakovic, T.; Dieplinger, B.; Dieplinger, H.; Erhart, G.; Kostner, G.M.; Herrmann, M.; März, W.; Grammer, T.B. Comparison of lipoprotein (a) serum concentrations measured by six commercially available immunoassays. Atherosclerosis 2019, 289, 206–213. [Google Scholar] [CrossRef]
- Marcovina, S.M.; Albers, J.J. Lipoprotein (a) measurements for clinical application. J. Lipid. Res. 2016, 57, 526–537. [Google Scholar] [CrossRef] [PubMed]
- Kronenberg, F.; Mora, S.; Stroes, E.S.G. Consensus and guidelines on lipoprotein(a)—Seeing the forest through the trees. Curr. Opin. Lipidol. 2022, 33, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.; Peden, J.F.; Hopewell, J.C.; Kyriakou, T.; Goel, A.; Heath, S.C.; Parish, S.; Barlera, S.; Franzosi, M.G.; Rust, S.; et al. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N. Engl. J. Med. 2009, 361, 2518–2528. [Google Scholar] [CrossRef] [PubMed]
- Németh, Á.; Daróczy, B.; Juhász, L.; Fülöp, P.; Harangi, M.; Paragh, G. Assessment of Associations Between Serum Lipoprotein (a) Levels and Atherosclerotic Vascular Diseases in Hungarian Patients With Familial Hypercholesterolemia Using Data Mining and Machine Learning. Front. Genet. 2022, 13, 849197. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, H.A.; Colhoun, H.M.; Johnson, T.; McKeigue, P.M.; Betteridge, D.J.; Durrington, P.N.; Fuller, J.H.; Livingstone, S.; Charlton-Menys, V.; Neil, A.; et al. Genome-wide association study of genetic determinants of LDL-c response to atorvastatin therapy: Importance of Lp(a). J. Lipid. Res. 2012, 53, 1000–1011. [Google Scholar] [CrossRef]
- Berg, K.; Dahlén, G.; Christophersen, B.; Cook, T.; Kjekshus, J.; Pedersen, T. Lp(a) lipoprotein level predicts survival and major coronary events in the Scandinavian Simvastatin Survival Study. Clin. Genet. 1997, 52, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Tsimikas, S.; Gordts, P.L.S.M.; Nora, C.; Yeang, C.; Witztum, J.L. Statin therapy increases lipoprotein(a) levels. Eur. Heart. J. 2020, 41, 2275–2284. [Google Scholar] [CrossRef]
- Hoover-Plow, J.; Huang, M. Lipoprotein(a) metabolism: Potential sites for therapeutic targets. Metabolism 2013, 62, 479–491. [Google Scholar] [CrossRef]
- Watts, G.F.; Chan, D.C.; Somaratne, R.; Wasserman, S.M.; Scott, R.; Marcovina, S.M.; Barrett, P.H.R. Controlled study of the effect of proprotein convertase subtilisin-kexin type 9 inhibition with evolocumab on lipoprotein(a) particle kinetics. Eur. Heart. J. 2018, 39, 2577–2585. [Google Scholar] [CrossRef]
- Awad, K.; Mikhailidis, D.P.; Katsiki, N.; Muntner, P.; Banach, M.; Lipid and Blood Pressure Meta-Analysis Collaboration (LBPMC) Group. Effect of Ezetimibe Monotherapy on Plasma Lipoprotein(a) Concentrations in Patients with Primary Hypercholesterolemia: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Drugs 2018, 78, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Sahebkar, A.; Simental-Mendía, L.E.; Pirro, M.; Banach, M.; Watts, G.F.; Sirtori, C.; Al-Rasadi, K.; Atkin, S.L. Impact of ezetimibe on plasma lipoprotein(a) concentrations as monotherapy or in combination with statins: A systematic review and meta-analysis of randomized controlled trials. Sci. Rep. 2018, 8, 17887. [Google Scholar] [CrossRef]
- Rizos, C.V.; Skoumas, I.; Rallidis, L.; Skalidis, E.; Tziomalos, K.; Garoufi, A.; Anagnostis, P.; Sfikas, G.; Kotsis, V.; Doumas, M.; et al. LDL cholesterol target achievement in heterozygous familial hypercholesterolemia patients according to 2019 ESC/EAS lipid guidelines: Implications for newer lipid-lowering treatments. Int. J. Cardiol. 2021, 345, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Koivisto, U.M.; Hubbard, A.L.; Mellman, I. A novel cellular phenotype for familial hypercholesterolemia due to a defect in polarized targeting of LDL receptor. Cell 2001, 105, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Beglova, N.; Blacklow, S.C. The LDL receptor: How acid pulls the trigger. Trends Biochem. Sci. 2005, 30, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Abifadel, M.; Varret, M.; Rabès, J.P.; Allard, D.; Ouguerram, K.; Devillers, M.; Cruaud, C.; Benjannet, S.; Wickham, L.; Erlich, D.; et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat. Genet. 2003, 34, 154–156. [Google Scholar] [CrossRef]
- Berge, K.E.; Ose, L.; Leren, T.P. Missense mutations in the PCSK9 gene are associated with hypocholesterolemia and possibly increased response to statin therapy. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1094–1100. [Google Scholar] [CrossRef]
- Chemello, K.; Beeské, S.; Trang Tran, T.T.; Blanchard, V.; Villard, E.F.; Poirier, B.; Le Bail, J.C.; Dargazanli, G.; Ho-Van-Guimbal, S.; Boulay, D.; et al. Lipoprotein(a) Cellular Uptake Ex Vivo and Hepatic Capture In Vivo Is Insensitive to PCSK9 Inhibition With Alirocumab. JACC Basic Transl. Sci. 2020, 5, 549–557. [Google Scholar] [CrossRef]
- O’Donoghue, M.L.; Fazio, S.; Giugliano, R.P.; Stroes, E.S.G.; Kanevsky, E.; Gouni-Berthold, I.; Im, K.; Lira Pineda, A.; Wasserman, S.M.; Češka, R.; et al. Lipoprotein(a), PCSK9 Inhibition, and Cardiovascular Risk. Circulation 2019, 139, 1483–1492. [Google Scholar] [CrossRef]
- Bittner, V.A.; Szarek, M.; Aylward, P.E.; Bhatt, D.L.; Diaz, R.; Edelberg, J.M.; Fras, Z.; Goodman, S.G.; Halvorsen, S.; Hanotin, C.; et al. Effect of Alirocumab on Lipoprotein(a) and Cardiovascular Risk After Acute Coronary Syndrome. J. Am. Coll. Cardiol. 2020, 75, 133–144. [Google Scholar] [CrossRef]
- Shapiro, M.D.; Minnier, J.; Tavori, H.; Kassahun, H.; Flower, A.; Somaratne, R.; Fazio, S. Relationship Between Low-Density Lipoprotein Cholesterol and Lipoprotein(a) Lowering in Response to PCSK9 Inhibition With Evolocumab. J. Am. Heart. Assoc. 2019, 8, e010932. [Google Scholar] [CrossRef]
- Raal, F.J.; Giugliano, R.P.; Sabatine, M.S.; Koren, M.J.; Blom, D.; Seidah, N.G.; Honarpour, N.; Lira, A.; Xue, A.; Chiruvolu, P.; et al. PCSK9 inhibition-mediated reduction in Lp(a) with evolocumab: An analysis of 10 clinical trials and the LDL receptor’s role. J. Lipid. Res. 2016, 57, 1086–1096. [Google Scholar] [CrossRef]
- Villard, E.F.; Thedrez, A.; Blankenstein, J.; Croyal, M.; Tran, T.T.; Poirier, B.; Le Bail, J.C.; Illiano, S.; Nobécourt, E.; Krempf, M.; et al. PCSK9 Modulates the Secretion But Not the Cellular Uptake of Lipoprotein(a) Ex Vivo: An Effect Blunted by Alirocumab. JACC Basic Transl. Sci. 2016, 1, 419–427. [Google Scholar] [CrossRef]
- Boffa, M.B.; Koschinsky, M.L. Update on lipoprotein(a) as a cardiovascular risk factor and mediator. Curr. Atheroscler. Rep. 2013, 15, 360. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.A.; Honarpour, N.; Wasserman, S.M.; Xu, F.; Scott, R.; Raal, F.J. Effect of the proprotein convertase subtilisin/kexin 9 monoclonal antibody, AMG 145, in homozygous familial hypercholesterolemia. Circulation 2013, 128, 2113–2120. [Google Scholar] [CrossRef] [PubMed]
- Saavedra, Y.G.; Dufour, R.; Davignon, J.; Baass, A. PCSK9 R46L, lower LDL, and cardiovascular disease risk in familial hypercholesterolemia: A cross-sectional cohort study. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2700–2705. [Google Scholar] [CrossRef]
- Saavedra, Y.G.L.; Dufour, R.; Baass, A. Familial hypercholesterolemia: PCSK9 InsLEU genetic variant and prediabetes/diabetes risk. J. Clin. Lipidol. 2015, 9, 786–793.e781. [Google Scholar] [CrossRef]
- Shapiro, M.D.; Tavori, H.; Fazio, S. PCSK9: From Basic Science Discoveries to Clinical Trials. Circ. Res. 2018, 122, 1420–1438. [Google Scholar] [CrossRef]
- Warden, B.A.; Fazio, S.; Shapiro, M.D. The PCSK9 revolution: Current status, controversies, and future directions. Trends Cardiovasc. Med. 2020, 30, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Raal, F.J.; Kallend, D.; Ray, K.K.; Turner, T.; Koenig, W.; Wright, R.S.; Wijngaard, P.L.J.; Curcio, D.; Jaros, M.J.; Leiter, L.A.; et al. Inclisiran for the Treatment of Heterozygous Familial Hypercholesterolemia. N. Engl. J. Med. 2020, 382, 1520–1530. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.K.; Wright, R.S.; Kallend, D.; Koenig, W.; Leiter, L.A.; Raal, F.J.; Bisch, J.A.; Richardson, T.; Jaros, M.; Wijngaard, P.L.J.; et al. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N. Engl. J. Med. 2020, 382, 1507–1519. [Google Scholar] [CrossRef]
- Kastelein, J.J.; Wedel, M.K.; Baker, B.F.; Su, J.; Bradley, J.D.; Yu, R.Z.; Chuang, E.; Graham, M.J.; Crooke, R.M. Potent reduction of apolipoprotein B and low-density lipoprotein cholesterol by short-term administration of an antisense inhibitor of apolipoprotein B. Circulation 2006, 114, 1729–1735. [Google Scholar] [CrossRef]
- Santos, R.D.; Raal, F.J.; Catapano, A.L.; Witztum, J.L.; Steinhagen-Thiessen, E.; Tsimikas, S. Mipomersen, an antisense oligonucleotide to apolipoprotein B-100, reduces lipoprotein(a) in various populations with hypercholesterolemia: Results of 4 phase III trials. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Merki, E.; Graham, M.J.; Mullick, A.E.; Miller, E.R.; Crooke, R.M.; Pitas, R.E.; Witztum, J.L.; Tsimikas, S. Antisense oligonucleotide directed to human apolipoprotein B-100 reduces lipoprotein(a) levels and oxidized phospholipids on human apolipoprotein B-100 particles in lipoprotein(a) transgenic mice. Circulation 2008, 118, 743–753. [Google Scholar] [CrossRef]
- Samaha, F.F.; McKenney, J.; Bloedon, L.T.; Sasiela, W.J.; Rader, D.J. Inhibition of microsomal triglyceride transfer protein alone or with ezetimibe in patients with moderate hypercholesterolemia. Nat. Clin. Pract. Cardiovasc. Med. 2008, 5, 497–505. [Google Scholar] [CrossRef]
- Cuchel, M.; Meagher, E.A.; du Toit Theron, H.; Blom, D.J.; Marais, A.D.; Hegele, R.A.; Averna, M.R.; Sirtori, C.R.; Shah, P.K.; Gaudet, D.; et al. Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: A single-arm, open-label, phase 3 study. Lancet 2013, 381, 40–46. [Google Scholar] [CrossRef]
- Cannon, C.P.; Shah, S.; Dansky, H.M.; Davidson, M.; Brinton, E.A.; Gotto, A.M.; Stepanavage, M.; Liu, S.X.; Gibbons, P.; Ashraf, T.B.; et al. Safety of anacetrapib in patients with or at high risk for coronary heart disease. N. Engl. J. Med. 2010, 363, 2406–2415. [Google Scholar] [CrossRef]
- Teramoto, T.; Shirakawa, M.; Kikuchi, M.; Nakagomi, M.; Tamura, S.; Surks, H.K.; McCrary Sisk, C.; Numaguchi, H. Efficacy and safety of the cholesteryl ester transfer protein inhibitor anacetrapib in Japanese patients with dyslipidemia. Atherosclerosis 2013, 230, 52–60. [Google Scholar] [CrossRef]
- Nelson, A.J.; Sniderman, A.D.; Ditmarsch, M.; Dicklin, M.R.; Nicholls, S.J.; Davidson, M.H.; Kastelein, J.J.P. Cholesteryl Ester Transfer Protein Inhibition Reduces Major Adverse Cardiovascular Events by Lowering Apolipoprotein B Levels. Int. J. Mol. Sci. 2022, 23, 9417. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Mistry, A.; Reynolds, J.M.; Lloyd, D.B.; Griffor, M.C.; Perry, D.A.; Ruggeri, R.B.; Clark, R.W.; Qiu, X. Crystal structures of cholesteryl ester transfer protein in complex with inhibitors. J. Biol. Chem. 2012, 287, 37321–37329. [Google Scholar] [CrossRef] [PubMed]
- Ford, J.; Lawson, M.; Fowler, D.; Maruyama, N.; Mito, S.; Tomiyasu, K.; Kinoshita, S.; Suzuki, C.; Kawaguchi, A.; Round, P.; et al. Tolerability, pharmacokinetics and pharmacodynamics of TA-8995, a selective cholesteryl ester transfer protein (CETP) inhibitor, in healthy subjects. Br. J. Clin. Pharmacol. 2014, 78, 498–508. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Ditmarsch, M.; Kastelein, J.J.; Rigby, S.P.; Kling, D.; Curcio, D.L.; Alp, N.J.; Davidson, M.H. Lipid lowering effects of the CETP inhibitor obicetrapib in combination with high-intensity statins: A randomized phase 2 trial. Nat. Med. 2022, 28, 1672–1678. [Google Scholar] [CrossRef]
- Chennamsetty, I.; Kostner, K.M.; Claudel, T.; Vinod, M.; Frank, S.; Weiss, T.S.; Trauner, M.; Kostner, G.M. Nicotinic acid inhibits hepatic APOA gene expression: Studies in humans and in transgenic mice. J. Lipid. Res. 2012, 53, 2405–2412. [Google Scholar] [CrossRef]
- Kamanna, V.S.; Kashyap, M.L. Mechanism of action of niacin. Am. J. Cardiol. 2008, 101, 20B–26B. [Google Scholar] [CrossRef]
- Croyal, M.; Ouguerram, K.; Passard, M.; Ferchaud-Roucher, V.; Chétiveaux, M.; Billon-Crossouard, S.; de Gouville, A.C.; Lambert, G.; Krempf, M.; Nobécourt, E. Effects of Extended-Release Nicotinic Acid on Apolipoprotein (a) Kinetics in Hypertriglyceridemic Patients. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 2042–2047. [Google Scholar] [CrossRef]
- Sahebkar, A.; Reiner, Ž.; Simental-Mendía, L.E.; Ferretti, G.; Cicero, A.F. Effect of extended-release niacin on plasma lipoprotein(a) levels: A systematic review and meta-analysis of randomized placebo-controlled trials. Metabolism 2016, 65, 1664–1678. [Google Scholar] [CrossRef]
- Boden, W.E.; Probstfield, J.L.; Anderson, T.; Chaitman, B.R.; Desvignes-Nickens, P.; Koprowicz, K.; McBride, R.; Teo, K.; Weintraub, W.; Investigators, A.-H. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N. Engl. J. Med. 2011, 365, 2255–2267. [Google Scholar] [CrossRef]
- Cenarro, A.; Puzo, J.; Ferrando, J.; Mateo-Gallego, R.; Bea, A.M.; Calmarza, P.; Jarauta, E.; Civeira, F. Effect of Nicotinic acid/Laropiprant in the lipoprotein(a) concentration with regard to baseline lipoprotein(a) concentration and LPA genotype. Metabolism 2014, 63, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Albers, J.J.; Slee, A.; O’Brien, K.D.; Robinson, J.G.; Kashyap, M.L.; Kwiterovich, P.O.; Xu, P.; Marcovina, S.M. Relationship of apolipoproteins A-1 and B, and lipoprotein(a) to cardiovascular outcomes: The AIM-HIGH trial (Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglyceride and Impact on Global Health Outcomes). J. Am. Coll. Cardiol. 2013, 62, 1575–1579. [Google Scholar] [CrossRef]
- Bruckert, E.; Labreuche, J.; Amarenco, P. Meta-analysis of the effect of nicotinic acid alone or in combination on cardiovascular events and atherosclerosis. Atherosclerosis 2010, 210, 353–361. [Google Scholar] [CrossRef]
- Akaike, M.; Azuma, H.; Kagawa, A.; Matsumoto, K.; Hayashi, I.; Tamura, K.; Nishiuchi, T.; Iuchi, T.; Takamori, N.; Aihara, K.; et al. Effect of aspirin treatment on serum concentrations of lipoprotein(a) in patients with atherosclerotic diseases. Clin. Chem. 2002, 48, 1454–1459. [Google Scholar] [CrossRef]
- Brunner, C.; Lobentanz, E.M.; Pethö-Schramm, A.; Ernst, A.; Kang, C.; Dieplinger, H.; Müller, H.J.; Utermann, G. The number of identical kringle IV repeats in apolipoprotein(a) affects its processing and secretion by HepG2 cells. J. Biol. Chem. 1996, 271, 32403–32410. [Google Scholar] [CrossRef] [PubMed]
- Azuma, H.; Yamaguchi, H.; Mima, N.; Shirakawa, M.; Miyamoto, K.; Kanagawa, Y.; Watanabe, A.; Shigekiyo, T.; Saito, S. An in vitro system for identifying agents capable of changing serum lipoprotein(a) concentration by regulating the transcriptional activity of the apolipoprotein(a) gene promoter. Biochem. Biophys. Res. Commun. 1996, 227, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Kagawa, A.; Azuma, H.; Akaike, M.; Kanagawa, Y.; Matsumoto, T. Aspirin reduces apolipoprotein(a) (apo(a)) production in human hepatocytes by suppression of apo(a) gene transcription. J. Biol. Chem. 1999, 274, 34111–34115. [Google Scholar] [CrossRef] [PubMed]
- Tsimikas, S. What Is the Role of Aspirin in Primary Prevention in Patients With Elevated Lp(a)? Cardiometab. Syndr. J. 2023, 3, 41–51. [Google Scholar] [CrossRef]
- Ridker, P.M.; Cook, N.R.; Lee, I.M.; Gordon, D.; Gaziano, J.M.; Manson, J.E.; Hennekens, C.H.; Buring, J.E. A randomized trial of low-dose aspirin in the primary prevention of cardiovascular disease in women. N. Engl. J. Med. 2005, 352, 1293–1304. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.; Ference, B.A.; Staley, J.R.; Freitag, D.F.; Mason, A.M.; Nielsen, S.F.; Willeit, P.; Young, R.; Surendran, P.; Karthikeyan, S.; et al. Association of LPA Variants With Risk of Coronary Disease and the Implications for Lipoprotein(a)-Lowering Therapies: A Mendelian Randomization Analysis. JAMA Cardiol. 2018, 3, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Milionis, H.J.; Efstathiadou, Z.; Tselepis, A.D.; Bairaktari, E.T.; Tsironis, L.D.; Tsatsoulis, A.; Elisaf, M.S. Lipoprotein (a) levels and apolipoprotein (a) isoform size in patients with subclinical hypothyroidism: Effect of treatment with levothyroxine. Thyroid 2003, 13, 365–369. [Google Scholar] [CrossRef]
- Ladenson, P.W.; Kristensen, J.D.; Ridgway, E.C.; Olsson, A.G.; Carlsson, B.; Klein, I.; Baxter, J.D.; Angelin, B. Use of the thyroid hormone analogue eprotirome in statin-treated dyslipidemia. N. Engl. J. Med. 2010, 362, 906–916. [Google Scholar] [CrossRef]
- Grover, G.J.; Egan, D.M.; Sleph, P.G.; Beehler, B.C.; Chiellini, G.; Nguyen, N.H.; Baxter, J.D.; Scanlan, T.S. Effects of the thyroid hormone receptor agonist GC-1 on metabolic rate and cholesterol in rats and primates: Selective actions relative to 3,5,3′-triiodo-L-thyronine. Endocrinology 2004, 145, 1656–1661. [Google Scholar] [CrossRef]
- Thompson, G.R.; Lowenthal, R.; Myant, N.B. Plasma exchange in the management of homozygous familial hypercholesterolæmia. Lancet 1975, 305, 1208–1211. [Google Scholar] [CrossRef] [PubMed]
- Julius, U.; Tselmin, S.; Schatz, U.; Fischer, S.; Birkenfeld, A.L.; Bornstein, S.R. Actual situation of lipoprotein apheresis in patients with elevated lipoprotein(a) levels. Atheroscler. Suppl. 2019, 40, 1–7. [Google Scholar] [CrossRef]
- Lamina, C.; Kronenberg, F.; The Lp(a)-GWAS-Consortium. Estimation of the Required Lipoprotein(a)-Lowering Therapeutic Effect Size for Reduction in Coronary Heart Disease Outcomes: A Mendelian Randomization Analysis. JAMA Cardiol. 2019, 4, 575–579. [Google Scholar] [CrossRef]
- Parish, S.; Hopewell, J.C.; Hill, M.R.; Marcovina, S.; Valdes-Marquez, E.; Haynes, R.; Offer, A.; Pedersen, T.R.; Baigent, C.; Collins, R.; et al. Impact of Apolipoprotein(a) Isoform Size on Lipoprotein(a) Lowering in the HPS2-THRIVE Study. Circ. Genom. Precis. Med. 2018, 11, e001696. [Google Scholar] [CrossRef]
- Hohenstein, B.; Julius, U.; Lansberg, P.; Jaeger, B.; Mellwig, K.P.; Weiss, N.; Graehlert, X.; Roeder, I.; Ramlow, W. Rationale and design of MultiSELECt: A European Multicenter Study on the Effect of Lipoprotein(a) Elimination by lipoprotein apheresis on Cardiovascular outcomes. Atheroscler. Suppl. 2017, 30, 180–186. [Google Scholar] [CrossRef]
- Korneva, V.A.; Kuznetsova, T.Y.; Julius, U. Modern Approaches to Lower Lipoprotein(a) Concentrations and Consequences for Cardiovascular Diseases. Biomedicines 2021, 9, 1271. [Google Scholar] [CrossRef]
- Thau, H.; Neuber, S.; Emmert, M.Y.; Nazari-Shafti, T.Z. Targeting Lipoprotein(a): Can RNA Therapeutics Provide the Next Step in the Prevention of Cardiovascular Disease? Cardiol. Ther. 2024, 13, 39–67. [Google Scholar] [CrossRef]
- Malick, W.A.; Goonewardena, S.N.; Koenig, W.; Rosenson, R.S. Clinical Trial Design for Lipoprotein(a)-Lowering Therapies: JACC Focus Seminar 2/3. J. Am. Coll. Cardiol. 2023, 81, 1633–1645. [Google Scholar] [CrossRef]
- Nissen, S.E.; Wolski, K.; Balog, C.; Swerdlow, D.I.; Scrimgeour, A.C.; Rambaran, C.; Wilson, R.J.; Boyce, M.; Ray, K.K.; Cho, L.; et al. Single Ascending Dose Study of a Short Interfering RNA Targeting Lipoprotein(a) Production in Individuals With Elevated Plasma Lipoprotein(a) Levels. JAMA 2022, 327, 1679–1687. [Google Scholar] [CrossRef]
- O’Donoghue, M.L.; G López, J.A.; Knusel, B.; Gencer, B.; Wang, H.; Wu, Y.; Kassahun, H.; Sabatine, M.S. Study design and rationale for the Olpasiran trials of Cardiovascular Events And lipoproteiN(a) reduction-DOSE finding study (OCEAN(a)-DOSE). Am. Heart. J. 2022, 251, 61–69. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, M.L.; Rosenson, R.S.; Gencer, B.; López, J.A.G.; Lepor, N.E.; Baum, S.J.; Stout, E.; Gaudet, D.; Knusel, B.; Kuder, J.F.; et al. Small Interfering RNA to Reduce Lipoprotein(a) in Cardiovascular Disease. N. Engl. J. Med. 2022, 387, 1855–1864. [Google Scholar] [CrossRef]
- Milosavljevic, M.N.; Stefanovic, S.M.; Pejcic, A.V. Potential Novel RNA-Targeting Agents for Effective Lipoprotein(a) Lowering: A Systematic Assessment of the Evidence From Completed and Ongoing Developmental Clinical Trials. J. Cardiovasc. Pharmacol. 2023, 82, 1–12. [Google Scholar] [CrossRef]
- Willeit, P.; Ridker, P.M.; Nestel, P.J.; Simes, J.; Tonkin, A.M.; Pedersen, T.R.; Schwartz, G.G.; Olsson, A.G.; Colhoun, H.M.; Kronenberg, F.; et al. Baseline and on-statin treatment lipoprotein(a) levels for prediction of cardiovascular events: Individual patient-data meta-analysis of statin outcome trials. Lancet 2018, 392, 1311–1320. [Google Scholar] [CrossRef]
- Ray, K.K.; Vallejo-Vaz, A.J.; Ginsberg, H.N.; Davidson, M.H.; Louie, M.J.; Bujas-Bobanovic, M.; Minini, P.; Eckel, R.H.; Cannon, C.P. Lipoprotein(a) reductions from PCSK9 inhibition and major adverse cardiovascular events: Pooled analysis of alirocumab phase 3 trials. Atherosclerosis 2019, 288, 194–202. [Google Scholar] [CrossRef]
- Toth, P.P.; Jones, S.R.; Monsalvo, M.L.; Elliott-Davey, M.; López, J.A.G.; Banach, M. Effect of Evolocumab on Non-High-Density Lipoprotein Cholesterol, Apolipoprotein B, and Lipoprotein(a): A Pooled Analysis of Phase 2 and Phase 3 Studies. J. Am. Heart. Assoc. 2020, 9, e014129. [Google Scholar] [CrossRef]
- Ray, K.K.; Kallend, D.; Koenig, W.; Leiter, L.; Raal, F.; Wright, S.; Wijngaard, P.; Kastelein, J.J.P. 6 monthly inclisiran and atherogenic lipoprotein reductions in Orion-11. J. Am. Coll. Cardiol. 2020, 75, 1853. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Ruotolo, G.; Brewer, H.B.; Wang, M.D.; Liu, L.; Willey, M.B.; Deeg, M.A.; Krueger, K.A.; Nissen, S.E. Evacetrapib alone or in combination with statins lowers lipoprotein(a) and total and small LDL particle concentrations in mildly hypercholesterolemic patients. J. Clin. Lipidol. 2016, 10, 519–527.e514. [Google Scholar] [CrossRef] [PubMed]
- Thomas, T.; Zhou, H.; Karmally, W.; Ramakrishnan, R.; Holleran, S.; Liu, Y.; Jumes, P.; Wagner, J.A.; Hubbard, B.; Previs, S.F.; et al. CETP (Cholesteryl Ester Transfer Protein) Inhibition With Anacetrapib Decreases Production of Lipoprotein(a) in Mildly Hypercholesterolemic Subjects. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1770–1775. [Google Scholar] [CrossRef] [PubMed]
- Tsimikas, S.; Karwatowska-Prokopczuk, E.; Gouni-Berthold, I.; Tardif, J.C.; Baum, S.J.; Steinhagen-Thiessen, E.; Shapiro, M.D.; Stroes, E.S.; Moriarty, P.M.; Nordestgaard, B.G.; et al. Lipoprotein(a) Reduction in Persons with Cardiovascular Disease. N. Engl. J. Med. 2020, 382, 244–255. [Google Scholar] [CrossRef] [PubMed]
- Khoshandam, M.; Soltaninejad, H.; Mousazadeh, M.; Hamidieh, A.A.; Hosseinkhani, S. Clinical applications of the CRISPR/Cas9 genome-editing system: Delivery options and challenges in precision medicine. Genes Dis. 2024, 11, 268–282. [Google Scholar] [CrossRef] [PubMed]
- Musunuru, K.; Chadwick, A.C.; Mizoguchi, T.; Garcia, S.P.; DeNizio, J.E.; Reiss, C.W.; Wang, K.; Iyer, S.; Dutta, C.; Clendaniel, V.; et al. In vivo CRISPR base editing of PCSK9 durably lowers cholesterol in primates. Nature 2021, 593, 429–434. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.G.; Mazzola, A.M.; Braun, M.C.; Platt, C.; Vafai, S.B.; Kathiresan, S.; Rohde, E.; Bellinger, A.M.; Khera, A.V. Efficacy and Safety of an Investigational Single-Course CRISPR Base-Editing Therapy Targeting. Circulation 2023, 147, 242–253. [Google Scholar] [CrossRef]
- Wulff, A.B.; Nordestgaard, B.G.; Langsted, A. Novel Therapies for Lipoprotein(a): Update in Cardiovascular Risk Estimation and Treatment. Curr. Atheroscler. Rep. 2024. [Google Scholar] [CrossRef]
- Nicholls, S.J.; Nissen, S.E.; Fleming, C.; Urva, S.; Suico, J.; Berg, P.H.; Linnebjerg, H.; Ruotolo, G.; Turner, P.K.; Michael, L.F. Muvalaplin, an Oral Small Molecule Inhibitor of Lipoprotein(a) Formation: A Randomized Clinical Trial. JAMA 2023, 330, 1042–1053. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Drugs | Effect on the Concentration of Lipoprotein(a) |
|---|---|
| Statins | −13% decrease (95% CI 10–15%) in the CARDS study [16]; −15% decrease (95% CI 13–17%) in the 4S study [17]; No significant effect [83] |
| Ezetimibe | No effect [22] |
| Niacin | −22.9% dose-independent decrease (95% CI 18.5–22.9%) [56] |
| PCSK9 inhibitors | −26.9% decrease in FOURIER study (evolocumab) [5] −25.6% decrease in all phase 3 studies (alirocumab) [84] These data are also confirmed in meta-analyses [85] |
| Inclisiran | −18.6% decrease in ORION-11 study [86] |
| Mipomersen | −26.4% in phase 3 studies [43] |
| CETP inhibitors | Up to −40% decrease in a phase 2 study (evacetrapib) [87]; −34.1% decrease in a phase 2 study (anacetrapib) [88]; −33.8% decrease (5 mg/day obicetrapib) and −56.5% decrease (10 mg/day obicetrapib) [52] |
| Lp(a) lowering ASO and siRNA formulations | −80% decrease (pelacarsen 20 mg/week) and −72% decrease (pelacarsen 60mg/month) [89]; Placebo-adjusted −70.5% decrease (olpasiran 10 mg/12 weeks); −97.4% decrease (olpasiran 225mg/12 weeks); −100.5% decrease (olpasiran 225 mg/24 weeks [80]; Up to 86–95% decrease (SLA340 middle dose) and −98% decrease (SLA340 high dose) [79] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paragh, G.; Zilahi, P.; Kolozsvári, L.R.; Lőrincz, H.; Fülöp, P.; Harangi, M. Novel Therapeutic Approaches for the Management of Elevated Lipoprotein(a): From Traditional Agents to Future Treatment Options. Life 2024, 14, 374. https://doi.org/10.3390/life14030374
Paragh G, Zilahi P, Kolozsvári LR, Lőrincz H, Fülöp P, Harangi M. Novel Therapeutic Approaches for the Management of Elevated Lipoprotein(a): From Traditional Agents to Future Treatment Options. Life. 2024; 14(3):374. https://doi.org/10.3390/life14030374
Chicago/Turabian StyleParagh, György, Péter Zilahi, László Róbert Kolozsvári, Hajnalka Lőrincz, Péter Fülöp, and Mariann Harangi. 2024. "Novel Therapeutic Approaches for the Management of Elevated Lipoprotein(a): From Traditional Agents to Future Treatment Options" Life 14, no. 3: 374. https://doi.org/10.3390/life14030374
APA StyleParagh, G., Zilahi, P., Kolozsvári, L. R., Lőrincz, H., Fülöp, P., & Harangi, M. (2024). Novel Therapeutic Approaches for the Management of Elevated Lipoprotein(a): From Traditional Agents to Future Treatment Options. Life, 14(3), 374. https://doi.org/10.3390/life14030374