

Platelet, Antiplatelet Therapy and Metabolic Dysfunction-Associated Steatotic Liver Disease: A Narrative Review

 ,
, 
 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Coagulation Cascade in Non-Alcoholic Fatty Liver Disease
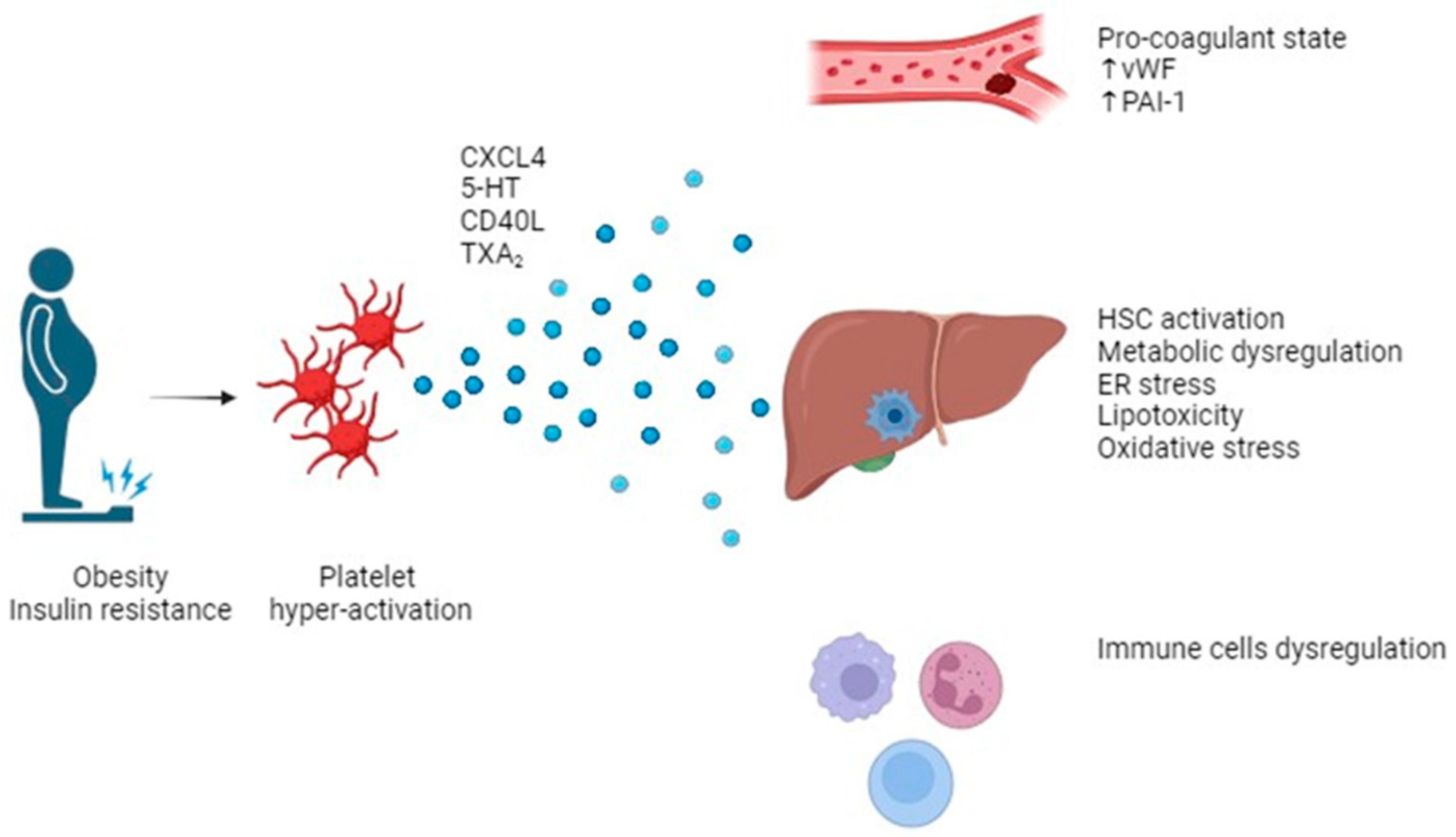
3. Role of Platelets in MASLD (NAFLD)
3.1. Platelets, Metabolism Dysregulation, and Liver Disease
3.2. Platelet & Liver
4. Platelet and Predictive Score of Liver Fibrosis
5. Antiplatelet Therapy Effects on Liver in MASLD (NAFLD) Patients
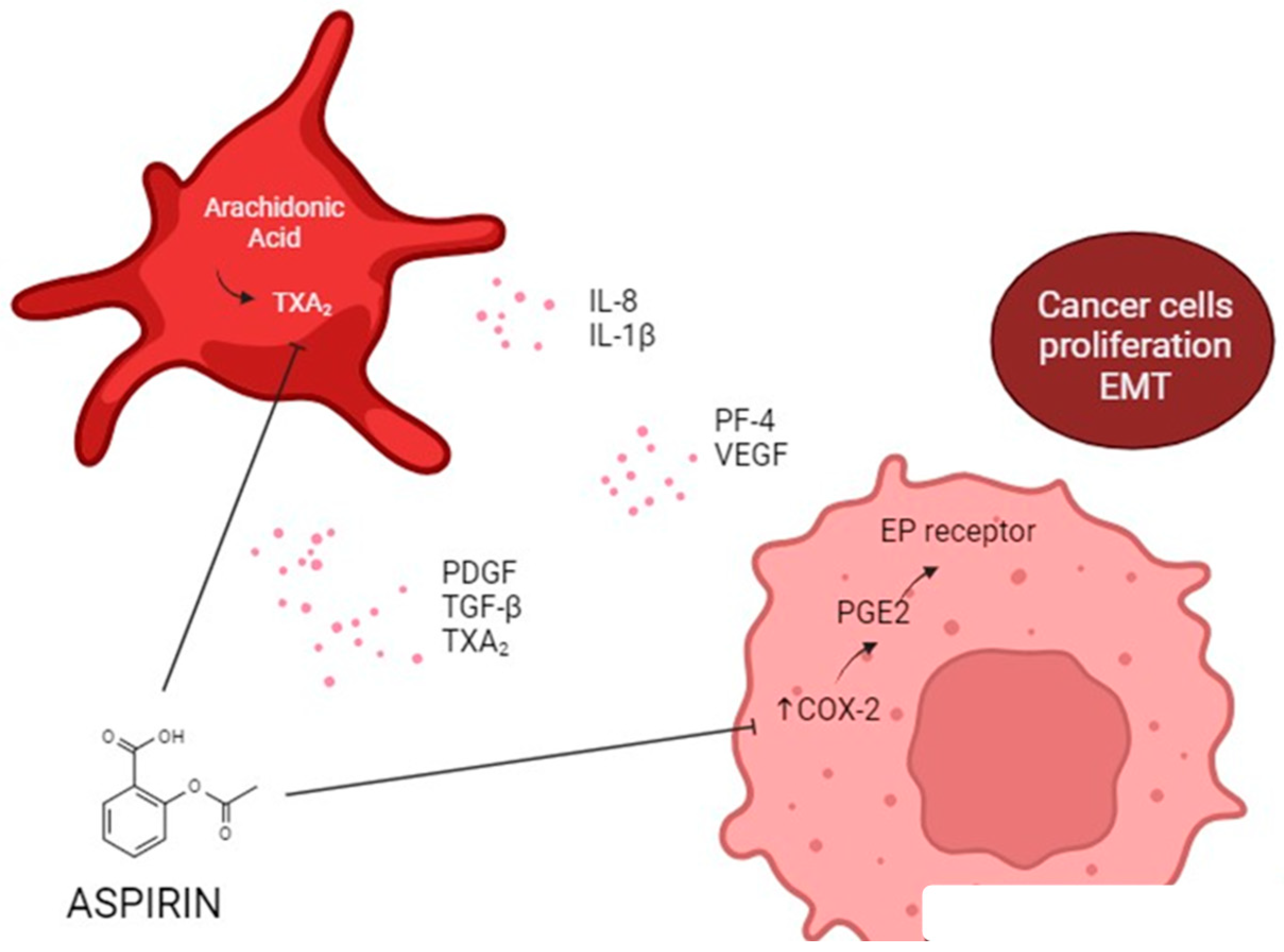
6. Antiplatelet Therapy and Cancer
Molecular Mechanisms
7. Platelets and Cardiovascular Risk in MASLD (NAFLD) Patients
8. Implications for Antiplatelet Therapy in Primary Prevention
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Grundy, S.M.; Cleeman, J.I.; Daniels, S.R.; Donato, K.A.; Eckel, R.H.; Franklin, B.A.; Gordon, D.J.; Krauss, R.M.; Savage, P.J.; Smith, S.C., Jr.; et al. Diagnosis and management of the metabolic syndrome: An American Heart Association/National Heart, Lung, and Blood Institute Scientific Statement. Circulation 2005, 112, 2735–2752. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.F.; Schattenberg, J.M.; et al. A new definition for metabolic dysfunction-associated fatty liver disease: An international expert consensus statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Rinella, M.E.; Lazarus, J.V.; Ratziu, V.; Francque, S.M.; Sanyal, A.J.; Kanwal, F.; Romero, D.; Abdelmalek, M.F.; Anstee, Q.M.; Arab, J.P.; et al. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. Hepatology 2023, 78, 1966–1986. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Petracca, G.; Beatrice, G.; Tilg, H.; Byrne, C.D.; Targher, G. Non-alcoholic fatty liver disease and risk of incident diabetes mellitus: An updated meta-analysis of 501 022 adult individuals. Gut 2021, 70, 962–969. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Liu, D.W.; Yan, H.Y.; Wang, Z.Y.; Zhao, S.H.; Wang, B. Obesity is an independent risk factor for non-alcoholic fatty liver disease: Evidence from a meta-analysis of 21 cohort studies. Obes. Rev. 2016, 17, 510–519. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Csermely, A.; Petracca, G.; Beatrice, G.; Corey, K.E.; Simon, T.G.; Byrne, C.D.; Targher, G. Non-alcoholic fatty liver disease and risk of fatal and non-fatal cardiovascular events: An updated systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 2021, 6, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.H.; Lee, H.A.; Kim, E.J.; Kim, H.Y.; Kim, H.C.; Ahn, S.H.; Lee, H.; Kim, S.U. Metabolic dysfunction-associated steatotic liver disease and risk of cardiovascular disease. Gut 2024, 73, 533–540. [Google Scholar] [CrossRef]
- Boccatonda, A.; Andreetto, L.; D’Ardes, D.; Cocco, G.; Rossi, I.; Vicari, S.; Schiavone, C.; Cipollone, F.; Guagnano, M.T. From NAFLD to MAFLD: Definition, Pathophysiological Basis and Cardiovascular Implications. Biomedicines 2023, 11, 883. [Google Scholar] [CrossRef]
- Targher, G.; Chonchol, M.; Miele, L.; Zoppini, G.; Pichiri, I.; Muggeo, M. Nonalcoholic fatty liver disease as a contributor to hypercoagulation and thrombophilia in the metabolic syndrome. Semin. Thromb. Hemost. 2009, 35, 277–287. [Google Scholar] [CrossRef]
- Northup, P.G.; Sundaram, V.; Fallon, M.B.; Reddy, K.R.; Balogun, R.A.; Sanyal, A.J.; Anstee, Q.M.; Hoffman, M.R.; Ikura, Y.; Caldwell, S.H. Hypercoagulation and thrombophilia in liver disease. J. Thromb. Haemost. 2008, 6, 2–9. [Google Scholar] [CrossRef]
- Northup, P.G.; Argo, C.K.; Shah, N.; Caldwell, S.H. Hypercoagulation and thrombophilia in nonalcoholic fatty liver disease: Mechanisms, human evidence, therapeutic implications, and preventive implications. Semin. Liver Dis. 2012, 32, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Wanless, I.R.; Wong, F.; Blendis, L.M.; Greig, P.; Heathcote, E.J.; Levy, G. Hepatic and portal vein thrombosis in cirrhosis: Possible role in development of parenchymal extinction and portal hypertension. Hepatology 1995, 21, 1238–1247. [Google Scholar] [PubMed]
- Anstee, Q.M.; Wright, M.; Goldin, R.; Thursz, M.R. Parenchymal extinction: Coagulation and hepatic fibrogenesis. Clin. Liver Dis. 2009, 13, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Lisman, T.; Kamphuisen, P.W.; Northup, P.G.; Porte, R.J. Established and new-generation antithrombotic drugs in patients with cirrhosis—Possibilities and caveats. J. Hepatol. 2013, 59, 358–366. [Google Scholar] [CrossRef] [PubMed]
- Kotronen, A.; Joutsi-Korhonen, L.; Sevastianova, K.; Bergholm, R.; Hakkarainen, A.; Pietiläinen, K.H.; Lundbom, N.; Rissanen, A.; Lassila, R.; Yki-Järvinen, H. Increased coagulation factor VIII, IX, XI and XII activities in non-alcoholic fatty liver disease. Liver Int. 2011, 31, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Verrijken, A.; Francque, S.; Mertens, I.; Prawitt, J.; Caron, S.; Hubens, G.; Van Marck, E.; Staels, B.; Michielsen, P.; Van Gaal, L. Prothrombotic factors in histologically proven nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology 2014, 59, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Tripodi, A.; Fracanzani, A.L.; Primignani, M.; Chantarangkul, V.; Clerici, M.; Mannucci, P.M.; Peyvandi, F.; Bertelli, C.; Valenti, L.; Fargion, S. Procoagulant imbalance in patients with non-alcoholic fatty liver disease. J. Hepatol. 2014, 61, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Lallukka, S.; Luukkonen, P.K.; Zhou, Y.; Isokuortti, E.; Leivonen, M.; Juuti, A.; Hakkarainen, A.; Orho-Melander, M.; Lundbom, N.; Olkkonen, V.M.; et al. Obesity/insulin resistance rather than liver fat increases coagulation factor activities and expression in humans. Thromb. Haemost. 2017, 117, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Potze, W.; Siddiqui, M.S.; Boyett, S.L.; Adelmeijer, J.; Daita, K.; Sanyal, A.J.; Lisman, T. Preserved hemostatic status in patients with non-alcoholic fatty liver disease. J. Hepatol. 2016, 65, 980–987. [Google Scholar] [CrossRef]
- Assy, N.; Bekirov, I.; Mejritsky, Y.; Solomon, L.; Szvalb, S.; Hussein, O. Association between thrombotic risk factors and extent of fibrosis in patients with non-alcoholic fatty liver diseases. World J. Gastroenterol. 2005, 11, 5834–5839. [Google Scholar] [CrossRef]
- Zanetto, A.; Campello, E.; Senzolo, M.; Simioni, P. The evolving knowledge on primary hemostasis in patients with cirrhosis: A comprehensive review. Hepatology 2024, 79, 460–481. [Google Scholar] [CrossRef]
- McCracken, E.; Monaghan, M.; Sreenivasan, S. Pathophysiology of the metabolic syndrome. Clin. Dermatol. 2018, 36, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Fricker, Z.P.; Pedley, A.; Massaro, J.M.; Vasan, R.S.; Hoffmann, U.; Benjamin, E.J.; Long, M.T. Liver Fat Is Associated with Markers of Inflammation and Oxidative Stress in Analysis of Data from the Framingham Heart Study. Clin. Gastroenterol. Hepatol. 2019, 17, 1157–1164.e4. [Google Scholar] [CrossRef] [PubMed]
- Santilli, F.; Blardi, P.; Scapellato, C.; Bocchia, M.; Guazzi, G.; Terzuoli, L.; Tabucchi, A.; Silvietti, A.; Lucani, B.; Gioffrè, W.R.; et al. Decreased plasma endogenous soluble RAGE, and enhanced adipokine secretion, oxidative stress and platelet/coagulative activation identify non-alcoholic fatty liver disease among patients with familial combined hyperlipidemia and/or metabolic syndrome. Vasc. Pharmacol. 2015, 72, 16–24. [Google Scholar] [CrossRef]
- Santilli, F.; Vazzana, N.; Bucciarelli, L.G.; Davì, G. Soluble forms of RAGE in human diseases: Clinical and therapeutical implications. Curr. Med. Chem. 2009, 16, 940–952. [Google Scholar] [CrossRef] [PubMed]
- Vazzana, N.; Santilli, F.; Cuccurullo, C.; Davì, G. Soluble forms of RAGE in internal medicine. Intern. Emerg. Med. 2009, 4, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Targher, G.; Bertolini, L.; Scala, L.; Zoppini, G.; Zenari, L.; Falezza, G. Non-alcoholic hepatic steatosis and its relation to increased plasma biomarkers of inflammation and endothelial dysfunction in non-diabetic men. Role of visceral adipose tissue. Diabet. Med. 2005, 22, 1354–1358. [Google Scholar] [CrossRef] [PubMed]
- Chávez-Tapia, N.C.; Rosso, N.; Uribe, M.; Bojalil, R.; Tiribelli, C. Kinetics of the inflammatory response induced by free fatty acid accumulation in hepatocytes. Ann. Hepatol. 2013, 13, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Chong, L.W.; Hsu, Y.C.; Lee, T.F.; Lin, Y.; Chiu, Y.T.; Yang, K.C.; Wu, J.C.; Huang, Y.T. Fluvastatin attenuates hepatic steatosis-induced fibrogenesis in rats through inhibiting paracrine effect of hepatocyte on hepatic stellate cells. BMC Gastroenterol. 2015, 15, 22. [Google Scholar] [CrossRef]
- Müller, C.; Gardemann, A.; Keilhoff, G.; Peter, D.; Wiswedel, I.; Schild, L. Prevention of free fatty acid-induced lipid accumulation, oxidative stress, and cell death in primary hepatocyte cultures by a Gynostemma pentaphyllum extract. Phytomedicine 2012, 19, 395–401. [Google Scholar] [CrossRef]
- Baker, R.G.; Hayden, M.S.; Ghosh, S. NF-κB, inflammation, and metabolic disease. Cell Metab. 2011, 13, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Yuan, M.; Frantz, D.F.; Melendez, P.A.; Hansen, L.; Lee, J.; Shoelson, S.E. Local and systemic insulin resistance resulting from hepatic activation of IKK-beta and NF-kappaB. Nat. Med. 2005, 11, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Metlakunta, A.; Dedousis, N.; Zhang, P.; Sipula, I.; Dube, J.J.; Scott, D.K.; O’Doherty, R.M. Depletion of liver Kupffer cells prevents the development of diet-induced hepatic steatosis and insulin resistance. Diabetes 2010, 59, 347–357. [Google Scholar] [CrossRef] [PubMed]
- Beavers, C.J.; Heron, P.; Smyth, S.S.; Bain, J.A.; Macaulay, T.E. Obesity and Antiplatelets-Does One Size Fit All? Thromb. Res. 2015, 136, 712–716. [Google Scholar] [CrossRef] [PubMed]
- Rotundo, L.; Persaud, A.; Feurdean, M.; Ahlawat, S.; Kim, H.S. The Association of leptin with severity of non-alcoholic fatty liver disease: A population-based study. Clin. Mol. Hepatol. 2018, 24, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Corsonello, A.; Perticone, F.; Malara, A.; De Domenico, D.; Loddo, S.; Buemi, M.; Ientile, R.; Corica, F. Leptin-dependent platelet aggregation in healthy, overweight and obese subjects. Int. J. Obes. Relat. Metab. Disord. 2003, 27, 566–573. [Google Scholar] [CrossRef]
- Wang, W.; Chen, J.; Mao, J.; Li, H.; Wang, M.; Zhang, H.; Li, H.; Chen, W. Genistein Ameliorates Non-alcoholic Fatty Liver Disease by Targeting the Thromboxane A(2) Pathway. J. Agric. Food Chem. 2018, 66, 5853–5859. [Google Scholar] [CrossRef]
- Russo, I.; Traversa, M.; Bonomo, K.; De Salve, A.; Mattiello, L.; Del Mese, P.; Doronzo, G.; Cavalot, F.; Trovati, M.; Anfossi, G. In central obesity, weight loss restores platelet sensitivity to nitric oxide and prostacyclin. Obesity 2010, 18, 788–797. [Google Scholar] [CrossRef] [PubMed]
- Grande, R.; Dovizio, M.; Marcone, S.; Szklanna, P.B.; Bruno, A.; Ebhardt, H.A.; Cassidy, H.; Ní Áinle, F.; Caprodossi, A.; Lanuti, P.; et al. Platelet-Derived Microparticles from Obese Individuals: Characterization of Number, Size, Proteomics, and Crosstalk with Cancer and Endothelial Cells. Front. Pharmacol. 2019, 10, 7. [Google Scholar] [CrossRef]
- Murakami, T.; Horigome, H.; Tanaka, K.; Nakata, Y.; Ohkawara, K.; Katayama, Y.; Matsui, A. Impact of weight reduction on production of platelet-derived microparticles and fibrinolytic parameters in obesity. Thromb. Res. 2007, 119, 45–53. [Google Scholar] [CrossRef]
- Burnouf, T.; Goubran, H.A.; Chou, M.L.; Devos, D.; Radosevic, M. Platelet microparticles: Detection and assessment of their paradoxical functional roles in disease and regenerative medicine. Blood Rev. 2014, 28, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Barry, O.P.; Pratico, D.; Lawson, J.A.; FitzGerald, G.A. Transcellular activation of platelets and endothelial cells by bioactive lipids in platelet microparticles. J. Clin. Investig. 1997, 99, 2118–2127. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.W.; Marlin, S.D.; Rothlein, R.; Toman, C.; Anderson, D.C. Cooperative interactions of LFA-1 and Mac-1 with intercellular adhesion molecule-1 in facilitating adherence and transendothelial migration of human neutrophils in vitro. J. Clin. Investig. 1989, 83, 2008–2017. [Google Scholar] [CrossRef] [PubMed]
- Forlow, S.B.; McEver, R.P.; Nollert, M.U. Leukocyte-leukocyte interactions mediated by platelet microparticles under flow. Blood 2000, 95, 1317–1323. [Google Scholar] [CrossRef] [PubMed]
- Cloutier, N.; Tan, S.; Boudreau, L.H.; Cramb, C.; Subbaiah, R.; Lahey, L.; Albert, A.; Shnayder, R.; Gobezie, R.; Nigrovic, P.A.; et al. The exposure of autoantigens by microparticles underlies the formation of potent inflammatory components: The microparticle-associated immune complexes. EMBO Mol. Med. 2013, 5, 235–249. [Google Scholar] [CrossRef]
- Braig, D.; Nero, T.L.; Koch, H.G.; Kaiser, B.; Wang, X.; Thiele, J.R.; Morton, C.J.; Zeller, J.; Kiefer, J.; Potempa, L.A.; et al. Transitional changes in the CRP structure lead to the exposure of proinflammatory binding sites. Nat. Commun. 2017, 8, 14188. [Google Scholar] [CrossRef]
- Barale, C.; Bonomo, K.; Frascaroli, C.; Morotti, A.; Guerrasio, A.; Cavalot, F.; Russo, I. Platelet function and activation markers in primary hypercholesterolemia treated with anti-PCSK9 monoclonal antibody: A 12-month follow-up. Nutr. Metab. Cardiovasc. Dis. 2020, 30, 282–291. [Google Scholar] [CrossRef]
- Yamazaki, M.; Uchiyama, S.; Xiong, Y.; Nakano, T.; Nakamura, T.; Iwata, M. Effect of remnant-like particle on shear-induced platelet activation and its inhibition by antiplatelet agents. Thromb. Res. 2005, 115, 211–218. [Google Scholar] [CrossRef]
- Gerrits, A.J.; Gitz, E.; Koekman, C.A.; Visseren, F.L.; van Haeften, T.W.; Akkerman, J.W. Induction of insulin resistance by the adipokines resistin, leptin, plasminogen activator inhibitor-1 and retinol binding protein 4 in human megakaryocytes. Haematologica 2012, 97, 1149–1157. [Google Scholar] [CrossRef]
- Simon, T.G.; Henson, J.; Osganian, S.; Masia, R.; Chan, A.T.; Chung, R.T.; Corey, K.E. Daily Aspirin Use Associated with Reduced Risk for Fibrosis Progression in Patients with Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2019, 17, 2776–2784.e4. [Google Scholar] [CrossRef]
- Davì, G.; Catalano, I.; Averna, M.; Notarbartolo, A.; Strano, A.; Ciabattoni, G.; Patrono, C. Thromboxane biosynthesis and platelet function in type II diabetes mellitus. N. Engl. J. Med. 1990, 322, 1769–1774. [Google Scholar] [CrossRef]
- Tang, W.H.; Stitham, J.; Gleim, S.; Di Febbo, C.; Porreca, E.; Fava, C.; Tacconelli, S.; Capone, M.; Evangelista, V.; Levantesi, G.; et al. Glucose and collagen regulate human platelet activity through aldose reductase induction of thromboxane. J. Clin. Investig. 2011, 121, 4462–4476. [Google Scholar] [CrossRef]
- Watala, C. Blood platelet reactivity and its pharmacological modulation in (people with) diabetes mellitus. Curr. Pharm. Des. 2005, 11, 2331–2365. [Google Scholar] [CrossRef] [PubMed]
- Anfossi, G.; Russo, I.; Massucco, P.; Mattiello, L.; Doronzo, G.; De Salve, A.; Trovati, M. Impaired synthesis and action of antiaggregating cyclic nucleotides in platelets from obese subjects: Possible role in platelet hyperactivation in obesity. Eur. J. Clin. Investig. 2004, 34, 482–489. [Google Scholar] [CrossRef]
- Santilli, F.; Marchisio, M.; Lanuti, P.; Boccatonda, A.; Miscia, S.; Davì, G. Microparticles as new markers of cardiovascular risk in diabetes and beyond. Thromb. Haemost. 2016, 116, 220–234. [Google Scholar] [CrossRef]
- Santilli, F.; Davì, G.; Consoli, A.; Cipollone, F.; Mezzetti, A.; Falco, A.; Taraborelli, T.; Devangelio, E.; Ciabattoni, G.; Basili, S.; et al. Thromboxane-dependent CD40 ligand release in type 2 diabetes mellitus. J. Am. Coll. Cardiol. 2006, 47, 391–397. [Google Scholar] [CrossRef]
- Sookoian, S.; Castaño, G.O.; Burgueño, A.L.; Rosselli, M.S.; Gianotti, T.F.; Mallardi, P.; Martino, J.S.; Pirola, C.J. Circulating levels and hepatic expression of molecular mediators of atherosclerosis in nonalcoholic fatty liver disease. Atherosclerosis 2010, 209, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Poggi, M.; Engel, D.; Christ, A.; Beckers, L.; Wijnands, E.; Boon, L.; Driessen, A.; Cleutjens, J.; Weber, C.; Gerdes, N.; et al. CD40L deficiency ameliorates adipose tissue inflammation and metabolic manifestations of obesity in mice. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2251–2260. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.; Stitham, J.; Jin, Y.; Liu, R.; Lee, S.H.; Du, J.; Atteya, G.; Gleim, S.; Spollett, G.; Martin, K.; et al. Aldose reductase-mediated phosphorylation of p53 leads to mitochondrial dysfunction and damage in diabetic platelets. Circulation 2014, 129, 1598–1609. [Google Scholar] [CrossRef]
- Dulai, P.S.; Singh, S.; Patel, J.; Soni, M.; Prokop, L.J.; Younossi, Z.; Sebastiani, G.; Ekstedt, M.; Hagstrom, H.; Nasr, P.; et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: Systematic review and meta-analysis. Hepatology 2017, 65, 1557–1565. [Google Scholar] [CrossRef]
- Malehmir, M.; Pfister, D.; Gallage, S.; Szydlowska, M.; Inverso, D.; Kotsiliti, E.; Leone, V.; Peiseler, M.; Surewaard, B.G.J.; Rath, D.; et al. Platelet GPIbα is a mediator and potential interventional target for NASH and subsequent liver cancer. Nat. Med. 2019, 25, 641–655. [Google Scholar] [CrossRef] [PubMed]
- Heijnen, H.; van der Sluijs, P. Platelet secretory behaviour: As diverse as the granules … or not? J. Thromb. Haemost. 2015, 13, 2141–2151. [Google Scholar] [CrossRef] [PubMed]
- Taus, F.; Meneguzzi, A.; Castelli, M.; Minuz, P. Platelet-Derived Extracellular Vesicles as Target of Antiplatelet Agents. What Is the Evidence? Front. Pharmacol. 2019, 10, 1256. [Google Scholar] [CrossRef] [PubMed]
- van der Meijden, P.E.J.; Heemskerk, J.W.M. Platelet biology and functions: New concepts and clinical perspectives. Nat. Rev. Cardiol. 2019, 16, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Xia, M.; Xue, Y.; Ma, F.; Cui, A.; Sun, Y.; Han, Y.; Xu, X.; Zhang, F.; Hu, Z.; et al. Thrombospondin 1 improves hepatic steatosis in diet-induced insulin-resistant mice and is associated with hepatic fat content in humans. EBioMedicine 2020, 57, 102849. [Google Scholar] [CrossRef] [PubMed]
- Grundy, S.M.; Cleeman, J.I.; Merz, C.N.; Brewer, H.B., Jr.; Clark, L.T.; Hunninghake, D.B.; Pasternak, R.C.; Smith, S.C., Jr.; Stone, N.J. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III guidelines. Circulation 2004, 110, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Werner, M.; Driftmann, S.; Kleinehr, K.; Kaiser, G.M.; Mathé, Z.; Treckmann, J.W.; Paul, A.; Skibbe, K.; Timm, J.; Canbay, A.; et al. All-In-One: Advanced preparation of Human Parenchymal and Non-Parenchymal Liver Cells. PLoS ONE 2015, 10, e0138655. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.J.; Adili, A.; Piotrowitz, K.; Abdullah, Z.; Boege, Y.; Stemmer, K.; Ringelhan, M.; Simonavicius, N.; Egger, M.; Wohlleber, D.; et al. Metabolic activation of intrahepatic CD8+ T cells and NKT cells causes nonalcoholic steatohepatitis and liver cancer via cross-talk with hepatocytes. Cancer Cell 2014, 26, 549–564. [Google Scholar] [CrossRef]
- Gawaz, M.; Langer, H.; May, A.E. Platelets in inflammation and atherogenesis. J. Clin. Investig. 2005, 115, 3378–3384. [Google Scholar] [CrossRef]
- Kirschbaum, M.; Karimian, G.; Adelmeijer, J.; Giepmans, B.N.; Porte, R.J.; Lisman, T. Horizontal RNA transfer mediates platelet-induced hepatocyte proliferation. Blood 2015, 126, 798–806. [Google Scholar] [CrossRef]
- Xu, Y.; Li, W.; Liang, G.; Peng, J.; Xu, X. Platelet microparticles-derived miR-25-3p promotes the hepatocyte proliferation and cell autophagy via reducing B-cell translocation gene 2. J. Cell. Biochem. 2020, 121, 4959–4973. [Google Scholar] [CrossRef]
- Kurokawa, T.; Ohkohchi, N. Platelets in liver disease, cancer and regeneration. World J. Gastroenterol. 2017, 23, 3228–3239. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, N.; Murata, S.; Maruyama, T.; Tamura, T.; Nozaki, R.; Kawasaki, T.; Fukunaga, K.; Oda, T.; Sasaki, R.; Homma, M.; et al. Platelet-derived adenosine 5’-triphosphate suppresses activation of human hepatic stellate cell: In vitro study. Hepatol. Res. 2012, 42, 91–102. [Google Scholar] [CrossRef]
- Salem, N.A.; Hamza, A.; Alnahdi, H.; Ayaz, N. Biochemical and Molecular Mechanisms of Platelet-Rich Plasma in Ameliorating Liver Fibrosis Induced by Dimethylnitrosurea. Cell Physiol. Biochem. 2018, 47, 2331–2339. [Google Scholar] [CrossRef]
- Zaldivar, M.M.; Pauels, K.; von Hundelshausen, P.; Berres, M.L.; Schmitz, P.; Bornemann, J.; Kowalska, M.A.; Gassler, N.; Streetz, K.L.; Weiskirchen, R.; et al. CXC chemokine ligand 4 (Cxcl4) is a platelet-derived mediator of experimental liver fibrosis. Hepatology 2010, 51, 1345–1353. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, N.I.; Messiha, B.A.S.; Salehc, I.G.; Abo-Saif, A.A.; Abdel-Bakky, M.S. Interruption of platelets and thrombin function as a new approach against liver fibrosis induced experimentally in rats. Life Sci. 2019, 231, 116522. [Google Scholar] [CrossRef]
- Kinnman, N.; Francoz, C.; Barbu, V.; Wendum, D.; Rey, C.; Hultcrantz, R.; Poupon, R.; Housset, C. The myofibroblastic conversion of peribiliary fibrogenic cells distinct from hepatic stellate cells is stimulated by platelet-derived growth factor during liver fibrogenesis. Lab. Investig. 2003, 83, 163–173. [Google Scholar] [CrossRef]
- Joshi, N.; Kopec, A.K.; Ray, J.L.; Cline-Fedewa, H.; Groeneveld, D.J.; Lisman, T.; Luyendyk, J.P. Von Willebrand factor deficiency reduces liver fibrosis in mice. Toxicol. Appl. Pharmacol. 2017, 328, 54–59. [Google Scholar] [CrossRef] [PubMed]
- Rohrbach, T.; Maceyka, M.; Spiegel, S. Sphingosine kinase and sphingosine-1-phosphate in liver pathobiology. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 543–553. [Google Scholar] [CrossRef]
- Ghafoory, S.; Varshney, R.; Robison, T.; Kouzbari, K.; Woolington, S.; Murphy, B.; Xia, L.; Ahamed, J. Platelet TGF-β1 deficiency decreases liver fibrosis in a mouse model of liver injury. Blood Adv. 2018, 2, 470–480. [Google Scholar] [CrossRef]
- Yoshida, S.; Ikenaga, N.; Liu, S.B.; Peng, Z.W.; Chung, J.; Sverdlov, D.Y.; Miyamoto, M.; Kim, Y.O.; Ogawa, S.; Arch, R.H.; et al. Extrahepatic platelet-derived growth factor-β, delivered by platelets, promotes activation of hepatic stellate cells and biliary fibrosis in mice. Gastroenterology 2014, 147, 1378–1392. [Google Scholar] [CrossRef] [PubMed]
- Miele, L.; Alberelli, M.A.; Martini, M.; Liguori, A.; Marrone, G.; Cocomazzi, A.; Vecchio, F.M.; Landolfi, R.; Gasbarrini, A.; Grieco, A.; et al. Nonalcoholic fatty liver disease (NAFLD) severity is associated to a nonhemostatic contribution and proinflammatory phenotype of platelets. Transl. Res. 2021, 231, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, A.; Adams, D.H.; Watson, S.P.; Lalor, P.F. Platelets: No longer bystanders in liver disease. Hepatology 2016, 64, 1774–1784. [Google Scholar] [CrossRef] [PubMed]
- Milovanovic Alempijevic, T.; Stojkovic Lalosevic, M.; Dumic, I.; Jocic, N.; Pavlovic Markovic, A.; Dragasevic, S.; Jovicic, I.; Lukic, S.; Popovic, D.; Milosavljevic, T. Diagnostic Accuracy of Platelet Count and Platelet Indices in Noninvasive Assessment of Fibrosis in Nonalcoholic Fatty Liver Disease Patients. Can. J. Gastroenterol. Hepatol. 2017, 2017, 6070135. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Zhao, C.; Shen, C.; Wang, Y. Cytokeratin 18, alanine aminotransferase, platelets and triglycerides predict the presence of nonalcoholic steatohepatitis. PLoS ONE 2013, 8, e82092. [Google Scholar] [CrossRef] [PubMed]
- Ozhan, H.; Aydin, M.; Yazici, M.; Yazgan, O.; Basar, C.; Gungor, A.; Onder, E. Mean platelet volume in patients with non-alcoholic fatty liver disease. Platelets 2010, 21, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Shin, W.Y.; Jung, D.H.; Shim, J.Y.; Lee, H.R. The association between non-alcoholic hepatic steatosis and mean platelet volume in an obese Korean population. Platelets 2011, 22, 442–446. [Google Scholar] [CrossRef] [PubMed]
- Madan, S.A.; John, F.; Pitchumoni, C.S. Nonalcoholic Fatty Liver Disease and Mean Platelet Volume: A Systemic Review and Meta-analysis. J. Clin. Gastroenterol. 2016, 50, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Potze, W.; Siddiqui, M.S.; Sanyal, A.J. Vascular Disease in Patients with Nonalcoholic Fatty Liver Disease. Semin. Thromb. Hemost. 2015, 41, 488–493. [Google Scholar] [CrossRef]
- Papanas, N.; Symeonidis, G.; Maltezos, E.; Mavridis, G.; Karavageli, E.; Vosnakidis, T.; Lakasas, G. Mean platelet volume in patients with type 2 diabetes mellitus. Platelets 2004, 15, 475–478. [Google Scholar] [CrossRef]
- Schmidt, K.G.; Rasmussen, J.W.; Bekker, C.; Madsen, P.E. Kinetics and in vivo distribution of 111-In-labelled autologous platelets in chronic hepatic disease: Mechanisms of thrombocytopenia. Scand. J. Haematol. 1985, 34, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, M.; Fujii, H.; Sumida, Y.; Hyogo, H.; Itoh, Y.; Ono, M.; Eguchi, Y.; Suzuki, Y.; Aoki, N.; Kanemasa, K.; et al. Platelet count for predicting fibrosis in nonalcoholic fatty liver disease. J. Gastroenterol. 2011, 46, 1300–1306. [Google Scholar] [CrossRef] [PubMed]
- Park, K.S.; Lee, Y.S.; Park, H.W.; Seo, S.H.; Jang, B.G.; Hwang, J.Y.; Cho, K.B.; Hwang, J.S.; Ahn, S.H.; Kang, Y.N.; et al. Factors associated or related to with pathological severity of nonalcoholic fatty liver disease. Korean J. Intern. Med. 2004, 19, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, H.; Hashimoto, E.; Yatsuji, S.; Tokushige, K.; Shiratori, K. Hyaluronic acid levels can predict severe fibrosis and platelet counts can predict cirrhosis in patients with nonalcoholic fatty liver disease. J. Gastroenterol. Hepatol. 2006, 21, 1459–1465. [Google Scholar] [CrossRef] [PubMed]
- Fierbinteanu-Braticevici, C.; Dina, I.; Petrisor, A.; Tribus, L.; Negreanu, L.; Carstoiu, C. Noninvasive investigations for non alcoholic fatty liver disease and liver fibrosis. World J. Gastroenterol. 2010, 16, 4784–4791. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Konishi, H.; Kishino, M.; Yatsuji, S.; Tokushige, K.; Hashimoto, E.; Shiratori, K. Prevalence of esophagogastric varices in patients with non-alcoholic steatohepatitis. Hepatol. Res. 2008, 38, 572–579. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.G.; Lydecker, A.; Murray, K.; Tetri, B.N.; Contos, M.J.; Sanyal, A.J. Comparison of noninvasive markers of fibrosis in patients with nonalcoholic fatty liver disease. Clin. Gastroenterol. Hepatol. 2009, 7, 1104–1112. [Google Scholar] [CrossRef]
- Duran-Bertran, J.; Rusu, E.C.; Barrientos-Riosalido, A.; Bertran, L.; Mahmoudian, R.; Aguilar, C.; Riesco, D.; Martínez, S.; Ugarte Chicote, J.; Sabench, F.; et al. Platelet-associated biomarkers in nonalcoholic steatohepatitis: Insights from a female cohort with obesity. Eur. J. Clin. Investig. 2024, 54, e14123. [Google Scholar] [CrossRef]
- Jialal, I.; Jialal, G.; Adams-Huet, B. The platelet to high density lipoprotein -cholesterol ratio is a valid biomarker of nascent metabolic syndrome. Diabetes Metab. Res. Rev. 2021, 37, e3403. [Google Scholar] [CrossRef]
- Calzadilla-Bertot, L.; Jeffrey, G.P.; Wang, Z.; Huang, Y.; Garas, G.; Wallace, M.; de Boer, B.; George, J.; Eslam, M.; Phu, A.; et al. Predicting liver-related events in NAFLD: A predictive model. Hepatology 2023, 78, 1240–1251. [Google Scholar] [CrossRef]
- Liu, Y.; Nong, L.; Jia, Y.; Tan, A.; Duan, L.; Lu, Y.; Zhao, J. Aspirin alleviates hepatic fibrosis by suppressing hepatic stellate cells activation via the TLR4/NF-κB pathway. Aging 2020, 12, 6058–6066. [Google Scholar] [CrossRef] [PubMed]
- Li, C.J.; Yang, Z.H.; Shi, X.L.; Liu, D.L. Effects of aspirin and enoxaparin in a rat model of liver fibrosis. World J. Gastroenterol. 2017, 23, 6412–6419. [Google Scholar] [CrossRef] [PubMed]
- Börgeson, E.; Johnson, A.M.; Lee, Y.S.; Till, A.; Syed, G.H.; Ali-Shah, S.T.; Guiry, P.J.; Dalli, J.; Colas, R.A.; Serhan, C.N.; et al. Lipoxin A4 Attenuates Obesity-Induced Adipose Inflammation and Associated Liver and Kidney Disease. Cell Metab. 2015, 22, 125–137. [Google Scholar] [CrossRef] [PubMed]
- Paik, Y.H.; Kim, J.K.; Lee, J.I.; Kang, S.H.; Kim, D.Y.; An, S.H.; Lee, S.J.; Lee, D.K.; Han, K.H.; Chon, C.Y.; et al. Celecoxib induces hepatic stellate cell apoptosis through inhibition of Akt activation and suppresses hepatic fibrosis in rats. Gut 2009, 58, 1517–1527. [Google Scholar] [CrossRef] [PubMed]
- Fujita, K.; Nozaki, Y.; Wada, K.; Yoneda, M.; Endo, H.; Takahashi, H.; Iwasaki, T.; Inamori, M.; Abe, Y.; Kobayashi, N.; et al. Effectiveness of antiplatelet drugs against experimental non-alcoholic fatty liver disease. Gut 2008, 57, 1583–1591. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.G.; Feldbrügge, L.; Tapper, E.B.; Popov, Y.; Ghaziani, T.; Afdhal, N.; Robson, S.C.; Mukamal, K.J. Aspirin use is associated with lower indices of liver fibrosis among adults in the United States. Aliment. Pharmacol. Ther. 2016, 43, 734–743. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Shahzad, G.; Jawairia, M.; Bostick, R.M.; Mustacchia, P. Association between aspirin use and the prevalence of nonalcoholic fatty liver disease: A cross-sectional study from the Third National Health and Nutrition Examination Survey. Aliment. Pharmacol. Ther. 2014, 40, 1066–1073. [Google Scholar] [CrossRef] [PubMed]
- Thongtan, T.; Deb, A.; Vutthikraivit, W.; Laoveeravat, P.; Mingbunjerdsuk, T.; Islam, S.; Islam, E. Antiplatelet therapy associated with lower prevalence of advanced liver fibrosis in non-alcoholic fatty liver disease: A systematic review and meta-analysis. Indian. J. Gastroenterol. 2022, 41, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Karahan, O.I.; Kurt, A.; Yikilmaz, A.; Kahriman, G. New method for the detection of intraperitoneal free air by sonography: Scissors maneuver. J. Clin. Ultrasound 2004, 32, 381–385. [Google Scholar] [CrossRef]
- Murohara, T.; Horowitz, J.R.; Silver, M.; Tsurumi, Y.; Chen, D.; Sullivan, A.; Isner, J.M. Vascular endothelial growth factor/vascular permeability factor enhances vascular permeability via nitric oxide and prostacyclin. Circulation 1998, 97, 99–107. [Google Scholar] [CrossRef]
- Sánchez de Miguel, L.; de Frutos, T.; González-Fernández, F.; del Pozo, V.; Lahoz, C.; Jiménez, A.; Rico, L.; García, R.; Aceituno, E.; Millás, I.; et al. Aspirin inhibits inducible nitric oxide synthase expression and tumour necrosis factor-alpha release by cultured smooth muscle cells. Eur. J. Clin. Investig. 1999, 29, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Campbell, J.S.; Hughes, S.D.; Gilbertson, D.G.; Palmer, T.E.; Holdren, M.S.; Haran, A.C.; Odell, M.M.; Bauer, R.L.; Ren, H.P.; Haugen, H.S.; et al. Platelet-derived growth factor C induces liver fibrosis, steatosis, and hepatocellular carcinoma. Proc. Natl. Acad. Sci. USA 2005, 102, 3389–3394. [Google Scholar] [CrossRef] [PubMed]
- Prattali, R.R.; Barreiro, G.C.; Caliseo, C.T.; Fugiwara, F.Y.; Ueno, M.; Prada, P.O.; Velloso, L.A.; Saad, M.J.; Carvalheira, J.B. Aspirin inhibits serine phosphorylation of insulin receptor substrate 1 in growth hormone treated animals. FEBS Lett. 2005, 579, 3152–3158. [Google Scholar] [CrossRef] [PubMed]
- Tarantino, G.; Caputi, A. JNKs, insulin resistance and inflammation: A possible link between NAFLD and coronary artery disease. World J. Gastroenterol. 2011, 17, 3785–3794. [Google Scholar] [CrossRef] [PubMed]
- Clària, J.; Planagumà, A. Liver: The formation and actions of aspirin-triggered lipoxins. Prostaglandins Leukot. Essent. Fatty Acids 2005, 73, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Gastaldelli, A.; Kozakova, M.; Højlund, K.; Flyvbjerg, A.; Favuzzi, A.; Mitrakou, A.; Balkau, B. Fatty liver is associated with insulin resistance, risk of coronary heart disease, and early atherosclerosis in a large European population. Hepatology 2009, 49, 1537–1544. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.M.; Lee, Y.J.; Jang, Y.N.; Kim, H.M.; Seo, H.S.; Jung, T.W.; Jeong, J.H. Aspirin Improves Nonalcoholic Fatty Liver Disease and Atherosclerosis through Regulation of the PPARδ-AMPK-PGC-1α Pathway in Dyslipidemic Conditions. Biomed Res. Int. 2020, 2020, 7806860. [Google Scholar] [CrossRef]
- Singal, A.G.; El-Serag, H.B. Hepatocellular Carcinoma from Epidemiology to Prevention: Translating Knowledge into Practice. Clin. Gastroenterol. Hepatol. 2015, 13, 2140–2151. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef]
- Kanwal, F.; Kramer, J.R.; Mapakshi, S.; Natarajan, Y.; Chayanupatkul, M.; Richardson, P.A.; Li, L.; Desiderio, R.; Thrift, A.P.; Asch, S.M.; et al. Risk of Hepatocellular Cancer in Patients with Non-Alcoholic Fatty Liver Disease. Gastroenterology 2018, 155, 1828–1837.e2. [Google Scholar] [CrossRef]
- Mittal, S.; Sada, Y.H.; El-Serag, H.B.; Kanwal, F.; Duan, Z.; Temple, S.; May, S.B.; Kramer, J.R.; Richardson, P.A.; Davila, J.A. Temporal trends of nonalcoholic fatty liver disease-related hepatocellular carcinoma in the veteran affairs population. Clin. Gastroenterol. Hepatol. 2015, 13, 594–601.e1. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Stepanova, M.; Ong, J.P.; Jacobson, I.M.; Bugianesi, E.; Duseja, A.; Eguchi, Y.; Wong, V.W.; Negro, F.; Yilmaz, Y.; et al. Nonalcoholic Steatohepatitis Is the Fastest Growing Cause of Hepatocellular Carcinoma in Liver Transplant Candidates. Clin. Gastroenterol. Hepatol. 2019, 17, 748–755.e3. [Google Scholar] [CrossRef] [PubMed]
- Haldar, D.; Kern, B.; Hodson, J.; Armstrong, M.J.; Adam, R.; Berlakovich, G.; Fritz, J.; Feurstein, B.; Popp, W.; Karam, V.; et al. Outcomes of liver transplantation for non-alcoholic steatohepatitis: A European Liver Transplant Registry study. J. Hepatol. 2019, 71, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Chung, G.E.; Lee, J.H.; Oh, S.; Nam, J.Y.; Chang, Y.; Cho, H.; Ahn, H.; Cho, Y.Y.; Yoo, J.J.; et al. Antiplatelet therapy and the risk of hepatocellular carcinoma in chronic hepatitis B patients on antiviral treatment. Hepatology 2017, 66, 1556–1569. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.Y.; Hsu, Y.C.; Tseng, H.C.; Yu, S.H.; Lin, J.T.; Wu, M.S.; Wu, C.Y. Association of Daily Aspirin Therapy with Risk of Hepatocellular Carcinoma in Patients with Chronic Hepatitis B. JAMA Intern. Med. 2019, 179, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Simon, T.G.; Duberg, A.S.; Aleman, S.; Chung, R.T.; Chan, A.T.; Ludvigsson, J.F. Association of Aspirin with Hepatocellular Carcinoma and Liver-Related Mortality. N. Engl. J. Med. 2020, 382, 1018–1028. [Google Scholar] [CrossRef] [PubMed]
- Santilli, F.; Boccatonda, A.; Davì, G. Aspirin, platelets, and cancer: The point of view of the internist. Eur. J. Intern. Med. 2016, 34, 11–20. [Google Scholar] [CrossRef]
- Lai, Q.; De Matthaeis, N.; Finotti, M.; Galati, G.; Marrone, G.; Melandro, F.; Morisco, F.; Nicolini, D.; Pravisani, R.; Giannini, E.G. The role of antiplatelet therapies on incidence and mortality of hepatocellular carcinoma. Eur. J. Clin. Investig. 2023, 53, e13870. [Google Scholar] [CrossRef]
- Sahasrabuddhe, V.V.; Gunja, M.Z.; Graubard, B.I.; Trabert, B.; Schwartz, L.M.; Park, Y.; Hollenbeck, A.R.; Freedman, N.D.; McGlynn, K.A. Nonsteroidal anti-inflammatory drug use, chronic liver disease, and hepatocellular carcinoma. J. Natl. Cancer Inst. 2012, 104, 1808–1814. [Google Scholar] [CrossRef]
- Lee, C.H.; Hsu, C.Y.; Yen, T.H.; Wu, T.H.; Yu, M.C.; Hsieh, S.Y. Daily Aspirin Reduced the Incidence of Hepatocellular Carcinoma and Overall Mortality in Patients with Cirrhosis. Cancers 2023, 15, 2946. [Google Scholar] [CrossRef]
- Choi, M.C.; Min, E.K.; Lee, J.G.; Joo, D.J.; Kim, M.S.; Kim, D.G. Antiplatelet Drugs on the Recurrence of Hepatocellular Carcinoma after Liver Transplantation. Cancers 2022, 14, 5329. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.Y.; Hsu, Y.C.; Ho, H.J.; Lin, J.T.; Chen, Y.J.; Wu, C.Y. Daily aspirin associated with a reduced risk of hepatocellular carcinoma in patients with non-alcoholic fatty liver disease: A population-based cohort study. EClinicalMedicine 2023, 61, 102065. [Google Scholar] [CrossRef] [PubMed]
- Tan, R.Z.H.; Lockart, I.; Abdel Shaheed, C.; Danta, M. Systematic review with meta-analysis: The effects of non-steroidal anti-inflammatory drugs and anti-platelet therapy on the incidence and recurrence of hepatocellular carcinoma. Aliment. Pharmacol. Ther. 2021, 54, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Patrignani, P.; Patrono, C. Aspirin, platelet inhibition and cancer prevention. Platelets 2018, 29, 779–785. [Google Scholar] [CrossRef] [PubMed]
- Lloyd, K.E.; Hall, L.H.; Ziegler, L.; Foy, R.; Green, S.M.C.; MacKenzie, M.; Taylor, D.G.; Smith, S.G. Acceptability of aspirin for cancer preventive therapy: A survey and qualitative study exploring the views of the UK general population. BMJ Open 2023, 13, e078703. [Google Scholar] [CrossRef] [PubMed]
- Patrono, C. Cyclooxygenase Inhibitors and Cancer: The Missing Pieces. J. Pharmacol. Exp. Ther. 2023, 386, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Santilli, F.; Boccatonda, A.; Davì, G.; Cipollone, F. The Coxib case: Are EP receptors really guilty? Atherosclerosis 2016, 249, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Joharatnam-Hogan, N.; Hatem, D.; Cafferty, F.H.; Petrucci, G.; Cameron, D.A.; Ring, A.; Kynaston, H.G.; Gilbert, D.C.; Wilson, R.H.; Hubner, R.A.; et al. Thromboxane biosynthesis in cancer patients and its inhibition by aspirin: A sub-study of the Add-Aspirin trial. Br. J. Cancer 2023, 129, 706–720. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Fu, Q.; Diggs, L.P.; McVey, J.C.; McCallen, J.; Wabitsch, S.; Ruf, B.; Brown, Z.; Heinrich, B.; Zhang, Q.; et al. Platelets control liver tumor growth through P2Y12-dependent CD40L release in NAFLD. Cancer Cell 2022, 40, 986–998.e5. [Google Scholar] [CrossRef]
- Iannacone, M.; Sitia, G.; Isogawa, M.; Marchese, P.; Castro, M.G.; Lowenstein, P.R.; Chisari, F.V.; Ruggeri, Z.M.; Guidotti, L.G. Platelets mediate cytotoxic T lymphocyte-induced liver damage. Nat. Med. 2005, 11, 1167–1169. [Google Scholar] [CrossRef]
- Elzey, B.D.; Tian, J.; Jensen, R.J.; Swanson, A.K.; Lees, J.R.; Lentz, S.R.; Stein, C.S.; Nieswandt, B.; Wang, Y.; Davidson, B.L.; et al. Platelet-mediated modulation of adaptive immunity. A communication link between innate and adaptive immune compartments. Immunity 2003, 19, 9–19. [Google Scholar] [CrossRef]
- Vonderheide, R.H. Prospect of targeting the CD40 pathway for cancer therapy. Clin. Cancer Res. 2007, 13, 1083–1088. [Google Scholar] [CrossRef] [PubMed]
- Rachidi, S.; Metelli, A.; Riesenberg, B.; Wu, B.X.; Nelson, M.H.; Wallace, C.; Paulos, C.M.; Rubinstein, M.P.; Garrett-Mayer, E.; Hennig, M.; et al. Platelets subvert T cell immunity against cancer via GARP-TGFβ axis. Sci. Immunol. 2017, 2, eaai7911. [Google Scholar] [CrossRef]
- Sutti, S.; Jindal, A.; Locatelli, I.; Vacchiano, M.; Gigliotti, L.; Bozzola, C.; Albano, E. Adaptive immune responses triggered by oxidative stress contribute to hepatic inflammation in NASH. Hepatology 2014, 59, 886–897. [Google Scholar] [CrossRef]
- Vogt, A.; Sadeghlar, F.; Ayub, T.H.; Schneider, C.; Möhring, C.; Zhou, T.; Mahn, R.; Bartels, A.; Praktiknjo, M.; Kornek, M.T.; et al. Alpha-Fetoprotein- and CD40Ligand-Expressing Dendritic Cells for Immunotherapy of Hepatocellular Carcinoma. Cancers 2021, 13, 3375. [Google Scholar] [CrossRef] [PubMed]
- Elgueta, R.; Benson, M.J.; de Vries, V.C.; Wasiuk, A.; Guo, Y.; Noelle, R.J. Molecular mechanism and function of CD40/CD40L engagement in the immune system. Immunol. Rev. 2009, 229, 152–172. [Google Scholar] [CrossRef]
- Handelsman, Y.; Anderson, J.E.; Bakris, G.L.; Ballantyne, C.M.; Beckman, J.A.; Bhatt, D.L.; Bloomgarden, Z.T.; Bozkurt, B.; Budoff, M.J.; Butler, J.; et al. DCRM Multispecialty Practice Recommendations for the management of diabetes, cardiorenal, and metabolic diseases. J. Diabetes Complicat. 2022, 36, 108101. [Google Scholar] [CrossRef] [PubMed]
- Hermann, A.; Rauch, B.H.; Braun, M.; Schrör, K.; Weber, A.A. Platelet CD40 ligand (CD40L)--subcellular localization, regulation of expression, and inhibition by clopidogrel. Platelets 2001, 12, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Deczkowska, A.; David, E.; Ramadori, P.; Pfister, D.; Safran, M.; Li, B.; Giladi, A.; Jaitin, D.A.; Barboy, O.; Cohen, M.; et al. XCR1(+) type 1 conventional dendritic cells drive liver pathology in non-alcoholic steatohepatitis. Nat. Med. 2021, 27, 1043–1054. [Google Scholar] [CrossRef]
- Ezzaty Mirhashemi, M.; Shah, R.V.; Kitchen, R.R.; Rong, J.; Spahillari, A.; Pico, A.R.; Vitseva, O.; Levy, D.; Demarco, D.; Shah, S.; et al. The Dynamic Platelet Transcriptome in Obesity and Weight Loss. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 854–864. [Google Scholar] [CrossRef]
- Shalapour, S.; Lin, X.J.; Bastian, I.N.; Brain, J.; Burt, A.D.; Aksenov, A.A.; Vrbanac, A.F.; Li, W.; Perkins, A.; Matsutani, T.; et al. Inflammation-induced IgA+ cells dismantle anti-liver cancer immunity. Nature 2017, 551, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Spinosa, M.; Stine, J.G. Nonalcoholic Fatty Liver Disease-Evidence for a Thrombophilic State? Curr. Pharm. Des. 2020, 26, 1036–1044. [Google Scholar] [CrossRef] [PubMed]
- Byrne, C.D.; Targher, G. Non-alcoholic fatty liver disease-related risk of cardiovascular disease and other cardiac complications. Diabetes Obes. Metab. 2022, 24, 28–43. [Google Scholar] [CrossRef] [PubMed]
- Heffron, S.P.; Marier, C.; Parikh, M.; Fisher, E.A.; Berger, J.S. Severe obesity and bariatric surgery alter the platelet mRNA profile. Platelets 2019, 30, 967–974. [Google Scholar] [CrossRef]
- Chu, S.G.; Becker, R.C.; Berger, P.B.; Bhatt, D.L.; Eikelboom, J.W.; Konkle, B.; Mohler, E.R.; Reilly, M.P.; Berger, J.S. Mean platelet volume as a predictor of cardiovascular risk: A systematic review and meta-analysis. J. Thromb. Haemost. 2010, 8, 148–156. [Google Scholar] [CrossRef] [PubMed]
- Alkhouri, N.; Kistangari, G.; Campbell, C.; Lopez, R.; Zein, N.N.; Feldstein, A.E. Mean platelet volume as a marker of increased cardiovascular risk in patients with nonalcoholic steatohepatitis. Hepatology 2012, 55, 331. [Google Scholar] [CrossRef]
- Abeles, R.D.; Mullish, B.H.; Forlano, R.; Kimhofer, T.; Adler, M.; Tzallas, A.; Giannakeas, N.; Yee, M.; Mayet, J.; Goldin, R.D.; et al. Derivation and validation of a cardiovascular risk score for prediction of major acute cardiovascular events in non-alcoholic fatty liver disease; the importance of an elevated mean platelet volume. Aliment. Pharmacol. Ther. 2019, 49, 1077–1085. [Google Scholar] [CrossRef]
- Kilciler, G.; Genc, H.; Tapan, S.; Ors, F.; Kara, M.; Karadurmus, N.; Ercin, C.N.; Karslioglu, Y.; Kilic, S.; Bagci, S.; et al. Mean platelet volume and its relationship with carotid atherosclerosis in subjects with non-alcoholic fatty liver disease. Upsala J. Med. Sci. 2010, 115, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Sert, A.; Pirgon, O.; Aypar, E.; Yılmaz, H.; Dündar, B. Relationship between aspartate aminotransferase-to-platelet ratio index and carotid intima-media thickness in obese adolescents with non-alcoholic fatty liver disease. J. Clin. Res. Pediatr. Endocrinol. 2013, 5, 182–188. [Google Scholar] [CrossRef]
- Mechanick, J.I.; Apovian, C.; Brethauer, S.; Timothy Garvey, W.; Joffe, A.M.; Kim, J.; Kushner, R.F.; Lindquist, R.; Pessah-Pollack, R.; Seger, J.; et al. Clinical Practice Guidelines for the Perioperative Nutrition, Metabolic, and Nonsurgical Support of Patients Undergoing Bariatric Procedures—2019 Update: Cosponsored by American Association of Clinical Endocrinologists/American College of Endocrinology, The Obesity Society, American Society for Metabolic and Bariatric Surgery, Obesity Medicine Association, and American Society of Anesthesiologists. Obesity 2020, 28, O1–O58. [Google Scholar] [CrossRef]
- Kokoska, L.A.; Wilhelm, S.M.; Garwood, C.L.; Berlie, H.D. Aspirin for primary prevention of cardiovascular disease in patients with diabetes: A meta-analysis. Diabetes Res. Clin. Pract. 2016, 120, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Sun, A.; Zhang, P.; Wu, C.; Zhang, S.; Fu, M.; Wang, K.; Zou, Y.; Ge, J. Aspirin for primary prevention of cardiovascular events in patients with diabetes: A meta-analysis. Diabetes Res. Clin. Pract. 2010, 87, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Shan, Z.; Zhang, Y.; Chen, S.; Yang, W.; Bao, W.; Rong, Y.; Yu, X.; Hu, F.B.; Liu, L. Aspirin for primary prevention of cardiovascular events: Meta-analysis of randomized controlled trials and subgroup analysis by sex and diabetes status. PLoS ONE 2014, 9, e90286. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Lu, Y.; Ma, D.L.; Du, T.T.; Shao, S.Y.; Yu, X.F. A meta-analysis of salicylates for type 2 diabetes mellitus. J. Huazhong Univ. Sci. Technol. Med. Sci. 2013, 33, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Ferreiro, J.L.; Angiolillo, D.J. Challenges and perspectives of antiplatelet therapy in patients with diabetes mellitus and coronary artery disease. Curr. Pharm. Des. 2012, 18, 5273–5293. [Google Scholar] [CrossRef] [PubMed]
- Bundhun, P.K.; Qin, T.; Chen, M.H. Comparing the effectiveness and safety between triple antiplatelet therapy and dual antiplatelet therapy in type 2 diabetes mellitus patients after coronary stents implantation: A systematic review and meta-analysis of randomized controlled trials. BMC Cardiovasc. Disord. 2015, 15, 118. [Google Scholar] [CrossRef] [PubMed]
- Niu, P.P.; Guo, Z.N.; Jin, H.; Xing, Y.Q.; Yang, Y. Antiplatelet regimens in the long-term secondary prevention of transient ischaemic attack and ischaemic stroke: An updated network meta-analysis. BMJ Open 2016, 6, e009013. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Meng, H.; Xu, L.; Liu, J.; Kong, D.; Chen, P.; Gong, X.; Bai, J.; Zou, F.; Yang, Z.; et al. Efficacy and safety of cilostazol based triple antiplatelet treatment versus dual antiplatelet treatment in patients undergoing coronary stent implantation: An updated meta-analysis of the randomized controlled trials. J. Thromb. Thrombolysis 2015, 39, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Dindyal, S.; Kyriakides, C. A review of cilostazol, a phosphodiesterase inhibitor, and its role in preventing both coronary and peripheral arterial restenosis following endovascular therapy. Recent. Pat. Cardiovasc. Drug Discov. 2009, 4, 6–14. [Google Scholar] [CrossRef]
- Tang, W.H.; Lin, F.H.; Lee, C.H.; Kuo, F.C.; Hsieh, C.H.; Hsiao, F.C.; Hung, Y.J. Cilostazol effectively attenuates deterioration of albuminuria in patients with type 2 diabetes: A randomized, placebo-controlled trial. Endocrine 2014, 45, 293–301. [Google Scholar] [CrossRef]
- Gaziano, J.M.; Brotons, C.; Coppolecchia, R.; Cricelli, C.; Darius, H.; Gorelick, P.B.; Howard, G.; Pearson, T.A.; Rothwell, P.M.; Ruilope, L.M.; et al. Use of aspirin to reduce risk of initial vascular events in patients at moderate risk of cardiovascular disease (ARRIVE): A randomised, double-blind, placebo-controlled trial. Lancet 2018, 392, 1036–1046. [Google Scholar] [CrossRef] [PubMed]
- Bowman, L.; Mafham, M.; Wallendszus, K.; Stevens, W.; Buck, G.; Barton, J.; Murphy, K.; Aung, T.; Haynes, R.; Cox, J.; et al. Effects of Aspirin for Primary Prevention in Persons with Diabetes Mellitus. N. Engl. J. Med. 2018, 379, 1529–1539. [Google Scholar] [CrossRef] [PubMed]
- McNeil, J.J.; Nelson, M.R.; Woods, R.L.; Lockery, J.E.; Wolfe, R.; Reid, C.M.; Kirpach, B.; Shah, R.C.; Ives, D.G.; Storey, E.; et al. Effect of Aspirin on All-Cause Mortality in the Healthy Elderly. N. Engl. J. Med. 2018, 379, 1519–1528. [Google Scholar] [CrossRef]
- Santilli, F.; Simeone, P. Aspirin in primary prevention: The triumph of clinical judgement over complex equations. Intern. Emerg. Med. 2019, 14, 1217–1231. [Google Scholar] [CrossRef] [PubMed]
- Li, X.Y.; Li, L.; Na, S.H.; Santilli, F.; Shi, Z.; Blaha, M. Implications of the heterogeneity between guideline recommendations for the use of low dose aspirin in primary prevention of cardiovascular disease. Am. J. Prev. Cardiol. 2022, 11, 100363. [Google Scholar] [CrossRef] [PubMed]
- Merat, S.; Jafari, E.; Radmard, A.R.; Khoshnia, M.; Sharafkhah, M.; Nateghi Baygi, A.; Marshall, T.; Shiravi Khuzani, A.; Cheng, K.K.; Poustchi, H.; et al. Polypill for prevention of cardiovascular diseases with focus on non-alcoholic steatohepatitis: The PolyIran-Liver trial. Eur. Heart J. 2022, 43, 2023–2033. [Google Scholar] [CrossRef] [PubMed]
- Santilli, F.; Rocca, B.; De Cristofaro, R.; Lattanzio, S.; Pietrangelo, L.; Habib, A.; Pettinella, C.; Recchiuti, A.; Ferrante, E.; Ciabattoni, G.; et al. Platelet cyclooxygenase inhibition by low-dose aspirin is not reflected consistently by platelet function assays: Implications for aspirin “resistance”. J. Am. Coll. Cardiol. 2009, 53, 667–677. [Google Scholar] [CrossRef]
- Rocca, B.; Santilli, F.; Pitocco, D.; Mucci, L.; Petrucci, G.; Vitacolonna, E.; Lattanzio, S.; Mattoscio, D.; Zaccardi, F.; Liani, R.; et al. The recovery of platelet cyclooxygenase activity explains interindividual variability in responsiveness to low-dose aspirin in patients with and without diabetes. J. Thromb. Haemost. 2012, 10, 1220–1230. [Google Scholar] [CrossRef]
- Simeone, P.; Liani, R.; Tripaldi, R.; Ciotti, S.; Recchiuti, A.; Abbonante, V.; Porro, B.; Del Boccio, P.; Di Castelnuovo, A.; Lanuti, P.; et al. Reduced platelet glycoprotein Ibα shedding accelerates thrombopoiesis and COX-1 recovery: Implications for aspirin dosing regimen. Haematologica 2023, 108, 1141–1157. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| CD40 Pathway Anti-Tumor Activity | CD40 Pathway Pro-Tumor Activity |
|---|---|
| CD40L is related to a strong CD8+ T cells responses through CD40 licensing of dendritic cell [61,122,124,136,137,138,144]. | Platelet-derived CD40L is greater released in both NAFLD mouse models and patients with NASH [139] |
| Co-stimulation with CD40L-expressing dendritic cells (DC) significantly improves vaccination by inducing an early and strong Th1-shift in the tumor environment as well as higher tumor apoptosis [145]. | In NAFLD patients CD40L protein production is induced in megakaryocytes rather than NAFLD or tumors causing a transfer of CD40L pre-mRNA or mRNA into platelets [144]. |
| Platelet-derived CD40L can increase CD8+ T cell activation and their recruitment to liver in NAFLD [61,139,146,147] | IL-12 dependent increase in CD40L production in bone marrow megakaryocytes in NAFLD models [139] |
| Megakaryocytes harbored in lung can produce higher CD40L amount [139,147] | |
| Inhibition of the P2Y12 receptor on platelets can promote tumor growth via CD40L in mice with NAFLD [139] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boccatonda, A.; Del Cane, L.; Marola, L.; D’Ardes, D.; Lessiani, G.; di Gregorio, N.; Ferri, C.; Cipollone, F.; Serra, C.; Santilli, F.; et al. Platelet, Antiplatelet Therapy and Metabolic Dysfunction-Associated Steatotic Liver Disease: A Narrative Review. Life 2024, 14, 473. https://doi.org/10.3390/life14040473
Boccatonda A, Del Cane L, Marola L, D’Ardes D, Lessiani G, di Gregorio N, Ferri C, Cipollone F, Serra C, Santilli F, et al. Platelet, Antiplatelet Therapy and Metabolic Dysfunction-Associated Steatotic Liver Disease: A Narrative Review. Life. 2024; 14(4):473. https://doi.org/10.3390/life14040473
Chicago/Turabian StyleBoccatonda, Andrea, Lorenza Del Cane, Lara Marola, Damiano D’Ardes, Gianfranco Lessiani, Nicoletta di Gregorio, Claudio Ferri, Francesco Cipollone, Carla Serra, Francesca Santilli, and et al. 2024. "Platelet, Antiplatelet Therapy and Metabolic Dysfunction-Associated Steatotic Liver Disease: A Narrative Review" Life 14, no. 4: 473. https://doi.org/10.3390/life14040473
APA StyleBoccatonda, A., Del Cane, L., Marola, L., D’Ardes, D., Lessiani, G., di Gregorio, N., Ferri, C., Cipollone, F., Serra, C., Santilli, F., & Piscaglia, F. (2024). Platelet, Antiplatelet Therapy and Metabolic Dysfunction-Associated Steatotic Liver Disease: A Narrative Review. Life, 14(4), 473. https://doi.org/10.3390/life14040473