
Unveiling Gene Expression Dynamics during Early Embryogenesis in Cynoglossus semilaevis: A Transcriptomic Perspective

 , ,
, , {kind=link}
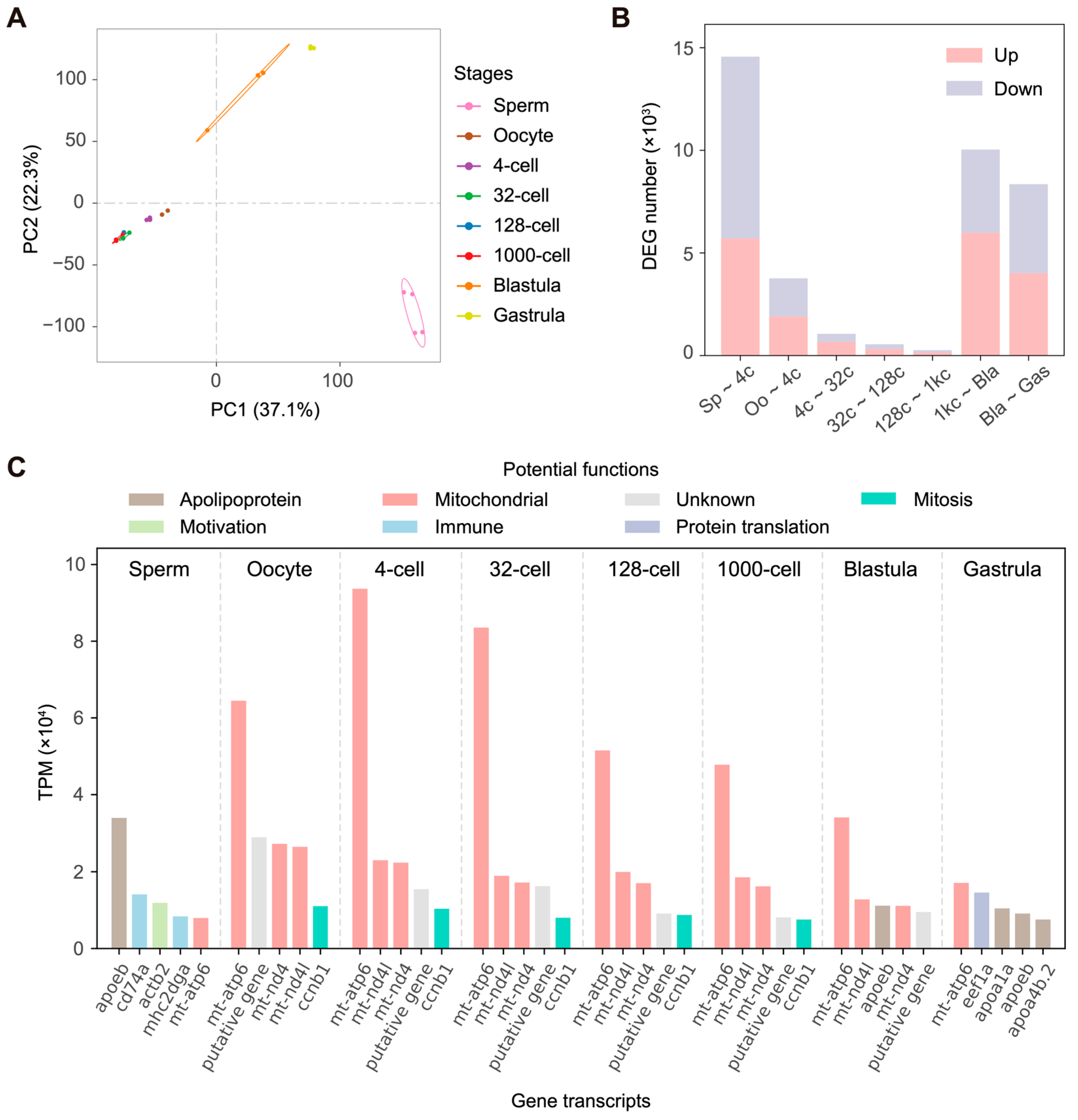{kind=link}
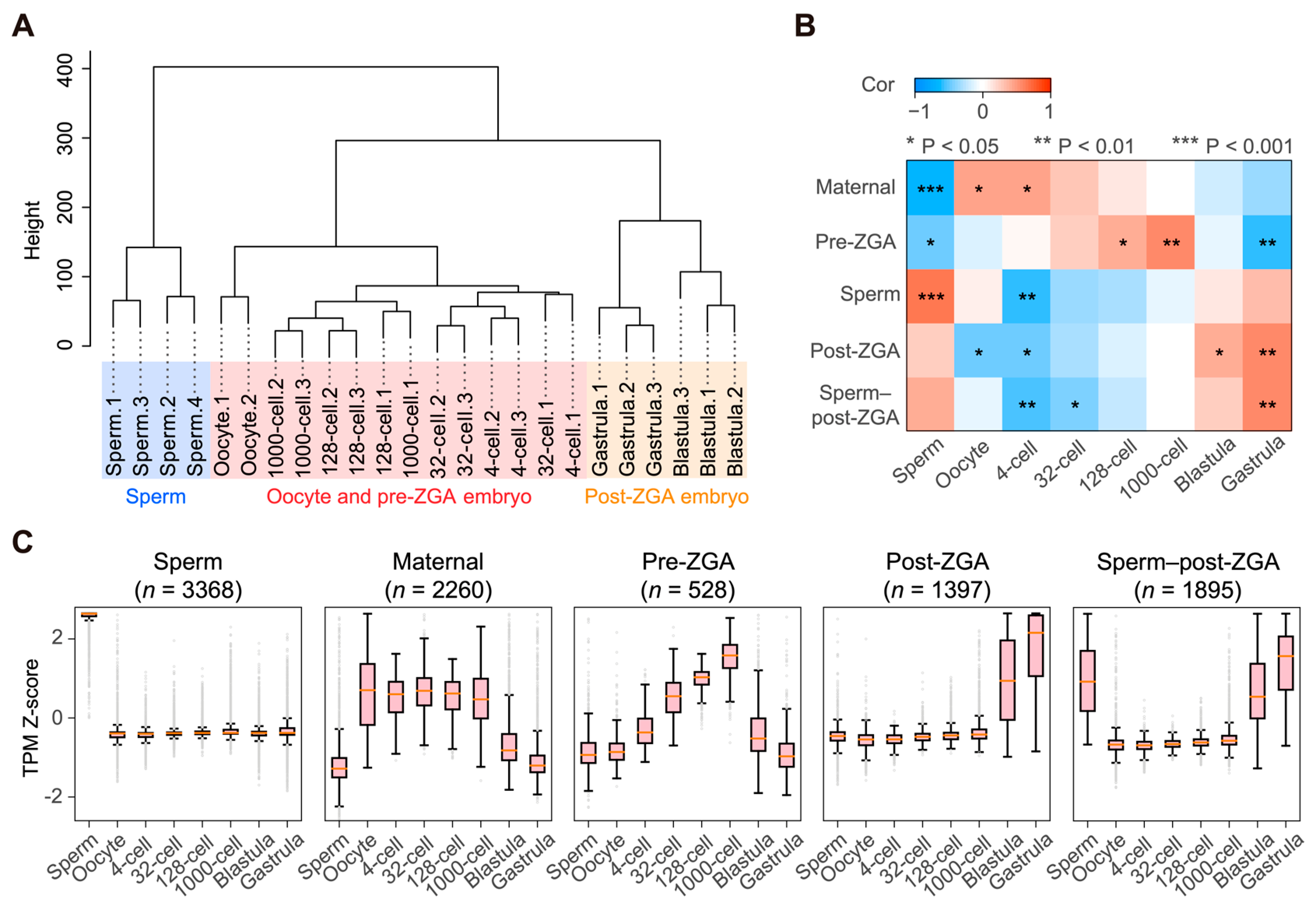{kind=link}
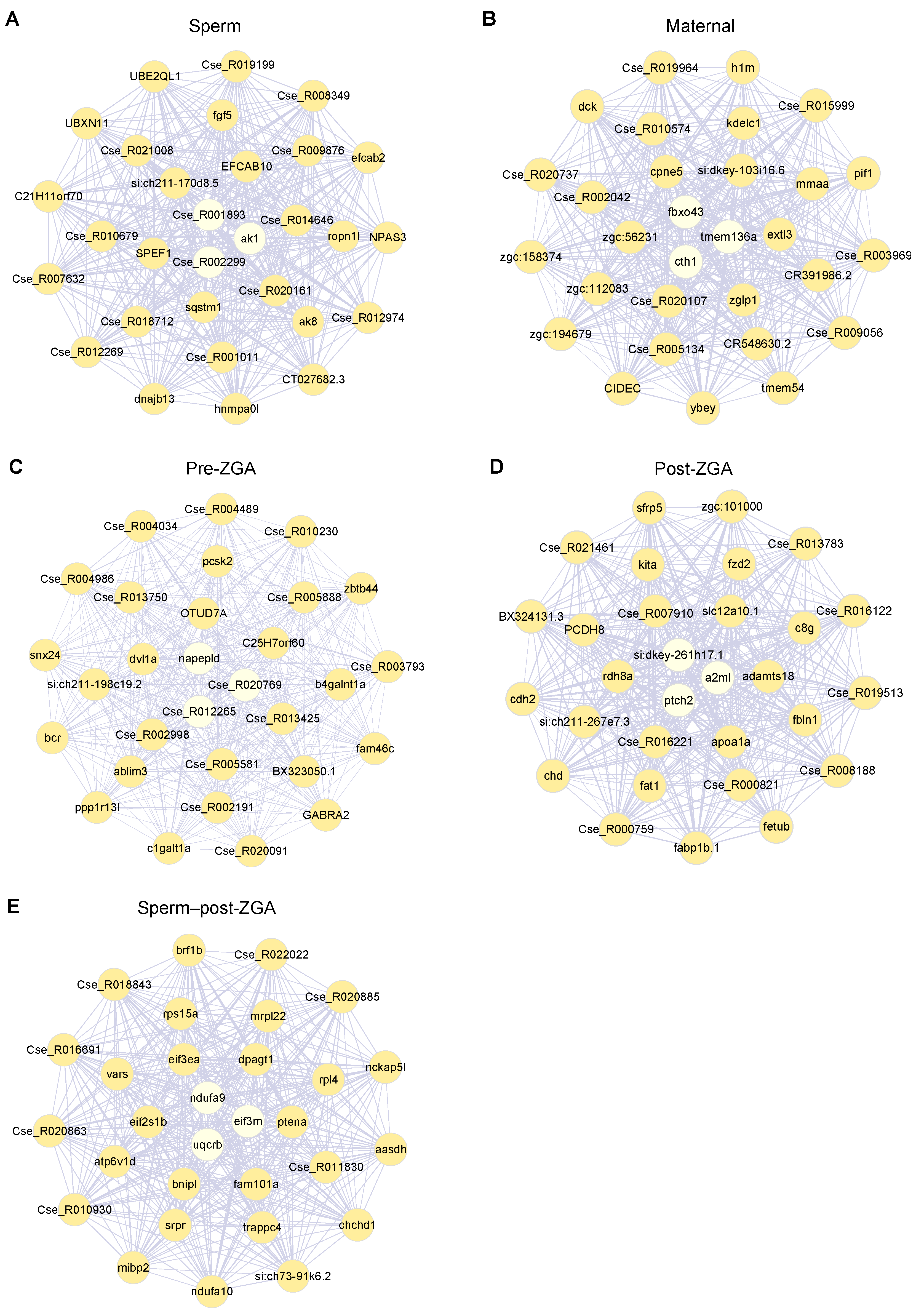{kind=link}
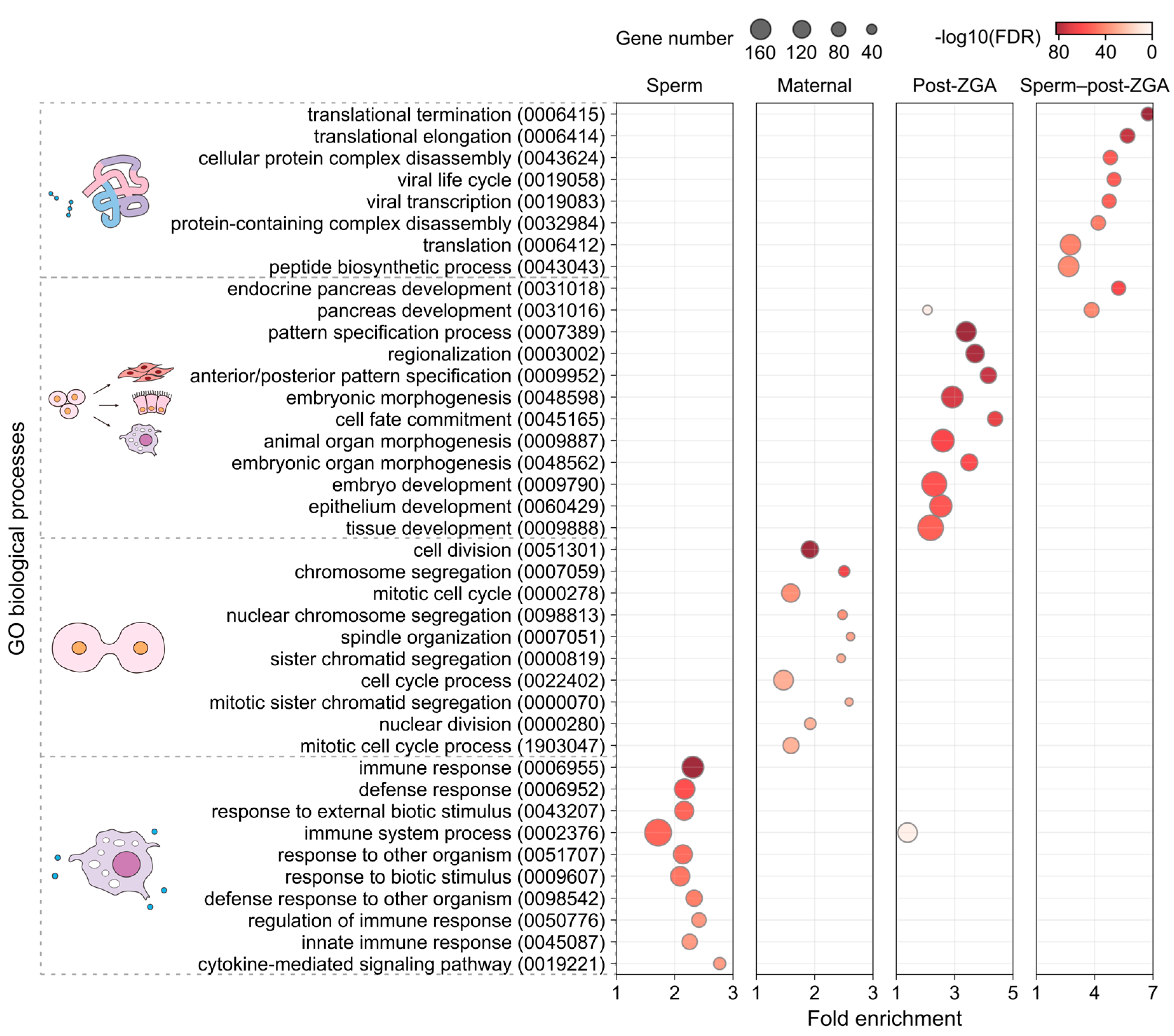{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling
2.2. RNA Extraction and RNA Sequencing
2.3. RNA-Seq Reads Filtering, Mapping, and Gene Expression Quantification
2.4. Differential Gene Expression Analysis
2.5. Gene Co-Expression Module Identification, Hub Genes Identification, and Functional Analysis
2.6. Homology Analysis between the Embryonic Transcriptome of Tongue Sole and Zebrafish
3. Results
3.1. RNA-Seq Data Exhibits High Sequencing Quality
3.2. Gene Expression Profile and Inferred Zygotic Genome Activation
3.3. Gene Co-Expression Modules and Corresponding Biological Functions
3.4. Comparative Analysis of Co-Expression Genes between Tongue Sole and Zebrafish
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhai, J.; Xiao, Z.; Wang, Y.; Wang, H. Human embryonic development: From peri-implantation to gastrulation. Trends Cell Biol. 2022, 32, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Pérez, J.A.; Hadjantonakis, A.-K. The Dynamics of Morphogenesis in the Early Mouse Embryo. Cold Spring Harb. Perspect. Biol. 2014, 7, a015867. [Google Scholar] [CrossRef] [PubMed]
- Arnold, S.J.; Robertson, E.J. Making a commitment: Cell lineage allocation and axis patterning in the early mouse embryo. Nat. Rev. Mol. Cell Biol. 2009, 10, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Jukam, D.; Shariati, S.A.M.; Skotheim, J.M. Zygotic Genome Activation in Vertebrates. Dev. Cell 2017, 42, 316–332. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.T.; Bonneau, A.R.; Giraldez, A.J. Zygotic genome activation during the maternal-to-zygotic transition. Annu. Rev. Cell Dev. Biol. 2014, 30, 581–613. [Google Scholar] [CrossRef] [PubMed]
- Fox, C.H.; Gibb, A.C.; Summers, A.P.; Bemis, W.E. Benthic walking, bounding, and maneuvering in flatfishes (Pleuronectiformes: Pleuronectidae): New vertebrate gaits. Zoology 2018, 130, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Holmes, R.A.; Gibson, R.N. A comparison of predatory behaviour in flatfish. Anim. Behav. 1983, 31, 1244–1255. [Google Scholar] [CrossRef]
- Janvier, P. Palaeontology: Squint of the fossil flatfish. Nature 2008, 454, 169–170. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhang, G.; Shao, C.; Huang, Q.; Liu, G.; Zhang, P.; Song, W.; An, N.; Chalopin, D.; Volff, J.-N.; et al. Whole-genome sequence of a flatfish provides insights into ZW sex chromosome evolution and adaptation to a benthic lifestyle. Nat. Genet. 2014, 46, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-L.; Li, J.; Deng, S.-P.; Tian, Y.-S.; Wang, Q.-Y.; Zhuang, Z.-M.; Sha, Z.-X.; Xu, J.-Y. Isolation of female-specific AFLP markers and molecular identification of genetic sex in half-smooth tongue sole (Cynoglossus semilaevis). Mar. Biotechnol. 2007, 9, 273–280. [Google Scholar] [CrossRef]
- Shao, C.-W.; Chen, S.-L.; Scheuring, C.F.; Xu, J.-Y.; Sha, Z.-X.; Dong, X.-L.; Zhang, H.-B. Construction of two BAC libraries from half-smooth tongue sole Cynoglossus semilaevis and identification of clones containing candidate sex-determination genes. Mar. Biotechnol. 2010, 12, 558–568. [Google Scholar] [CrossRef]
- Sha, Z.; Wang, S.; Zhuang, Z.; Wang, Q.; Wang, Q.; Li, P.; Ding, H.; Wang, N.; Liu, Z.; Chen, S. Generation and analysis of 10,000 ESTs from the half-smooth tongue sole Cynoglossus semilaevis and identification of microsatellite and SNP markers. J. Fish Biol. 2010, 76, 1190–1204. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Li, Y.; Zhao, Y.; Liu, Y.; Niu, Y.; Pang, R.; Miao, G.; Liao, X.; Shao, C.; Gao, F.; et al. Construction of a high-density microsatellite genetic linkage map and mapping of sexual and growth-related traits in half-smooth tongue sole (Cynoglossus semilaevis). PLoS ONE 2012, 7, e52097. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Shao, C.; Zhang, L.; Liu, K.; Gao, F.; Dong, Z.; Xu, P.; Chen, S. A first generation BAC-based physical map of the half-smooth tongue sole (Cynoglossus semilaevis) genome. BMC Genom. 2014, 15, 215. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wang, W.; Yi, Q.; Ma, L.; Zhou, X.; Zhao, H.; Wang, X.; Qi, J.; Yu, H.; Wang, Z.; Zhang, Q. Sequencing and characterization of the transcriptome of half-smooth tongue sole (Cynoglossus semilaevis). BMC Genom. 2014, 15, 470. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Lyu, L.; Wen, H.; Shi, B. Brain and Pituitary Transcriptome Analyses Reveal the Differential Regulation of Reproduction-Related LncRNAs and mRNAs in Cynoglossus semilaevis. Front. Genet. 2021, 12, 802953. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Lyu, L.; Zhang, D.; Li, J.; Wen, H.; Shi, B. Integrated lncRNA and mRNA Transcriptome Analyses in the Ovary of Cynoglossus semilaevis Reveal Genes and Pathways Potentially Involved in Reproduction. Front. Genet. 2021, 12, 671729. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Zheng, M.; Liu, J.; Liu, Y.; Lu, J.; Sun, X. Sexually Dimorphic Gene Expression Associated with Growth and Reproduction of Tongue Sole (Cynoglossus semilaevis) Revealed by Brain Transcriptome Analysis. Int. J. Mol. Sci. 2016, 17, 1402. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.-F.; Liu, Q.; Liu, K.-Q.; Wang, H.-Y.; Li, C.-H.; Wang, Q.; Shao, C.-W. Identification of global alternative splicing and sex-specific splicing via comparative transcriptome analysis of gonads of Chinese tongue sole (Cynoglossus semilaevis). Zool Res. 2022, 43, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Lin, G.; Gao, D.; Lu, J.; Sun, X. Transcriptome Profiling Reveals the Sexual Dimorphism of Gene Expression Patterns during Gonad Differentiation in the Half-Smooth Tongue Sole (Cynoglossus semilaevis). Mar. Biotechnol. 2021, 23, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Wang, W.; Zhang, Y.; Wang, L.; Cui, Y.; Li, H. Integrative analysis reveals pathways associated with sex reversal in Cynoglossus semilaevis. PeerJ 2020, 8, e8801. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Cui, Z.; Wang, N.; Zhang, M.; Wang, J.; Xu, X.; Liu, Y.; Chen, S. Transcriptomic analysis revealed gene expression profiles during the sex differentiation of Chinese tongue sole (Cynoglossus semilaevis). Comp. Biochem. Physiol. Part D Genom. Proteom. 2021, 40, 100919. [Google Scholar] [CrossRef] [PubMed]
- Shao, C.; Li, Q.; Chen, S.; Zhang, P.; Lian, J.; Hu, Q.; Sun, B.; Jin, L.; Liu, S.; Wang, Z.; et al. Epigenetic modification and inheritance in sexual reversal of fish. Genome Res. 2014, 24, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Wang, X.; Sha, Z.; Yang, C.; Liu, S.; Wang, N.; Chen, S.-L. Establishment and characterization of a testicular cell line from the half-smooth tongue sole, Cynoglossus semilaevis. Int. J. Biol. Sci. 2011, 7, 452–459. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.; Wang, T.Z.; Wang, N.; Liu, X.F.; Sha, Z.X.; Chen, S.L. Establishment and characterization of an ovarian cell line from half-smooth tongue sole Cynoglossus semilaevis. J. Fish Biol. 2015, 86, 46–59. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.L.; Wang, N.; Sha, Z.X.; Chen, S.L. Establishment, characterization of a new cell line from heart of half smooth tongue sole (Cynoglossus semilaevis). Fish Physiol. Biochem. 2010, 36, 1181–1189. [Google Scholar] [CrossRef] [PubMed]
- Sun, A.; Chen, S.-L.; Gao, F.-T.; Li, H.-L.; Liu, X.-F.; Wang, N.; Sha, Z.-X. Establishment and characterization of a gonad cell line from half-smooth tongue sole Cynoglossus semilaevis pseudomale. Fish Physiol. Biochem. 2015, 41, 673–683. [Google Scholar] [CrossRef]
- Zheng, Y.; Wang, N.; Xie, M.-S.; Sha, Z.-X.; Chen, S.-L. Establishment and characterization of a new fish cell line from head kidney of half-smooth tongue sole (Cynoglossus semilaevis). Fish Physiol. Biochem. 2012, 38, 1635–1643. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, Y.; Shi, C.; Huang, Z.; Zhang, Y.; Li, S.; Li, Y.; Ye, J.; Yu, C.; Li, Z.; et al. SOAPnuke: A MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 2018, 7, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Sirén, J.; Välimäki, N.; Mäkinen, V. Indexing Graphs for Path Queries with Applications in Genome Research. IEEE/ACM Trans. Comput. Biol. Bioinform. 2014, 11, 375–388. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Schurch, N.J.; Schofield, P.; Gierliński, M.; Cole, C.; Sherstnev, A.; Singh, V.; Wrobel, N.; Gharbi, K.; Simpson, G.G.; Owen-Hughes, T.; et al. How many biological replicates are needed in an RNA-seq experiment and which differential expression tool should you use? RNA 2016, 22, 839–851. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Yang, P.; Jiang, F.; Wei, Y.; Ma, Z.; Kang, L. De novo analysis of transcriptome dynamics in the migratory locust during the development of phase traits. PLoS ONE 2010, 5, e15633. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Beissbarth, T.; Speed, T.P. GOstat: Find statistically overrepresented Gene Ontologies within a group of genes. Bioinformatics 2004, 20, 1464–1465. [Google Scholar] [CrossRef] [PubMed]
- Lechner, M.; Findeiss, S.; Steiner, L.; Marz, M.; Stadler, P.F.; Prohaska, S.J. Proteinortho: Detection of (co-)orthologs in large-scale analysis. BMC Bioinform. 2011, 12, 124. [Google Scholar] [CrossRef] [PubMed]
- Levin, M.; Anavy, L.; Cole, A.G.; Winter, E.; Mostov, N.; Khair, S.; Senderovich, N.; Kovalev, E.; Silver, D.H.; Feder, M.; et al. The mid-developmental transition and the evolution of animal body plans. Nature 2016, 531, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Mateyak, M.K.; Kinzy, T.G. eEF1A: Thinking outside the ribosome. J. Biol. Chem. 2010, 285, 21209–21213. [Google Scholar] [CrossRef] [PubMed]
- Kimmel, C.B.; Ballard, W.W.; Kimmel, S.R.; Ullmann, B.; Schilling, T.F. Stages of embryonic development of the zebrafish. Dev. Dyn. 1995, 203, 253–310. [Google Scholar] [CrossRef] [PubMed]
- Hao, X.; Wang, Q.; Hou, J.; Liu, K.; Feng, B.; Shao, C. Temporal Transcriptome Analysis Reveals Dynamic Expression Profiles of Gametes and Embryonic Development in Japanese Flounder (Paralichthys olivaceus). Genes 2021, 12, 1561. [Google Scholar] [CrossRef] [PubMed]
- Niu, J.; Guan, J.; Li, R.; Li, X.; Zhai, J.; Qi, J.; He, Y. Cynoglossus semilaevis Rspo3 Regulates Embryo Development by Inhibiting the Wnt/β-Catenin Signaling Pathway. Int. J. Mol. Sci. 2018, 19, 1915. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Yang, F.; Li, R.; Li, X.; Wu, X.; Sun, Z.; Zhai, J.; He, Y.; Qi, J. Functional characterization of Cynoglossus semilaevis R-spondin2 and its role in muscle development during embryogenesis. Genes Genet. Syst. 2018, 93, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhu, H.; Liu, Y.; Qu, J.; Han, M.; Jin, C.; Zhang, Q.; Liu, J. Molecular characterization and expression profiles provide new insights into GATA5 functions in tongue sole (Cynoglossus semilaevis). Gene 2019, 708, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Deng, S.P.; Chen, S.L. cDNA cloning, tissues, embryos and larvae expression analysis of Sox10 in half-smooth tongue-sole, Cynoglossus semilaevis. Mar. Genom. 2008, 1, 109–114. [Google Scholar] [CrossRef]
- Bai, J.; Solberg, C.; Fernandes, J.M.O.; Johnston, I.A. Profiling of maternal and developmental-stage specific mRNA transcripts in Atlantic halibut Hippoglossus hippoglossus. Gene 2007, 386, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Aanes, H.; Winata, C.L.; Lin, C.H.; Chen, J.P.; Srinivasan, K.G.; Lee, S.G.P.; Lim, A.Y.M.; Hajan, H.S.; Collas, P.; Bourque, G.; et al. Zebrafish mRNA sequencing deciphers novelties in transcriptome dynamics during maternal to zygotic transition. Genome Res. 2011, 21, 1328–1338. [Google Scholar] [CrossRef] [PubMed]
- Kaitetzidou, E.; Xiang, J.; Antonopoulou, E.; Tsigenopoulos, C.S.; Sarropoulou, E. Dynamics of gene expression patterns during early development of the European seabass (Dicentrarchus labrax). Physiol. Genom. 2015, 47, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Kaitetzidou, E.; Katsiadaki, I.; Lagnel, J.; Antonopoulou, E.; Sarropoulou, E. Unravelling paralogous gene expression dynamics during three-spined stickleback embryogenesis. Sci. Rep. 2019, 9, 3752. [Google Scholar] [CrossRef] [PubMed]
- Soda, T.; Miyagawa, Y.; Fukuhara, S.; Tanaka, H. Physiological role of actin regulation in male fertility: Insight into actin capping proteins in spermatogenic cells. Reprod. Med. Biol. 2020, 19, 120–127. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, H.; Finkelstein, M. Actin cytoskeleton and sperm function. Biochem. Biophys. Res. Commun. 2018, 506, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Selvaraju, S.; Ramya, L.; Parthipan, S.; Swathi, D.; Binsila, B.K.; Kolte, A.P. Deciphering the complexity of sperm transcriptome reveals genes governing functional membrane and acrosome integrities potentially influence fertility. Cell Tissue Res. 2021, 385, 207–222. [Google Scholar] [CrossRef] [PubMed]
- Jodar, M.; Sendler, E.; Krawetz, S.A. The protein and transcript profiles of human semen. Cell Tissue Res. 2016, 363, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Sahoo, B.; Choudhary, R.K.; Sharma, P.; Choudhary, S.; Gupta, M.K. Significance and Relevance of Spermatozoal RNAs to Male Fertility in Livestock. Front. Genet. 2021, 12, 768196. [Google Scholar] [CrossRef] [PubMed]
- Schjenken, J.E.; Sharkey, D.J.; Green, E.S.; Chan, H.Y.; Matias, R.A.; Moldenhauer, L.M.; Robertson, S.A. Sperm modulate uterine immune parameters relevant to embryo implantation and reproductive success in mice. Commun. Biol. 2021, 4, 572. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Song, W.; Qu, J.; Liu, H.; Qi, J.; He, Y.; Niu, J. Transcriptome sequencing reveals ovarian immune response and development during female sperm storage in viviparous black rockfish (Sebastes schlegelii). Comp. Biochem. Physiol. Part D Genom. Proteom. 2023, 45, 101050. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheng, X.; Jiang, W.; Wang, Q.; Liu, K.; Dai, W.; Liu, Y.; Shao, C.; Li, Q. Unveiling Gene Expression Dynamics during Early Embryogenesis in Cynoglossus semilaevis: A Transcriptomic Perspective. Life 2024, 14, 505. https://doi.org/10.3390/life14040505
Cheng X, Jiang W, Wang Q, Liu K, Dai W, Liu Y, Shao C, Li Q. Unveiling Gene Expression Dynamics during Early Embryogenesis in Cynoglossus semilaevis: A Transcriptomic Perspective. Life. 2024; 14(4):505. https://doi.org/10.3390/life14040505
Chicago/Turabian StyleCheng, Xinyi, Wei Jiang, Qian Wang, Kaiqiang Liu, Wei Dai, Yuyan Liu, Changwei Shao, and Qiye Li. 2024. "Unveiling Gene Expression Dynamics during Early Embryogenesis in Cynoglossus semilaevis: A Transcriptomic Perspective" Life 14, no. 4: 505. https://doi.org/10.3390/life14040505
APA StyleCheng, X., Jiang, W., Wang, Q., Liu, K., Dai, W., Liu, Y., Shao, C., & Li, Q. (2024). Unveiling Gene Expression Dynamics during Early Embryogenesis in Cynoglossus semilaevis: A Transcriptomic Perspective. Life, 14(4), 505. https://doi.org/10.3390/life14040505